
Abstract
Ischemic stroke triggers endogenous angiogenic mechanisms, which correlates with longer survival in patients. As such, promoting angiogenesis appears to be a promising approach. Experimental studies investigated mostly the potent angiogenic factor vascular endothelial growth factor isoform-A (VEGF-A). However, VEGF-A increases the risk of destabilizing the brain microvasculature, thus hindering the translation of its usage in clinics. An attractive alternative VEGF isoform-B (VEGF-B) was recently reported to act as a survival factor rather than a potent angiogenic factor. In this study, we investigated the therapeutic potential of VEGF-B in ischemic stroke using different in vivo and in vitro approaches. We showed that the delayed intranasal administration of VEGF-B reduced neuronal damage and inflammation. Unexpectedly, VEGF-B stimulated the formation of stable brain microvasculature within the injured region by promoting the interaction between endothelial cells and pericytes. Our data indicate that the effects of VEGF-B were mediated via its specific receptor VEGF receptor-1 (VEGFR-1) that is predominately expressed in brain pericytes. Importantly, VEGF-B promoted the survival of pericytes, and not brain endothelial cells, by inducing expression of the anti-apoptotic protein B-cell lymphoma 2 (Bcl-2) and the main protein involved in energy homeostasis AMP-activated protein kinase α (AMPKα). Moreover, we showed that VEGF-B stimulated the pericytic release of factors stimulating a “reparative angiogenesis” that does not compromise microvasculature stability. Our study unraveled hitherto unknown role of VEGF-B/VEGFR-1 signaling in regulating the function of pericytes. Furthermore, our findings suggest that brain microvasculature stabilization via VEGF-B constitutes a safe therapeutic approach for ischemic stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Brain microvasculature has an intimate functional relationship with the brain parenchyma [1]. This relationship is governed by the neurovascular unit (NVU), which comprises tightly sealed endothelial cells, forming the blood-brain barrier (BBB), that interact with extracellular matrix proteins, pericytes, astrocyte end-feet, microglia, and neurons [2, 3]. The NVU is a site of dynamic and complex biochemical and cellular interactions that work in concert to enable proper brain homeostasis and functioning [2–4]. Pericytes are tightly associated to brain endothelial cells covering more than 80% of the brain microvasculature, which is the highest coverage rate observed among all other organs [5, 6]. These cells have been shown to possess broad cellular functions that include angiogenesis, microvasculature stability, BBB maintenance, and cerebral blood flow (CBF) regulation [5, 6]. Ischemic stroke triggers pericyte death and detachment from brain endothelial cells in the acute phase (minutes to hours after stroke onset), thus destabilizing the microvasculature and altering BBB properties [7–10]. These events contribute to the progression of secondary brain injury by increasing brain edema and exacerbating the inflammatory response in the sub-acute phase (hours to days after stroke onset) [7, 10, 11].
Ischemic stroke triggers the formation of new microvasculature in the peri-infarct region (area surrounding infarct core) to compensate for disruption of the CBF by activating several angiogenic mechanisms [12]. Importantly, neuronal survival is greatest in ischemic tissue undergoing angiogenesis, and the number of new microvasculature within the ischemic brain correlates with longer survival in stroke patients [13]. Based on these observations, several angiogenic factors have been investigated in experimental studies, namely, VEGF-A, which potently promotes angiogenesis [14]. These studies have demonstrated that VEGF-A can indeed promote neuroprotection and neurorestoration following ischemic stroke [14, 15]. Nonetheless, the mechanisms underlying the effects of VEGF-A remain elusive [14]. In this regard, we have previously demonstrated that the prophylactic intracerebral administration of VEGF-A increased the coverage of ischemic brain microvasculature by pericytes and enhanced their physical interaction with endothelial cells by increasing expression of the N-cadherin, a protein involved in cell-cell cross talk [9]. However, the administration of VEGF-A is a double-edged sword strategy, as it significantly increases the risk of exacerbating vascular permeability by further destabilizing the BBB [16], thereby hindering the translation of its usage into clinics.
Although promising, a key question that remains to be answered if this approach is to be translated to clinics is how the safety of pro-angiogenic strategies can be improved. An attractive possibility is to use VEGF-B that has been shown to act as a “survival,” rather than a potent “angiogenic” factor [17], thus offering new perspectives for ischemic stroke therapies. VEGF-A mediates most of its biological activity via VEGF receptor 2 (VEGFR-2), whereas VEGF-B specifically binds VEGFR-1 [18]. Compared with the other members of the VEGF family, VEGF-B has received much less attention. Thereby, the biological relevance of VEGF-B signaling through VEGFR-1 in several cell types is poorly understood. Very few experimental studies examined the role of VEGF-B in ischemic stroke, although VEGF-B expression has been reported to increase in the peri-infarct region after middle cerebral artery occlusion (MCAo) [19]. One study showed that the depletion of VEGF-B increased brain damage and neurological deficits in mice subjected to permanent MCAo [20]. Another study showed that the intracerebral injection of VEGF-B in VEGFB−/− mice before subjecting mice to permanent MCAo rescued neuronal survival within the peri-infarct region [21]. These studies demonstrated that VEGF-B signaling through VEGFR-1 inhibited the expression of genes encoding for pro-apoptotic proteins. In contrast to VEGFR-2 that is expressed mainly on endothelial cells, VEGFR-1 has been reported to be expressed in pericytes. Whether VEGF-B regulates the function of pericytes after ischemic stroke remains totally unknown. Furthermore, although the existing investigations outlined the protective role of VEGF-B once stroke occurs, its potential in a therapeutic perspective after ischemic stroke remains to be addressed.
In this study, we evaluated the therapeutic potential of VEGF-B administration in the sub-acute phase on neuronal damage, neurological deficits, angiogenesis, and microvasculature stability by deciphering the role of VEGF-B/VEGFR1 signaling in brain pericytes.
