
Abstract
The endothelial transient receptor potential cation channel subfamily V member 4 (TRPV4) plays a crucial role in vascular remodeling; however, TRPV4-mediated angiogenesis after ischemic neuronal death as a neurorestorative strategy has not yet been thoroughly examined. In this study, we first tested whether TRPV4 activation can improve functional recovery in rats subjected to transient brain ischemia. The possible mechanisms for TRPV4 activation-promoted functional recovery were explored. A TRPV4 agonist, 4α-phorbol 12,13-didecanoate (4α-PDD), was intravenously injected via the tail vein at 6 h and 1, 2, 3, 4 days after ischemic stroke. The treatment reduced infarct volume by almost 50% (14.7 ± 3.7 vs. 29.2 ± 6.2%; p < 0.0001) and improved functional outcomes (p = 0.03) on day 5. To explore the therapeutic mechanism, we measured endothelial nitric oxide synthase (eNOS) expression and phosphorylation, vascular endothelial growth factor A (VEGFA) signaling, and neural stem/progenitor cells (NPCs). TRPV4 activation significantly increased eNOS expression and phosphorylation (serine 1177) by more than 2-fold in the ischemic region. The expressions of VEGFA and VEGF receptor-2 were significantly higher in the treated animals, especially an increase of the proangiogenic VEGFA164a isoform while a decrease of the antiangiogenic VEGFA165b isoform. We evaluated angiogenesis by detecting microvessel density in ischemic region. Using the immunohistochemistry staining, we found that 4α-PDD treatment caused a 3.4-fold increase of microvessel density (p < 0.0001). In addition, NPC proliferation and migration in the ischemic hemisphere were increased by 3-fold and 5-fold, respectively. In conclusion, our data suggest that TRPV4 activation by 4α-PDD may improve poststroke functional improvement through angiogenesis and neurogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Stroke is the leading cause of adult disability, and also associated with a limited degree of functional recovery [1]. With the advancement of the medical technology in the past decades, more and more stroke patients have survived from the initial injury. Sixty percent of survivors have disabilities in arm or leg, and up to one third needs to stay in a nursing home or to assistant device for independent living [2, 3]. Development of effective treatment or new therapeutic strategies for ischemic stroke patients is therefore crucial.
Brain neurorestoration leads to considerable poststroke functional recovery [4, 5]. The brain attempts to repair itself after an ischemic stroke by neurogenesis and angiogenesis [6]. Neural stem/progenitor cells (NPCs) and endothelial progenitor cells (EPCs) play important roles in neurogenesis and angiogenesis, respectively [5]. After stroke, NPCs migrate to the ischemic boundary where angiogenesis takes place, and NPCs migration is closely associated with cerebral vessels. Suppression of angiogenesis substantially reduces migration of newly formed NPCs to the ischemic region [5]. In addition to guiding NPCs migration, activated endothelial cells (ECs) secrete vascular endothelial growth factor A (VEGFA) to increase neurogenesis [4].
The transient receptor potential vanilloid 4 (TRPV4) cation channel, a member of the TRP vanilloid subfamily, is widely expressed in a broad range of tissues [7]. Previous studies have shown that TRPV4 channels possess multiple activation and regulatory sites to integrate distinct physical and chemical stimuli, and TRPV4 is involved in a wide range of physiological functions, such as cell proliferation, survival, differentiation, migration, and adhesion [8,9,10]. TRPV4 in ECs is involved in endothelium-dependent vasorelaxation via Ca2+-influx and phosphorylation of endothelial nitric oxide synthase (eNOS) serine 1177 [11]. Endothelial TRPV4-mediated Ca2+ inflow also contributes to ECs proliferation and differentiation [10, 12].
Fluid shear stress (FSS) leads to the development of collateral flow conductance and the remodeling of collateral vessels [13, 14]. Endothelial TRPV4 plays a crucial role in vascular remodeling because it can transmit circumferential wall FSS to an active intracellular growth response [15]. In addition, VEGFA, blood–brain barrier (BBB) integrity, blood vessel growth, and vasodilatation are also increased by FSS [16], which are critical for neurogenesis and neuroplasticity. Therefore, this study aimed to evaluate whether TRPV4 activation by a TRPV4 agonist can promote poststroke functional recovery via angiogenesis and neurogenesis.
Materials and Methods
Animals
All experimental procedures were approved by the Institutional Animal Ethical Committee Kaohsiung Medical University and were conducted according to the Guide for the Care and Use of Laboratory Animal of the National Institute of Health. Sprague-Dawley rats (280–350 g) were subjected to transient cerebral ischemia by right transient middle cerebral artery occlusion (tMCAO). In brief, rats were immobilized with isoflurane for the intraperitoneal (i.p.) injection of Equithesin (4 ml/kg) and the body temperature was maintained at 37 ± 0.5 °C by a heating device. A midline incision along the ventral neck was made to expose the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA). A 5–0 silk suture was ligated on the CCA and the ECA. A small vascular clip was clamped between the CCA bifurcation and the ligature to prevent the backward flush of blood from ICA. Thereafter, a small incision was made on the CCA to permit the insertion of a 3–0 nylon filament with silicon modification on the tip. This nylon filament was advanced approximately 22 mm beyond the CCA bifurcation. Reperfusion was established by gently withdrawing the filament after 120 min of occlusion. Free access to food and water was allowed after recovery from anesthesia (Supplementary Fig. 1).
The neurological deficits were evaluated using the neurological deficits score 6 h post-tMCAO [17]. The neurological deficits scores are as follows: 0, no neurological symptoms; 1, unable to extend left forepaw fully; 2, reduced grip of the left forelimb; 3, torso turning to the left side when held by tail; 4, circling or walking to the left; 5, failure to walk without help; 6, no spontaneous activity or narcosis; and 7, dead. The rats with scores 2–5 were eligible for further studies and were randomly divided into groups.
Functional Assessment
The Garcia score (GS) was used to evaluate the functional recovery at 6 h and 3 and 5 days after tMCAO as described earlier [18]. The rats were evaluated by six tests: spontaneous activity, movement symmetry of four limbs, forepaw outstretching, climbing, body proprioception, and vibrissae touch tests. The score from each test was summed up to the GS score with the range from 0 to 18 (from maximal deficit to normal). Mild neurological dysfunction is defined as a score between 13 and 18, moderate neurological dysfunction as a score between 7 and 12, and severe neurological dysfunction as a score between 1 and 6.
Drug Treatment
The phorbol ester, 4α-phorbol-12,13-didecanoate (4α-PDD, Sigma-Aldrich), was used as a TRPV4 agonist. 4α-PDD was dissolved in DMSO (5% v/v) right before administration to animals and then IV injection (0.1 mg/kg/day). The initial dose (6 h) was given after rats were evaluated for the GS. To mimic the clinical setting, we decided to treat animals daily before the end of study (i.e., day 5). The daily drug dosage was derived from a previous study where continuous infusion was used [15]. Therefore, the treatment was given at 6 h and 1, 2, 3, and 4 days after tMCAO. Control animals received the same dosage DMSO without 4α-PDD.
Infarct Volume Measurement
Five days after tMCAO, stroke rats were sacrificed and their brains were dissected from the cranium and immersed in cold (4 °C) saline for 5 min. Each rat brain was cut into 2 mm coronal sections for a total of eight slices of coronal sections. The brain slices were stained with 0.1% 2,3,5-triphenyltetrazolium chloride, and the viable brain parenchyma was stained in red and the infarct region in pale white. The infarct area was calculated using ImageJ (NIH) to calculate the infarct area and the percentage of infarction. The percentage of infarct volume was determined according to an indirect method: Infarct volume = (area of contralateral hemisphere − area of normal region in the ipsilateral hemisphere) / area of contralateral hemisphere × 100%.
RNA Isolation and Quantitative Real-Time Reverse Transcription-PCR
Total RNA extraction from brain tissues was carried out using TRIzol according to the manufacturer’s instructions. Complementary DNA (cDNA) was synthesized from 1 μg total RNA using random primer and the MultiScribe reverse transcriptase kit.
For quantitative real-time PCR, specific primers for all rat eNOS, VEGFA, VEGFA164a, VEGFA165b, VEGF receptor-2 (VEGFR2), and GAPDH were designed (supplementary Table S1). Relative quantification of gene expression was performed with preoptimized conditions using the ABI 7900 real-time PCR machine (Applied Biosystems). PCRs were performed in duplicate using 5 μl 2× SYBR Green PCR Master Mix, 0.2 μl primer sets, 1 μl cDNA, and 3.6 μl nuclease-free H2O to yield a 10-μl reaction. The expression ratios were calculated as the normalized CT difference between control and sample, with adjustment for amplification efficiency relative to the expression level of GAPDH.