Materials and Methods
Animal Experiments
Experiments were performed according to the Canadian Council on Animal Care guidelines, as administered by the Laval University Animal Welfare Committee. Adult C57BL6/j male mice (3 months old) were subjected to focal ischemic stroke via the transient occlusion of the MCA using an intraluminal filament technique as described [22]. Briefly, under 1.5% isoflurane (30% O2, remainder N2O) anesthesia, a midline neck incision was made and the left common and external carotid arteries were isolated and ligated. A microvascular clip was placed on the internal carotid artery, and a 7–0 silicon-coated nylon monofilament (Doccol Corporation, MA, USA) was directed through the internal carotid artery until the origin of MCA. The monofilament was left in place for 45 min and then withdrawn. Throughout the surgery, the rectal temperature was maintained between 36 and 37 °C using a feedback-controlled heating system (Harvard Apparatus, QC, Canada). During the experiment, laser Doppler flow (LDF) was monitored using a flexible fiber optic probe (Moor Instruments Inc., DE, USA) attached to the skull overlying the core of the MCA territory. One set of mice (n = 6 per group) was intravenously treated 1 h after MCAo induction with (i) saline, (ii) VEGF-A (PeproTech, NJ, USA; 15 μg/kg), or (iii) VEGF-B (PeproTech, NJ, USA; 15 μg/kg). This dose of VEGF-A has been previously reported to induce brain vascular permeability after experimental ischemic stroke [23]; thereby, a similar dose of VEGF-B was used. Twenty-four hours after MCAo induction, mice were sacrificed via a transcardiac perfusion with ice-cold normal saline. Three hours before sacrifice, mice were intraperitoneally treated with 2% Evans blue solution (4 mL/kg).
Another set of mice (n = 6–9 per group) was intranasally treated starting at 24 h after MCAo induction with (i) saline or (ii) VEGF-B (0.16 mg/kg/dose) daily for three successive days (a total dose of 12 μg/mouse was delivered). Previous studies demonstrated that at this dose, VEGF-A was effective in promoting angiogenesis after experimental ischemic stroke [24]; thereby, a similar dose of VEGF-B was chosen. Twenty-four hours after last injection, mice were sacrificed via a transcardiac perfusion of 4% paraformaldehyde (PFA). Brains were removed and cut on microtome into 25-μm coronal sections that were kept in an anti-freeze solution at −20 °C for further use.
Assessment of Neurological Deficit
Neurological deficits were monitored 24 h after MCAo induction as previously described [25]. The sensorimotor performance of mice subjected to MCAo was assessed using a neurological score test that closely correlates the deficits with the severity of the histological injury. Neurological deficits were evaluated using the following score: 0 = normal function, 1 = flexion of torso and the contralateral forelimb on lifting of the animal by the tail, 2 = circling to the contralateral side but normal posture at rest, 3 = reclination to the contralateral side at rest, and 4 = absence of spontaneous motor activity.
Evaluation of Brain Vascular Permeability
Evans blue solution was dissolved in normal saline (2% w/v). A total volume of 4 mL/kg of 2% Evans blue solution was intraperitoneally injected in mice subjected to focal ischemic stroke 3 h before sacrifice mice by a transcardiac perfusion with 50 mL of ice-cold normal saline. Brains were removed and hemispheres were separated. Ipsilateral hemispheres were homogenized in 1100 μL of ×1X phosphate-buffered saline (PBS), sonicated, and centrifuged (30 min, 15,000 rpm, 4 °C). The supernatants were collected, and to each 500 μL, an equal amount of 50% trichloroacetic acid (TCA) was added. After an overnight incubation at 4 °C, samples were centrifuged (30 min, 15,000 rpm, 4 °C). Evans blue extravasation into the ischemic brain was quantified by measuring the absorbance using a microplate reader at 610-nm wavelength (SpectraMax 340PC, Molecular Devices, CA, USA). Data were analyzed using SOFTmax Pro3.1.1 software (Molecular Devices) and quantified according to a standard curve. The results are presented as (μg of Evans blue)/(g of brain tissue).
Analysis of Ischemic Injury and Immunoglobulin G Extravasation
Representative free-floating brain sections were mounted onto SuperFrost slides (Fisher Scientific, Ottawa, ON, Canada) and left thorough overnight drying under vacuum. Sections were next stained with 0.5% cresyl violet to assess the brain infarct size, as previously described [26]. Stained sections were digitized and the border between injured and non-injured healthy tissue was outlined with image analysis software (ImageJ; National Institutes of Health, Bethesda, MD, USA). The infarct size was measured by subtracting the area of ipsilateral hemisphere from that of contralateral hemisphere. Adjacent sections were processed for immunohistochemistry to stain extravasated serum immunoglobulin G (IgG), which was studied as an endogenous marker of BBB permeability, as previously described [26]. Briefly, mounted brain sections were washed with potassium phosphate-buffered saline (KPBS) (Sigma-Aldrich) and then incubated for 20 min in a permeabilization/blocking solution containing 4% normal goat serum (NGS), 1% bovine serum albumin (BSA) (Sigma-Aldrich), and 0.4% Triton X-100 (Sigma-Aldrich) in KPBS. Biotinylated anti-mouse IgG antibody (Santa Cruz Biotechnology) was incubated overnight at 4 °C. Staining was revealed using avidin peroxidase kit (Vectastain Elite; Vector Labs, Burlingame, CA, USA) by immersing brain sections for 1 h in avidin-biotin complex (ABC) mixture. Brain sections were washed and stained in diaminobenzidine (DAB) solution for 10 min and washed again. Slices were mounted onto slides, dried, dehydrated, and overlaid with coverslips using distyrene plasticizer xylene (DPX) mounting solutions. Brain sections were digitized and analyzed for areas exhibiting IgG extravasation using ImageJ software.
Fluoro-Jade B Staining
Fluoro-jade B (FJB) staining was used as an indicator of neuronal death as described [27]. Mounted free-floating brain sections were fixed with 4% PFA for 20 min. Fixed sections were then rinsed twice with KPBS for 5 min and processed through a cycle of dehydration/rehydration in ethanol at different dilutions as follows: 3 min in 50%, 1 min in 70%, 3 min in 100%, 1 min in 70%, 1 min in 50%, and 1 min in distilled water. Mounted sections were next treated for 10 min with 0.06% potassium permanganate (MP Biomedicals, Santa Ana, CA, USA), rinsed for 1 min with distilled water, and then incubated in 0.0004% FJB solution (EMD Millipore, Etobicoke, ON, Canada) containing 0.1% acetic acid and 2 μg/mL DAPI. Sections were dried overnight, immersed in xylene, and then coverslipped in anti-fade mounting solution (Sigma-Aldrich). Fluorescent images were taken using a Nikon C80i microscope (Nikon Instruments, Williston, VT, USA) equipped with a motorized stage (Ludl, Hawthorne, NY, USA) and QImaging® color camera (MBF 2000 R, Quantitative Imaging, Surrey, BC, Canada) using QCapture version 2.98.2 software (Quantitative Imaging).