Protein Isolation and Western Blotting
Brain tissues were homogenized in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, and 50 mM Tris) (GeneTex), and insoluble constituents were removed by centrifugation. Protein lysates were denatured and loaded onto a 4–12% SDS polyacrylamide gel. The separated proteins were then transferred onto a PVDF membrane (Merck Millipore) and blocked with 5% non-fat dry milk for 1 h at room temperature. The membrane was incubated overnight at 4 °C in 5% non-fat dry milk /PBST containing the primary antibodies. Primary antibodies against phospho-ser1177-eNOS (1:125, Abgent), VEGFA (1:125, EMD Millipore), VEGFA165b (8 μg/ml, EMD Millipore), VEGFR2 (1:25, Abcam), and GAPDH (0.25 μg/ml, EMD Millipore) were used. The membrane was incubated with the secondary antibody conjugated to horseradish peroxidase. The ECL non-radioactive detection system was used to detect the antibody–protein complexes by LAS-3000 imaging system (Fujifilm). Blot intensity was semi-quantitatively measured using ImageJ (NIH).
Immunohistochemistry
Rats were sacrificed at day 5 after tMCAO and perfused transcardially with 0.9% saline at 4 °C followed by 4% paraformaldehyde in phosphate buffer (0.1 mol/l, pH 7.4). The brains were removed, fixed in the above fixation for 8 h at 4 °C, and then immersed sequentially in 20 and 30% sucrose until sinking occurred. Coronal sections (10 μm thick) were selected from bregma 1.0 to −0.20 mm. Primary antibodies and dilutions used in immunostaining were CD31 (1:200, GeneTex) and Sox2 (1:200, Proteintech). For immunostaining, sections were first treated with 3% H2O2 for 20 min and incubated with block reagent for 1 h at room temperature, and were then incubated with primary antibody for 2 h, followed by incubation with biotinylated secondary antibody (1:200, BioTnA).
Immunostaining images were obtained with a TS100 Inverted Biological Microscope (Nikon). Every three coronal sections from bregma 1.0 to −0.20 mm of each rat brain following immunostaining were taken. Microvessel density as an index of angiogenesis, defined as follows: CD31-positive cells area / total surface area of each section × 100%, in the ischemic penumbra was counted in each of the five randomly magnified (×400) fields. The number of Sox2-positive cells in the SVZ was estimated using a ×400 magnification objective and in the ischemic penumbra using the three randomly magnified (×400) fields. Images were processed using ImageJ (NIH).
Statistical Analysis
Data are presented as means ± standard error of the mean (SEM). Statistical differences between groups were assessed by Mann-Whitney U test. p < 0.05 was considered statistically significant.
Results
TRPV4 Activation by 4α-PDD Reduces Infarct Volumes and Improves Functional Outcomes
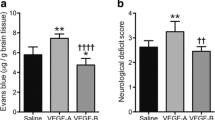
4α-PDD is a TRPV4 agonist. Based on the neurological deficits score measured at 6 h poststroke, a total of 49 rats were eligible for the experiments. Among these 49 rats, 13 rats died (5 rats in the treated group and 8 rats in control group) before the end of the study, which led to 18 rats in each group in the end of the study. The data on 18 rats of each group were used for analyses at all time points including 6 h, day 3, and day 5. The mortality rates were no significant difference between the two groups (supplementary Fig. 2). Compared to the control group, 4α-PDD treatment significantly reduced the infarct volume (14.7 ± 3.7 vs. 29.2 ± 6.2%; p < 0.0001) in stroke rats (Fig. 1a). In addition, 4α-PDD treatment significantly promoted functional outcomes on day 5 after tMCAO (p < 0.05; Fig. 1b). Among six GS tests, three tests were statistically different on day 5 (supplementary Table S2).

TRPV4 activation by 4α-PDD reduces the infarct volume and improves neurological recovery. a Quantitation of the infarct volume showed that 4α-PDD mediated TRPV4 activation significantly reduced infarct volume on day 5 post-tMCAO. All data are represented as means ± SEM. n = 9 per group. b tMCAO caused a markedly neurological deficit, and 4α-PDD-mediated TRPV4 activation improved the Garcia score on day 5 after tMCAO. All data are represented as means ± SEM. n = 18 per group
TRPV4 Activation by 4α-PDD Increases eNOS, VEGFA, and VEGFR2 Expressions
The mRNA expression level of eNOS was higher in the infarct hemisphere of 4α-PDD-treated rats than the placebo-treated rats by 2.7-fold (Fig. 2a). Similarly, the level of eNOS phosphorylation (serine 1177) of the infarct hemisphere was higher in 4α-PDD group than the control group by 2.49-fold (Fig. 2b, c).