Immunofluorescence Analysis
Mounted sections were first incubated for 20 min in a permeabilization/blocking solution containing 4% NGS, 1% BSA, and 0.4% Triton X-100 in KPBS and then overnight at 4 °C with different primary antibodies diluted in the same solution. The following primary antibodies were used: rabbit anti-desmin (1/250; Abcam, Cambridge, UK), rabbit anti-platelet-derived growth factor receptor-β (PDGFRβ; 1/250; Abcam), rat anti-CD31 (i.e., platelet endothelial cell adhesion molecule-1 (PECAM1); 1/500; BD Biosciences, San Jose, CA, USA), rabbit anti-ionized calcium binding adaptor molecule-1 (Iba1; 1/250; Wako Chemicals, Cape Charles, VA, USA), rabbit anti-VEGFR-1 (1/250; Abcam), and mouse anti-CD105 (i.e., endoglin (ENG); 1/100; Novus Biologicals, Littleton, CO, USA). Afterwards, sections were rinsed in KPBS, followed by a 2-h incubation with one of the following secondary antibodies: Alexa Fluor 488-conjugated goat anti-rabbit antibody (Invitrogen, Carlsbad, CA, USA), Alexa Fluor 488-conjugated goat anti-rat antibody (Invitrogen), Cy3 AffiniPure goat anti-rabbit IgG (H + L) (Jackson Immunoreasearch, West Grove, PA, USA), Cy3 AffiniPure goat anti-rat IgG (H + L) (Jackson Immunoreasearch), or Cy3 AffiniPure goat anti-mouse IgG (H + L) (Jackson Immunoreasearch). After washing with KPBS, sections were coverslipped with anti-fade mounting medium (Sigma-Aldrich). Epifluorescence images were taken using a Nikon C80i microscope equipped with a motorized stage (Ludl) and a QImaging® color camera (Quantitative Imaging) using QCapture version 2.98.2 software (Quantitative Imaging). Confocal laser-scanning microscopy was performed with a LSM 800 microscope equipped with the ZEN Imaging software (Carl Zeiss Canada, Toronto, ON, Canada).
Oxygen and Glucose Deprivation Induction
To investigate the responses of brain endothelial cells and pericytes challenged by ischemia/reperfusion-like conditions, cells were incubated in oxygen and glucose deprived (OGD) conditions. OGD was induced by incubating cells at 37 °C in a Dulbecco’s modified Eagle’s medium (DMEM)-glucose free medium (Multicell, Wisent, St-Bruno, QC, Canada) under hypoxic condition (1% O2) for 3 h (ischemia-like) using a Modular Incubator Chamber (Billups-Rothenberg Inc., Del Mar, CA, USA). Following the 3 h OGD, DMEM-glucose free medium was immediately replaced by DMEM glucose-normal medium and cells were incubated under normal oxygenation conditions to allow a re-oxygenation period (reperfusion-like) for either 1 or 24 h as specified in different experiments. As control, cells were incubated at 37 °C in DMEM glucose-normal medium (Multicell, Wisent) under normal oxygenation conditions (normoxia). Cells were either harvested for protein extraction or fixed at room temperature with 3.5% PFA for further analysis. To simplify, we used throughout the text OGD instead of OGD/re-oxygenation.
Cell Culture Experiments
We chose to use a primary human brain microvascular pericytes (HBMVPs) (ScienCell Research Laboratories, Carlsbad, CA, USA) to accurately investigate the function of pericytes as these cells are the main focus of this study. Additionally, we choose immortalized murine brain-derived endothelial cells (bEnd3) (ATCC, Manassas, VA, USA) to represent brain endothelial cells, as these cells have been shown to have similar barrier characteristics to primary brain microvascular endothelial cells (BEMCs) [28]. Moreover, bEnd3 cells are attractive candidates to represent the cellular component of the BBB and are amenable to numerous molecular interventions [28]. HBMVP and bEnd3 were cultured at 37 °C in 5% CO2, 95% air in DMEM normal-glucose medium (Multicell, Wisent) containing 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. In all experiments, cells were grown to approximately 80% confluence and subjected to a maximum of eight cell passages. To induce OGD, HBMVP were seeded at 1.5 × 105 cells/well, and bEnd3 were seeded at 2 × 105 cells/well in a 12-well plate (Corning, NY, USA). Cells were treated with saline, 4 or 16 ng of VEGF-B (PeproTech). As screening experiments showed that VEGF-B at 16 ng was more efficient, this dose was adopted to perform the functional studies. At the end of each experiment, cell culture medium was collected, and cells were either harvested for protein extraction or fixed at room temperature with 3.5% PFA for further analysis.
Western Blot Analysis
HBMVP and bEnd3 in different conditions were harvested, and whole cell lysates were prepared with NP40 lysis buffer supplemented with 1% protease inhibitor cocktail (Sigma-Aldrich) and 1% phosphatase inhibitor cocktail (Sigma-Aldrich), as specified before [29]. Total protein content for each sample was determined using the bicinchoninic acid (BCA) method (QuantiPro Assay Kit, Sigma-Aldrich) [29]. Protein samples (20 μg) were mixed with ×5 sodium dodecyl sulfate (SDS)-loading buffer and heated for 5 min at 95 °C. Samples were subjected to 8, 10, or 12% SDS polyacrylamide gel electrophoresis (SDS-PAGE) and subjected to electrophoresis using Mini-PROTEAN® Tetra Cell (Bio-Rad, Hercules, CA). After migration, resolved protein bands were transferred onto a 0.45-mm polyvinylidene fluoride (PVDF) membrane (EMD Millipore) for 1 h on ice using Mini Trans-Blot® Electrophoretic Transfer Cell (Bio-Rad). The PVDF membrane was rinsed three times with a 0.1 M Tris-buffered saline solution containing 0.5% Tween-20 (TBS-Tween; Sigma-Aldrich) and blocked in TBS-Tween with 5% (w/v) skim milk for 30 min at room temperature. The PVDF membrane was then incubated overnight at 4 °C with different primary antibodies diluted at 1/1000 in TBS-T solution. The following antibodies were used: rabbit anti-VEGFR-1 (Santa Cruz Biotechnology), goat anti-VEGFR-2 (R&D Systems, Minneapolis, MN, USA), rabbit anti-AMP-activated protein kinase-α (AMPKα; Cell Signaling Technology, Danvers, MA), rabbit anti-B-cell lymphoma-2 (Bcl-2; Santa Cruz Biotechnology), and mouse anti-β-actin (EMD Millipore). Primary antibodies were detected with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson Immunoreasearch) that were diluted 1/500 in TBS-T and revealed by enhanced chemiluminescence plus (ECL) solution (Bio-Rad, Hercules, CA, USA). β-actin was used to ensure equal protein loading. Blots were revealed and immediately digitized using Thermo Scientific myECL Imager (ThermoFisher Scientific, Waltham, MA USA). Digitized blots were densitometrically analyzed with ImageJ software, corrected for protein loading by means of β-actin, and expressed as relative values comparing different groups.