TRPV4 activation by 4α-PDD promotes eNOS expression and function. a 4α-PDD-mediated TRPV4 activation significantly increased eNOS mRNA level in the infarct hemisphere (n = 9 per group). b Representative images show that 4α-PDD treatment caused enhanced expression of eNOS phosphorylation (serine 1177). c Semi-quantitative analysis revealed greater eNOS phosphorylation (serine 1177) expression after 4α-PDD-mediated TRPV4 activation (n = 6 per group). All data are represented as means ± SEM
We further tested whether VEGFA was also increased by TRPV4 activation, and the result showed that VEGFA level in infarct hemisphere was significantly higher by 2.7-fold in the 4α-PDD group than the control group (Fig. 3a). Two VEGFA isoforms, pro-angiogenic VEGFA164a isoform and antiangiogenic VEGFA165b isoform, were specifically measured. We found that the increase of VEGFA mRNA was primarily caused by the pro-angiogenic isoform VEGFA164a (a 2.8-fold increase) in the infarct hemisphere of 4α-PDD-treated rats (Fig. 3a), while no significant change of VEGFA165b level (Fig. 3a). Furthermore, VEGFR2 mRNA was also significantly increased by 2.6-fold in the infarct hemisphere of 4α-PDD-treated rats (Fig. 3a). Consistently, the protein amounts of VEGFA and VEGFR2 were increased by 4α-PDD treatment, while VEGFA165b protein reduced significantly (Fig. 3b). There were 1.39-fold increases of VEGFA protein, 5.38-fold increase of VEGFR2 protein, and 0.43-fold decrease of VEGFA165b protein (Fig. 3c). These data suggests that 4α-PDD treatment can activate TRPV4 to influence VEGFA-VEGFR2 expression. Because of no commercially available antibody for VEGFA164a, no VEGFA164a protein data could be presented in Fig. 3.

TRPV4 activation by 4α-PDD enhances VEGFA-VEGFR2 expression. a mRNA levels of VEGFA, VEGFA164a, and VEGFR2 were elevated after 4α-PDD treatment. 4α-PDD treatment non-significantly increased VEGFA165b (n = 9 per group). b Representative images show that 4α-PDD treatment caused higher expression of VEGFA and VEGFR2 protein and lower expression of VEGFA165b protein. c Semi-quantitative analysis of VEGFA and VEGFR2 protein increased after 4α-PDD treatment. VEGFA165b protein level reduced significantly in the 4α-PDD group (n = 6 per group). All data are represented as means ± SEM
TRPV4 Activation by 4α-PDD Promotes the Angiogenesis Around Ischemic Region
Microvessels were identified by the CD31 monoclonal antibody in the penumbra around the infarct region. In the control group, the density of microvessels in the penumbra was significantly lower than that in the 4α-PDD group in penumbra (2.08 ± 1.48% in control group vs. 7.16 ± 3.44% in 4α-PDD group; p < 0.0001; Fig. 4).

TRPV4 activation by 4α-PDD increases microvessel density. a Representative images show that the 4α-PDD treatment caused more intensive microvessels (CD31+ endothelial cells, arrows) in peri-lesional area than the control group on day 5 post-tMCAO. b Quantification of microvessel density, represented as percentage of CD31+-stained cells, significantly increased after 4α-PDD treatment. All data are represented as means ± SEM. n = 9 per group
TRPV4 Activation by 4α-PDD Enhances Poststroke Neurogenesis
We used Sox2 as the marker to identify neural stem cells. The number of Sox2+ cells in the ipsilateral subventricular zone (SVZ) of the lateral ventricle was significantly higher (Fig. 5a, b) in the 4α-PDD group than the control group (53.22 ± 18.69 vs. 16.22 ± 9.0; p < 0.001) on day 5. In addition, the 4α-PDD treatment increased Sox2+ cells in the peri-infarct area (15.44 ± 9.27 in 4α-PDD group vs. 3.07 ± 2.88 in control group; p < 0.0001) (Fig. 5c, d).