Cell Viability, Loss, and Count
HBMVP cell viability was assessed using the 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H–tetrazolium-5-carboxanilide (XTT) assay according to the manufacturer’s procedure (Cell Signaling Technology). Cells were plated at 1.5 × 105 cells/well in 12-well plates under either normoxia or OGD conditions. Cells were stimulated either with sterile saline solution or VEGF-B (16 ng) during OGD induction and the re-oxygenation period. One hundred microliter of cell culture medium representing each condition was incubated with 50 μL of XTT detection solution for 3 h at 37 °C. Cell viability was determined by reading absorbance at 450 nm using a microtiter plate reader and analyzed with the SOFTmax Pro3.1.1 software. Independent similar experiments were performed but by plating cells onto 15-mm coverslips for adherent cells (Electron Microscopy Sciences, Hatfield, PA). Cells were fixed with 3.5% PFA at room temperature and incubated with 1/250 Alexa Fluor®546 phalloidin (ThermoFisher Scientific). Epifluorescence images were taken using a Nikon C80i microscope equipped with a Microfire CCD color camera. HBMVP and bEnd3 cell death/viability ratio 24 h after OGD induction (i.e., 24-h re-oxygenation) was assessed using a viability/cytotoxicity assay according to the manufacturer’s procedure (Biotium, Fremont, CA, USA). The assay employs two probes that detect intracellular esterase activity in live cells and compromised plasma membrane integrity in dead cells. The esterase substrate calcein AM stains live cells green, while the membrane-impermeable DNA dye ethidium homodimer III (EthD-III) stains dead cells red. Cells were plated at 1.5 × 105 cells/well in 12-well plates under either normoxia or OGD conditions. Cell viability/death ratio was measured using EVOS® FL cell imaging system (ThermoFisher Scientific) equipped with highly sensitive Sony ICX445 monochrome CCD camera. In independent similar experiments, absolute cell count was additionally performed using the same cell imaging system. The number of cells under different conditions was quantified and corrected to the number of cells seeded at the beginning of each experiment. The count of viable/dead cells and absolute cell count were performed using the EVOS FL Auto software that includes a built-in program to perform cell count.
Cytokine Profiling Analysis
For cytokine expression studies, NP40 HBMVP lysates were used. Protein expression was determined using the Proteome Profiler™ Array (R&D Systems) that allows assessing the simultaneous relative expression levels of 36 human cytokines and chemokines. The experiment was performed according to the manufacturer’s protocol. Briefly, a total amount of 200 μg per sample was mixed with biotinylated detection antibodies and then incubated with the array membrane which contains immobilized capture antibodies for the detection of a broad set of inflammatory cytokines and chemokines. Signals were visualized by streptavidin-HRP and chemiluminiscent detection. Membranes were revealed and immediately digitized using Thermo Scientific myECL Imager (ThermoFisher Scientific). Digitized blots were densitometrically analyzed with ImageJ software by measuring for each data spot the mean pixel intensity corrected for the background signal. Data were analyzed with mean values from duplicates and related to the mean of three internal control values on each membrane, as previously described [30].
Statistics
Results are expressed as mean ± standard deviation (SD). For comparisons between two groups, Student’s t tests were used. For comparisons of multiple groups, one-way analysis of variance (ANOVA) followed by the Tukey’s post hoc tests were used. P values lower than 0.05 were considered significant. All statistical analyses were performed using GraphPad Prism version 6 for Windows (GraphPad Software, San Diego, CA, USA).
Results
Early Administration of VEGF-B, and Not VEGF-A, Reduces the Leakage of Ischemic Brain Vasculature
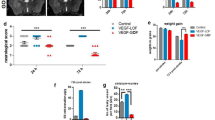
It is well documented that the intravenous administration of VEGF-A early in the acute phase of ischemic stroke increases tissue damage and neurological deficits by exacerbating the vascular permeability due to an excessive pro-angiogenic response [16]. As VEGF-B is suggested to act as a survival rather than an angiogenic factor [17], the effects of its systemic early administration on vascular permeability and neurological deficits were first evaluated. In contrast to VEGF-A, a single intravenous dose of VEGF-B administered 1 h after MCAo induction decreased the levels of the exogenous tracer Evans blue in the ipsilateral hemisphere (P < 0.0001), indicating that the permeability of the brain vasculature was preserved (Fig. 1a). Furthermore, in contrast to VEGF-A, VEGF-B did not exacerbate the neurological deficits of mice 24 h after MCAo induction (P < 0.01), indicating that the histological injury did not worsen (Fig. 1b). In mice, the half-life of VEGF in the blood circulation is very short (approximately 3 min) [31], suggesting that the reported effects of VEGF-B were essentially mediated by targeting the ischemic brain vasculature rather than acting directly on cells within the brain parenchyma.

VEGF-B is a safe alternative for VEGF-A in ischemic stroke therapy. a Evans blue quantification shows reduced extravasation into the ischemic brain following early systemic administration of the VEGF-B. VEGF-A increases extravasation of the Evans blue into the ischemic hemisphere. b Neurological score analysis shows that systemic early administration of the VEGF-B does not exacerbate neurological deficit of mice subjected to ischemic stroke, whereas VEGF-A worsens the neurological deficit of these mice. VEGF-A vascular growth factor isoform-A, VEGF-B vascular growth factor isoform-B. Data are mean ± SD (n = 6 animals per group). *P < 0.05/**P < 0.01 VEGF-B treated group compared with saline-treated group, †† P < 0.01/†††† P < 0.0001 VEGF-B-treated group compared with VEGF-A-treated group (ANOVA followed by Tukey’s multiple comparison test)
Delayed Intranasal Administration of VEGF-B Decreases the Structural Damage of the Ischemic Brain
After evaluating the efficacy and safety of VEGF-B, the effects of its delayed intranasal administration in the sub-acute phase, during which destabilization and permeability of the ischemic brain microvasculature peak, were investigated. VEGF-B was administered intranasally to increase the translational potential of the expected findings as this route of administration can be used in human and to reduce the possible adverse effects of the repetitive VEGF systemic administration on hemodynamics [32]. The intranasal administration of the VEGF-B in the sub-acute phase decreased infarct size (P < 0.05) (Fig. 2a) and IgG extravasation (P < 0.05) (Fig. 2b) and reduced the number of degenerating neurons (FJB+ cells) in the ipsilateral hemisphere (P < 0.05) (Fig. 2c). Interestingly, attenuation of the neuronal degeneration was accompanied by a decreased activation of microglial cells (Iba1+ cells), indicating a reduced inflammatory response in the ipsilateral hemisphere (P < 0.05) (Fig. 2d). Laser-scan confocal microscopy analysis showed that microvasculature in the ipsilateral hemisphere of saline-treated animals has odd bubble-like morphology presenting several constricted regions, outlining severe dysfunction, whereas in VEGF-B-treated groups, the majority of ischemic microvasculature presents normal morphology (Fig. 2e). These results indicate that the effects of VEGF-B were essentially mediated by targeting the microvasculature. Nonetheless, to confirm the specificity of the effects of the intranasal administration of VEGF-B on the integrity of brain microvasculature, we administered half of the dose used before and found that this low dose did not decrease the levels of Evans blue in the ipsilateral hemisphere, indicating that the permeability of the brain microvasculature was not affected (saline 4.852 ± 0.5422 vs VEGFBlow dose 5.022 ± 0.5535 μg of Evans blue/g of brain tissue; P = 0.83).

VEGF-B attenuates brain injury after focal ischemic stroke. a Infarct size analysis shows a reduction of ischemic injury after sub-acute intranasal VEGF-B treatment. b Immunohistochemistry analysis demonstrates a reduced IgG extravasation into the ischemic brain after sub-acute intranasal VEGF-B treatment. c FJB representative images and analysis outline a reduced number of degenerating neuron in the ipsilateral hemisphere after sub-acute intranasal VEGF-B treatment. d VEGF-B treatment decreases the activation of microglial cells (Iba1+ staining) in the ipsilateral hemisphere. e Laser-scan confocal analysis examining the morphology of the brain microvasculature (CD31+ staining; green) in the ipsilateral hemisphere shows that the majority of brain microvasculature in the saline-treated animals displays odd bubble-like morphology presenting several constricted regions (white arrows), while the majority in VEGF-B-treated animals presents normal microvasculature morphology. VEGF-B vascular growth factor isoform-B, IgG immunoglobulin G, FJB fluoro-jade B, Iba1 ionized calcium-binding adaptor molecule-1. Data are mean ± SD (n = 9 animals per group). *P < 0.05 compared with saline-treated group (standard two-tailed unpaired t tests). Epifluorescent images (c) were acquired with a ×20 objective. Laser-scan confocal images (e) were acquired with a ×40 objective. Scale bar = 100 μm
Microvasculature Density and Coverage by Pericytes in the Ischemic Brain Increase After VEGF-B Delayed Intranasal Delivery
Based on the previous findings, the effects of VEGF-B on the dynamics of ischemic brain microvasculature were next analyzed. Unexpectedly, the intranasal sub-acute administration of VEGF-B increased the density of CD31+ brain microvasculature in the ipsilateral hemisphere (P < 0.001) (Fig. 3a). This result was not expected as VEGF-B was not supposed to have an angiogenic activity. Nonetheless, VEGF-B did not affect the number of CD31+ brain microvasculature in the contralateral hemisphere (data not shown). On the other hand, the absolute abundance of pericytes (desmin+ immunofluorescent staining) (P < 0.05) (Fig. 3b) and the coverage of brain microvasculature by these cells (PDGFRβ+/CD31+ immunofluorescent staining) (P < 0.05) (Fig. 3c) in the ipsilateral hemisphere significantly increased following VEGF-B administration. Next, we performed structural analysis by looking closer to the morphology and behavior of PDGFRβ+ pericytes in the ipsilateral hemisphere. We found that in the saline-treated animals, these cells are highly dissociated from brain endothelial cells and are activated by adopting a ramified-like structure forming several cellular protrusions towards brain parenchyma (Fig. 4). Interestingly, VEGF-B treatment potently restored the morphology of PDGFRβ+ pericytes of the ipsilateral hemisphere (Fig. 4). As VEGF-B signals specifically through VEGFR-1, the expression of this receptor in pericytes located in the ipsilateral hemisphere was assessed. In line with previous reports, brain endothelial cells do not appear to express VEGFR-1 in the mouse brain tissue samples used in this study (data not shown), indicating that the angiogenic activity is not directly mediated by the action of VEGF-B on brain endothelial cells. To overcome technical challenges related to the specificity of antibodies against VEGFR-1 and their compatibility with those against the markers of pericytes in mouse brain tissue, we decided to comprehensively evaluate in the next experiment expression pattern and regulation of the VEGFR-1 in brain endothelial cells and brain pericytes using in vitro cell-based assays.