TRPV4 activation by 4α-PDD increases NPCs proliferation and migration. a, c Representative images show that in the 4α-PDD group, intensive NPCs (Sox2+ cells, arrows) were found in the ipsilateral SVZ (a) and peri-infarct area (c) on day 5 post-tMCAO. In contrast, in control group, the density of NPCs was much lower than the 4α-PDD group. b, d Quantification showed that 4α-PDD treatment significantly enhanced Sox2+ cells in the ipsilateral SVZ (b) and peri-infarct area (d). All data are represented as means ± SEM. n = 9 per group
Discussion
This present study shows that TRPV4 activation by 4α-PDD can reduce brain infarction, augment angiogenesis, and promote neurogenesis leading to better functional recovery. Our findings are schematically summarized in Fig. 6. The beneficial effects of 4α-PDD treatment may be mediated by several pathways including upregulation of eNOS to increase NO levels, an increase of VEGFA–VEGFR2 pathway to promote neovascularization and activation of NPCs for neurogenesis. TRPV4 is highly expressed in ECs but its role in poststroke angiogenesis and neurogenesis has been barely explored. Although TRPV4 has been considered to be activated by FSS, the present study showed that a chemical stimulation can mimic FSS effect on activation of TRPV4. Our result indicates an opportunity of developing TRPV4 stimulant to treat acute ischemic stroke.

Graphic representation of the effects of TRPV4
Several regulatory pathways contribute to the pro-angiogenic effects of TRPV4. Neovascularization as indicated by the increase of microvessel density could be attributed to upregulation of pro-angiogenic genes (eNOS, VEGFA, and VEGFR2). First, NO can stimulate EC proliferation and migration, and mediate progenitor cell mobilization, all of which cause neovascularization [19, 20]. Secondly, TRPV4 activation increases both VEGFA and VEGFR2 simultaneously, which are ligand and receptor, respectively. VEGFA and VEGFR2 are important determinants in pro-angiogenic signaling and play key roles in promoting angiogenesis after stroke [5, 21]. Thirdly, TRPV4 activation elevates the proangiogenic VEGFA splice isoform (VEGFA164a) while reduces the antiangiogenic VEGFA165b splice isoform. Because of the critical role of VEGFA in ischemic diseases, there were several clinical trials of VEGFA therapy. However, these clinical interventions revealed limited efficacy in stroke [22] and in PAD [23]. The unsatisfied results from clinical trials may be partially due to VEGFA contains pro- and antiangiogenic isoforms [22]. Recent works also have identified that although higher circulating levels of total VEGFA are found in PAD, pro-angiogenic VEGFA164a is reduced while antiangiogenic VEGFA165b is elevated [24]. Notably, VEGFA165b competes with VEGFA164a for the binding to VEGFR2 [25]. TRPV4 activation was also shown to facilitate proliferating and sprouting of ECs. The activation of TRPV4 may change Ca2+-dependent signaling in human brain ECs to affect angiogenesis [10], and can also influence EPC proliferation [12].
The exact mechanism for NPCs activation by TRPV4 activation is not clear. Ca2+ influx through TRPV4 may also stimulate ECs to produce and release certain factors (i.e. NO and VEGFA), which consequently facilitate neurotrophic activities [20, 26,27,28]. NO has been shown to stimulate epidermal growth factor receptor to increase NPCs proliferation [29, 30] and neuroblast migration [20]. On the contrary, eNOS-deficient mice exhibit reduced poststroke NPCs proliferation [31]. Further works are warranted to elucidate how TRPV4 activation increases neurogenesis.
This is the first demonstration of the importance of TRPV4 activation in stroke treatment. Compared to intra-cerebroventricular activation of TRPV4 [32, 33], our approach provides neuroprotection and a more clinical feasible route. Using growth factors to treat acute stroke may lead to uncontrolled vessel growth, BBB leakage, and neuronal damage [34], while our study using TRPV4 activation does not show such unwanted effects. Furthermore, given that endothelial TRPV4 activation is one mechanism underlying FSS effects, chemical induced- TRPV4 activation may become an alternative for patients who have difficult for exercise in the acute stroke.
In conclusion, TRPV4 activation increases recovery of neurological function, decreases brain infarction size, and enhances the angiogenesis and neurogenesis in ischemic stroke rats. These effects may be mediated through upregulation of the eNOS, VEGFA164a, and VEGFR2. The results implied the potential clinical usefulness of TRPV4 activation in ischemic stroke. Further studies to replicate the results and explore safety issues are warranted before it can be applied to clinical practice.