VEGF-B increases the density of brain microvasculature and their coverage by pericytes. a CD31 (endothelial marker) immunofluorescent representative images (green) and respective stereological quantification show increased brain microvasculature density in the ipsilateral hemisphere after sub-acute intranasal VEGF-B treatment. b Desmin (pericyte marker) immunofluorescent representative images (red) and respective stereological quantification exhibit an increased absolute abundance of pericytes in the ipsilateral hemisphere after sub-acute intranasal VEGF-B treatment. c CD31 (endothelial marker)/PDGFRβ (pericyte marker) immunofluorescent representative images and respective stereological quantification of co-localization show an increase in the number of brain microvasculature (CD31+ staining; green) covered by pericytes (PDGFRβ+ staining; red) in the ipsilateral hemisphere after sub-acute intranasal VEGF-B treatment. VEGF-B vascular growth factor isoform-B, PDGFRβ platelet-derived growth factor receptor-β. Data are mean ± SD (n = 5–8 animals per group). *P < 0.05/***P < 0.001 compared with saline-treated group (standard two-tailed unpaired t tests). Laser-scan confocal images were acquired with a ×10 objective (a, c) or ×20 (b). Scale bar = 100 μm

Association of brain endothelial cells and pericytes increases following VEGF-B administration. Laser-scan confocal analysis of the triple immunofluorescent staining for CD31 (endothelial marker; green)/PDGFRβ (pericyte marker; red)/DAPI (nucleus marker; blue) shows that in the ipsilateral hemisphere of saline-treated animals (upper panel), pericytes (red) are activated presenting a ramified-like morphology and tend to detach from endothelial cells (green) by protruding towards the parenchyma (white arrows). VEGF-B (lower panel) reduces the activation of pericytes that are now more firmly attached to endothelial cells and enhances the ensheathment of brain microvasculature by pericytes (white arrows). VEGF-B vascular growth factor isoform-B, PDGFRβ platelet-derived growth factor receptor-β. VEGFR-1 vascular endothelial growth factor receptor-1, DAPI 4′,6-diamidino-2-phenylindole. Laser-scan confocal images were acquired with a ×40 objective. Scale bar = 50 μm
Brain Endothelial Cells and Pericytes Are Differentially Affected by the OGD Conditions
The previous observations suggest that VEGF-B seems to act on brain pericytes rather than brain endothelial cells. To elucidate this point, and to further characterize the role of VEGF-B/VEGFR-1 signaling in the brain pericytes, several cell-based assays under OGD conditions that mimic ischemia-like conditions were used. First, the effects of ischemic conditions on the cells from the microvasculature—brain endothelial cells and pericytes—were dissected. For this purpose, HBMVP and bEnd3 were exposed to OGD. The cell viability/cytotoxicity assay showed that the ratio of HBMPV death was significantly higher compared to the death of bEnd3 1 h after OGD induction (Fig. 5a). More precisely, brain pericyte cell death was around 80% (P < 0.05), whereas brain endothelial cell death was around 50% (P < 0.05) (Fig. 5a). Furthermore, the capacity of cells to recover and proliferate 24 h following OGD induction was specifically investigated. Absolute cell count analysis demonstrated that HBMVP were enable to recover 24 h after OGD induction (P < 0.05 compared to normoxia) (Fig. 5b), thus translating a profound cell dysfunction, whereas bEnd3 recover much more efficiently 24 h after OGD induction (P = 0.707 compared to normoxia) (Fig. 5c), thus translating mild cell dysfunction.

OGD differentially affects the survival and death of brain pericytes and brain endothelial cells. a Cell viability/cytotoxicity assay shows that the ratio of cell death (presented as percentage of initially seeded cells for each condition) is higher in HBMVP (brain pericytes) 1 h after exposure to OGD compared to bEnd3 (brain endothelial cells). b Absolute cell count analysis indicates that HBMVP loss persists 24 h after exposure to OGD. c Absolute cell count analysis indicates that bEnd3 recuperates 24 h after exposure to OGD. Absolute cell count was corrected with the number of seeded cells. NO normoxia, OGD oxygen and glucose deprivation, HBMVP human brain microvascular pericytes. Data are mean ± SD (n = 3 independent experiments per condition). *P < 0.05 death of OGD-exposed HBMVP compared with OGD-exposed bEnd3, † P < 0.05 survival of OGD-exposed HBMVP compared with OGD-exposed, bEnd3 saline-treated group (standard two-tailed unpaired t tests)
VEGFR-1, and Not VEGFR-2, Is Highly Expressed in Brain Microvascular Pericytes and Is Upregulated Under OGD Conditions
The in vivo results are suggesting that VEGFR-1 is predominately expressed in desmin-like expressing cells; thereby, protein expression of the VEGFR-1 was analyzed in HBMVP. Protein expression analysis showed that OGD rapidly—1 h after induction—increased the expression level of VEGFR-1 in HBMVP (P < 0.01) (Fig. 6a). Furthermore, expression of the soluble form of VEGFR-1 (sVEGFR-1), which is involved in regulating the angiogenic response by neutralizing the potent pro-angiogenic VEGF-A, was induced under OGD conditions (Fig. 6b). It has been shown that in contrast to VEGFR-2, which is mainly expressed in brain endothelial cells, expression of the VEGFR-1 is regulated by hypoxia, and this regulation seems to be cell type dependent [33]. Importantly, protein expression analysis demonstrated that VEGFR-2 is constitutively expressed in bEnd3 but not in HBMVP (Fig. 6c). Additionally, the protein expression level of VEGFR-2 in bEnd3 remained unchanged after OGD challenge (Fig. 6c). Although protein analysis demonstrated that VEGFR-1 is expressed in brain endothelial cells, its basal expression level was significantly lower compared to brain pericytes under normoxic conditions (P < 0.05) but was accentuated under OGD conditions (P < 0.01). Moreover, expression level of the VEGFR-1 in bEnd3 remained unchanged under ODG conditions, indicating lack of efficient responsiveness to hypoxia (P = 0.344) (Fig. 6d). To our knowledge, this is the first report to show rapid upregulation of VEGFR-1 protein expression and the induction of sVEGFR-1 in brain pericytes exposed to OGD.

VEGFR-1 expression is high and dynamic in brain pericytes exposed to OGD conditions. Western blot analysis using whole cell lysates shows that OGD conditions increase a expression of the VEGFR-1 in HBMVP (brain pericytes) and specifically induce b expression of the soluble form of VEGFR-1 (sVEGFR-1). c Western blot analysis using whole cell lysates shows that HBMVP do not express VEGFR-2 and its expression level remains unchanged under OGD conditions, whereas VEGFR-2 is highly expressed in bEnd3 (brain endothelial cells) and its expression level remains unchanged as well under OGD conditions. d Western blot analysis using whole cell lysates indicates that expression of the VEGFR-1 is significantly higher in HBMVP compared to bEnd3 (four times higher) and that its expression level increases in HBMVP cells exposed to OGD conditions but remains unchanged in bEnd3 cells. NO normoxia, OGD oxygen and glucose deprivation, HBMVP human brain microvascular pericytes, VEGF-B vascular growth factor isoform-B, VEGFR-1 vascular endothelial growth factor receptor-1. Data are mean ± SD (n = 3 independent experiments per condition). **P < 0.01 OGD-exposed HBMVP compared with NO-exposed HBMVP, † P < 0.05 NO-exposed HBMVP compared with NO-exposed bEnd3, ## P < 0.01 OGD-exposed HBMVP compared with OGD-exposed bEnd3
Survival and Function of Brain Microvascular Pericytes Challenged by the OGD Conditions Are Potently Ameliorated by VEGF-B
The previous in vivo findings suggest that VEGF-B increased the abundance of pericytes that ensheath brain endothelial cells and restored the overall structure and stability of brain microvasculature. The specific enhanced expression of VEGFR-1 in pericytes increased upon hypoxia, thus enhancing its bioavailability to bind and mediate the biological effects of VEGF-B in these cells. However, the underlying mechanisms remain unknown. For this purpose, HBMVP were exposed to OGD and were stimulated with 4 and 16 ng of VEGF-B. The dose of VEGF-B at 16 ng was more efficient; thereby, this concentration was used in performing the experiments. As shape translates the overall health of cells, the morphological changes associated to actin organization of HBMVP maintained under different conditions were first analyzed. OGD induced a rapid disruption of actin cytoskeleton in HBMVP (Fig. 7a), outlining cell dysfunction and death. Importantly, VEGF-B potently restored the morphology of HBMVP exposed to OGD (Fig. 7a). Next, cell survival and death were quantitatively analyzed. XTT viability assay showed that VEGF-B significantly enhanced the survival of HBMVP exposed to OGD (P < 0.001) (Fig. 7b) and potently rescued absolute cell loss (P < 0.01) (Fig. 7c). Moreover, VEGF-B restored expression of the anti-apoptotic protein Bcl-2 in HBMVP exposed to OGD (P < 0.01) and even induced protein expression pattern to higher levels in comparison to normoxia (P < 0.05) (Fig. 7d). As OGD alters glucose metabolism and activate hypoxia-induced pathways, activation (i.e., phosphorylation) of the AMPKα, an enzyme playing key roles in cellular energy homeostasis, was next investigated. Strikingly, VEGF-B significantly enhanced the phosphorylation of AMPKα (P < 0.05) (Fig. 7e), outlining a previously unknown role of VEGF-B/VEGFR-1 signaling in modulating energy-related mechanisms in brain pericytes. To our knowledge, this is the first report to outline a link between VEGF-B and AMPKα in pericytes. Importantly, the protein levels of Bcl-2 remained unchanged in bEnd3 cells challenged by OGD and stimulated by VEGF-B (P = 0.817) (Fig. 8a). The phosphorylation level of AMPKα has a tendency to slightly increase under OGD conditions but remained unchanged following VEGF-B stimulation (P = 0.105) (Fig. 8b). This indicates that VEGFR-1 seemingly fulfills different roles in brain endothelial cells in comparison to brain pericytes.

Survival and function of brain pericytes challenged by the OGD conditions are potently ameliorated by VEGF-B. a Representative images of F-actin staining using Alexa Fluor®546 phalloidin shows normal actin cytoskeleton morphology in NO conditions, disrupted morphology in OGD conditions, and restored morphology in OGD conditions following VEGF-B stimulation. b XTT cell viability assay shows that VEGF-B significantly enhanced the survival of HBMVP (brain pericytes) exposed to OGD. c Absolute cell count analysis shows that VEGF-B potently rescues cell loss induced by the OGD conditions. d Western blot analysis using whole cell lysates shows that VEGF-B restores the expression of Bcl-2 in HBMVP exposed to the OGD conditions. e Western blot analysis using whole cell lysates demonstrates that VEGF-B significantly enhances the phosphorylation (i.e., activation) of AMPKα in HBMVP exposed to the OGD conditions. XTT 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H–tetrazolium-5-carboxanilide, NO normoxia, OGD oxygen and glucose deprivation, HBMVP human brain microvascular pericytes. VEGF-B vascular growth factor isoform-B. VEGFR-1 vascular endothelial growth factor receptor-1, Bcl-2 B-cell lymphoma 2. AMPKα AMP-activated protein kinase-α. Data are mean ± SD (n = 3 independent experiments per condition for all except for XTT cell viability assay, n = 12 wells per condition). *P < 0.05/****P < 0.0001 compared with NO-exposed cells. † P < 0.05/†† P < 0.01/††† P < 0.001 compared with OGD-exposed cells treated with VEGF-B. Epifluorescent images were acquired with a ×20 objective. Scale bar = 100 μm

VEGF-B does not affect Bcl-2 expression and AMPKα phosphorylation in brain endothelial cells exposed to the OGD conditions. a Western blot analysis using whole cell lysates shows that expression level of Bcl-2 remains unchanged in bEnd3 exposed to the OGD. b Western blot analysis using whole cell lysates shows that VEGF-B does not alter the phosphorylation of AMPKα in bEnd3 exposed to the OGD conditions. NO normoxia, OGD oxygen and glucose deprivation, VEGF-B vascular growth factor isoform-B, Bcl-2 B-cell lymphoma 2, AMPKα AMP-activated protein kinase-α. Data are mean ± SD (n = 3 independent experiments per condition)
VEGF-B Stimulation Potentiates the Capacity of Brain Microvascular Pericytes Challenged by OGD to Promote Following Reparative Angiogenesis
After elucidating its role in regulating the survival of pericytes under ischemic conditions, how VEGF-B stimulated angiogenesis and preserved the function of endothelial cells was addressed. Upon ischemic stroke, pericytes have been reported to acquire an inflammatory profile affecting vascular remodeling [34, 35]. Thereby, cytokine/chemokine profiler assays were performed. We found that VEGF-B induced expression of the interleukin-8 (IL-8) in HBMVP exposed to OGD conditions (P < 0.0001) (Fig. 9a). IL-8 is a cytokine that has been demonstrated to be directly involved in enhancing endothelial cell survival and proliferation [36]. Moreover, OGD reduce expression of the plasminogen activator inhibitor-1 (PAI-1) (P < 0.0001) (Fig. 9b), which plays important roles in controlling the angiogenic response as well by stabilizing the vascular network [37]. Importantly, VEGF-B potently restored stimulated expression of the PAI-1 in HBMVP exposed to OGD (P < 0.0001) (Fig. 9b). In parallel, the profiler assay showed that OGD induced expression of the stromal-cell derived factor-1 (SDF-1) in HBMVP exposed to ODG (P < 0.05) and effect that was further accentuated by VEGF-B (P < 0.001) (Fig. 9c). SDF-1 is a chemokine that plays a major role in the recruitment and retention of endothelial progenitor cells (EPCs) to the angiogenic niches within the ischemic tissue [38]. Finally, OGD significantly reduced expression of macrophage migration inhibitory factor (MIF) (P < 0.0001), which is a pleiotropic cytokine with chemokine-like functions [39] (Fig. 9d). Stimulation of the HBMVP exposed to OGD with VEGF-B attenuated this effect by increasing expression of the MIF (P < 0.01) (Fig. 9d). MIF has been recently shown to play major roles in neo-angiogenesis by activating endothelial cells and stimulating the recruitment of EPCs to angiogenic vasculature [40]. Additionally, MIF has been reported to have a protective effect under ischemic conditions [41, 42]. Finally, to translate the biological significance of these observations in vivo, immunofluorescent analysis demonstrated that VEGF-B intranasal administration increased the number of stable neo-angiogenic CD105+ microvasculature (Fig. 9e).