Abbreviations
- 4α-PDD:
-
4α-Phorbol 12,13-didecanoate
- BBB:
-
Blood–brain barrier
- EPCs:
-
Endothelial progenitor cells
- ECs:
-
Endothelial cells
- FSS:
-
Fluid shear stress
- GS:
-
Garcia score
- eNOS:
-
Endothelial nitric oxide synthase
- NPCs:
-
Neural stem/progenitor cells
- NO:
-
Nitric oxide
- SVZ:
-
Subventricular zone
- tMCAO:
-
Transient middle carotid artery occlusion
- TRPV4:
-
Transient receptor potential vanilloid 4
- VEGFA:
-
Vascular endothelial growth factor A
- VEGFR2:
-
VEGF receptor-2
References
Mozaffarian D, Benjamin EJ, Go AS et al (2016) Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation 133:e38–e360
Meyer S, Verheyden G, Brinkmann N et al (2015) Functional and motor outcome 5 years after stroke is equivalent to outcome at 2 months: Follow-up of the collaborative evaluation of rehabilitation in stroke across Europe. Stroke 46:1613–1619
Cowman S, Royston M, Hickey A et al (2010) Stroke and nursing home care: a national survey of nursing homes. BMC Geriatr 10:4
Teng H, Zhang ZG, Wang L et al (2008) Coupling of angiogenesis and neurogenesis in cultured endothelial cells and neural progenitor cells after stroke. J Cereb Blood Flow Metab 28:764–771
Zhang ZG, Chopp M (2009) Neurorestorative therapies for stroke: underlying mechanisms and translation to the clinic. Lancet Neurol 8:491–500
Kahle MP, Bix GJ (2013) Neuronal restoration following ischemic stroke: influences, barriers, and therapeutic potential. Neurorehabil Neural Repair 27:469–478
Everaerts W, Nilius B, Owsianik G (2010) The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol 103:2–17
Troidl C, Troidl K, Schierling W et al (2009) Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J Cell Mol Med 13:2613–2621
Zaninetti R, Fornarelli A, Ciarletta M et al (2011) Activation of TRPV4 channels reduces migration of immortalized neuroendocrine cells. J Neurochem 116:606–615
Hatano N, Suzuki H, Itoh Y et al (2013) TRPV4 partially participates in proliferation of human brain capillary endothelial cells. Life Sci 92:317–324
Randhawa PK, Jaggi AS (2015) TRPV1 and TRPV4 channels: potential therapeutic targets for ischemic conditioning-induced cardioprotection. Eur J Pharmacol 746:180–185
Dragoni S, Guerra G, Fiorio Pla A et al (2015) A functional transient receptor potential vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J Cell Physiol 230:95–104
Schmidt W, Endres M, Dimeo F et al (2013) Train the vessel, gain the brain: physical activity and vessel function and the impact on stroke prevention and outcome in cerebrovascular disease. Cerebrovasc Dis 35:303–312
Liu J, Wang Y, Akamatsu Y et al (2014) Vascular remodeling after ischemic stroke: mechanisms and therapeutic potentials. Prog Neurobiol 115:138–156
Schierling W, Troidl K, Apfelbeck H et al (2011) Cerebral arteriogenesis is enhanced by pharmacological as well as fluid-shear-stress activation of the Trpv4 calcium channel. Eur J Vasc Endovasc Surg 41:589–596
Wragg JW, Durant S, McGettrick HM et al (2014) Shear stress regulated gene expression and angiogenesis in vascular endothelium. Microcirculation 21:290–300
Zhang L, Hu X, Luo J et al (2013) Physical exercise improves functional recovery through mitigation of autophagy, attenuation of apoptosis and enhancement of neurogenesis after MCAO in rats. BMC Neurosci 14:1
Ke Z, Yip S, Li L et al (2011) The effects of voluntary, involuntary, and forced exercises on brain-derived neurotrophic factor and motor function recovery: a rat brain ischemia model. PLoS One 6:e16643
Zhu J, Song W, Li L et al (2016) Endothelial nitric oxide synthase: a potential therapeutic target for cerebrovascular diseases. Mol Brain 9:30
Gertz K, Endres M (2008) eNOS and stroke: prevention, treatment and recovery. Future Neurol 3:537–550
Gao Y, Zhao Y, Pan J et al (2014) Treadmill exercise promotes angiogenesis in the ischemic penumbra of rat brains through caveolin-1/VEGF signaling pathways. Brain Res 1585:83–90
Mac Gabhann F, Qutub AA, Annex BH et al (2010) Systems biology of pro-angiogenic therapies targeting the VEGF system. Wiley Interdiscip Rev Syst Biol Med 2:694–707
Rajagopalan S, Mohler ER, Lederman RJ et al (2003) Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation 108:1933–1938
Kikuchi R, Nakamura K, MacLauchlan S et al (2014) An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat Med 20:1464–1471
Harper SJ, Bates DO (2008) VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat Rev Cancer 8:880–887
Sawada M, Sawamoto K (2013) Mechanisms of neurogenesis in the normal and injured adult brain. Keio J Med 62:13–28
Saha B, Peron S, Murray K et al (2013) Cortical lesion stimulates adult subventricular zone neural progenitor cell proliferation and migration to the site of injury. Stem Cell Res 11:965–977
Christie KJ, Turnley AM (2013) Regulation of endogenous neural stem/progenitor cells for neural repair-factors that promote neurogenesis and gliogenesis in the normal and damaged brain. Front Cell Neurosci 6:70
Carreira BP, Morte MI, Inácio  et al (2010) Nitric oxide stimulates the proliferation of neural stem cells bypassing the epidermal growth factor receptor. Stem Cells 28:1219–1230
Zhang RL, Zhang Z, Zhang L et al (2006) Delayed treatment with sildenafil enhances neurogenesis and improves functional recovery in aged rats after focal cerebral ischemia. J Neurosci Res 83:1213–1219
Chen J, Zacharek A, Zhang C et al (2005) Endothelial nitric oxide synthase regulates brain derived neurotrophic factor expression and neurogenesis after stroke in mice. J Neurosci 25:2366–2375
Jie P, Lu Z, Hong Z et al (2016) Activation of transient receptor potential vanilloid 4 is involved in neuronal injury in middle cerebral artery occlusion in mice. Mol Neurobiol 53:8–17
Li L, Qu W, Zhou L et al (2013) Activation of transient receptor potential vanilloid 4 increases NMDA-activated current in hippocampal pyramidal neurons. Front Cell Neurosci 7:17
Navaratna D, Guo S, Arai K et al (2009) Mechanisms and targets for angiogenic therapy after stroke. Cell Adhes Migr 3:216–223
Acknowledgements
We thank JY. Wang (Division of Colorectal Surgery, Department of Surgery, Kaohsiung Medical University) for technical support. This work was supported by the following funding: Ministry of Science and Technology (Taiwan, R.O.C.; MOST 103-2314-B-075-076-MY3, MOST 104-2745-B-037-001, MOST 105-2314-B-039-050, MOST 103-2314-B-037-027-MY2) and National Health Research Institutes (Taiwan, R.O.C.; NHRI-EX106-10605PI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All experimental procedures were approved by the Institutional Animal Ethical Committee Kaohsiung Medical University and were conducted according to the Guide for the Care and Use of Laboratory Animal of the National Institute of Health.
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic Supplementary Material
Supplementary Fig. 1
Schematic illustration of the experimental design. Transient middle cerebral artery occlusion (tMCAO) was inducted by 2-h occlusion in right internal carotid artery. 4a-phorbol-12,13-didecanoate (4α-PDD) was used as an agonist for endothelial transient receptor potential vanilloid 4.* represents the test points for neurological deficits score (NDS). ** represents the test points for infarct volume. # represents the test points for Garcia score (GS). (GIF 15 kb)
Supplementary Fig. 2
Delayed deaths are not observed with 4α-PDD treatment. Mortality was assessed during 5 d after tMCAO in 4α-PDD group (n = 23), and control group (treated with DMSO, n = 26). Mortality at day 5 was not significantly different. (GIF 34 kb)
Supplementary Table S1
(DOCX 13 kb)
Supplementary Table S2
(DOCX 14 kb)
Rights and permissions
About this article

Cite this article
Chen, CK., Hsu, PY., Wang, TM. et al. TRPV4 Activation Contributes Functional Recovery from Ischemic Stroke via Angiogenesis and Neurogenesis. Mol Neurobiol 55, 4127–4135 (2018). https://doi.org/10.1007/s12035-017-0625-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0625-0