Stimulation of brain pericytes with VEGF-B restores cell capacity to produce molecules involved in maintaining angiogenesis. a Cytokine/chemokine profiler assay demonstrates that VEGF-B induces protein expression of IL-8 in HBMVP (brain pericytes) exposed to the OGD conditions. b Cytokine/chemokine profiler assay shows that OGD reduces the expression of PAI-1 in HBMVP exposed to the OGD conditions and that VEGF-B restores protein expression. c Cytokine/chemokine profiler assay shows that OGD increases expression of SDF-1 in HBMVP exposed to the OGD conditions and that VEGF-B further increases its protein expression. d Cytokine/chemokine profiler assay shows that OGD reduces expression of MIF in HBMVP exposed to the OGD conditions and that VEGF-B restores its protein expression. e Representative images of CD105 (proliferating endothelial cell marker) immunofluorescent staining outlines an increase in the number of CD105+ brain microvasculature in the ipsilateral hemisphere of mice subjected to ischemic stroke and treated intranasally in the sub-acute phase with VEGF-B. Data are mean ± SD (n = 3 independent experiments per condition). *P < 0.05/****P < 0.0001 compared with NO-exposed cells, †† P < 0.01/††† P < 0.001/†††† P < 0.0001 compared with OGD-exposed cells treated with VEGF-B. Laser-scan confocal images were acquired with a ×20 objective. Scale bar = 100 μm
Discussion
This study highlights for the first time the potential of VEGF-B therapy in ischemic stroke. Using in vivo approaches, we showed that local (i.e., intranasal) VEGF-B administration after the post-acute phase (24 h post-stroke for 3 successive days) enabled recovery of the damaged tissue by promoting the ischemic microvasculature formation and stabilization. Epifluorescence and laser-scan microscopy investigations demonstrated that VEGF-B restored the structure of ischemic brain microvasculature and enhanced their coverage by pericytes. Additionally, we showed that VEGF-B enhanced the association of brain endothelial cells with pericytes. Using in vitro approaches, we unraveled a novel and previously unrecognized role of VEGF-B in regulating the function of brain pericytes challenged by ischemia-like conditions. Cell-based investigations demonstrated that VEGF-B’s specific receptor, VEGFR-1, not VEGF-A’s receptor VEGFR-2, is highly expressed in pericytes, and its expression is induced by hypoxic conditions. Activation of VEGFR-1 signaling by VEGF-B enhanced the survival of pericytes exposed to ischemia-like conditions by stimulating pro-survival and energy-generating pathways. These effects were specific to brain pericytes, as VEGF-B was virtually neutral in brain endothelial cells. Finally, we demonstrated that VEGF-B-stimulated pericytes induced expression of soluble factors involved in promoting endothelial cell survival, proliferation, and microvasculature stability.
Therapeutic angiogenesis constitutes a promising approach in ischemic stroke treatment [15]. All experimental studies evaluated the potent pro-angiogenic isoform VEGF-A [14]. Although it yielded beneficial effects upon local and/or delayed administration, it significantly increased vascular permeability, which was more pronounced upon systemic and/or early administration. These reports dampened the enthusiasm regarding this approach in ischemic stroke therapies. Recent reports demonstrated that the isoform VEGF-B acts as a survival rather than an angiogenic factor, thus offering new therapeutic perspectives for ischemic stroke [17]. We showed here that in contrast to VEGF-A, the systemic early administration of VEGF-B decreased brain vascular leakage and did not worsen the neurological deficits of mice subjected to focal transient MCAo, indicating that VEGF-B-based therapies are safe. To increase the translational potential of our study, VEGF-B was next delivered intranasally [43], an administration route that can be used in human to safely deliver pharmacological molecules into the brain [44]. VEGF-B reduced neuronal degeneration, attenuated inflammation, and decreased extravasation of serum IgG into the ischemic hemisphere. Laser-scan microscopy analysis showed that VEGF-B promoted the integrity of ischemic brain microvasculature following VEGF-B administration. Unexpectedly, VEGF-B significantly increased the number of brain microvasculature within the ischemic tissue, outlining a potentiation of the post-stroke angiogenic process.
Angiogenesis is a highly dynamic process that involves finely tuned interactions between endothelial cells and pericytes [5, 45]. Angiogenic microvasculature is initially unstable and subsequently become stabilized through the association with pericytes located in the perivascular space [5]. Besides, the capability of brain microvasculature to orchestrate the exchange between the blood circulation and the brain parenchyma in response to tissue needs tightly depends upon the proper interactions between brain endothelial cells with pericytes [5, 46]. Interestingly, VEGF-B significantly ameliorated the number of ischemic brain microvasculature covered by pericytes, translating an enhanced interaction with the brain endothelial cells. Laser-scan microscopy analysis further showed that in VEGF-B-treated animals, the pericytes within the ischemic tissue stay firmly attached to the endothelium and have fewer tendencies to leave their perivascular location. These results indicate that VEGF-B mediated its effects by acting specifically and directly on pericytes within the ischemic brain microvasculature.
In the brain, VEGFR-2 is highly and constitutively expressed in endothelial cells under physiological conditions [18]. Under pathological conditions, such as ischemic stroke, VEGFR-2 was detected in some neurons and glial cells [18]. However, the cell type expressing VEGFR-1 is still controversial and not fully elucidated. While some reports have suggested that VEGFR-1 is expressed in motor neurons, endothelial cells, and astrocytes [18], other reports showed different expression pattern [9, 47]. This discrepancy may be explained by the fact that protein expression of the VEGFR-1 is physiologically weak in the adult normal brain and that its expression is hypoxia-inducible in cell type-specific manner [33]. In different vascular systems, namely, the retinal microvasculature, pericytes have been shown to predominately express VEGFR-1 [48]. While performing immunofluorescent staining, we faced major technical challenges concerning the efficiency and specificity of existing VEGFR-1 antibodies (five different antibodies cited in major publications were used) that work in mouse brain tissue. Nonetheless, to overcome this technical issue, we used primary brain pericyte cell culture to investigate VEGFR-1 signaling.
Cell-based investigations showed that OGD severely compromised the survival of pericytes and to a lesser extent, brain endothelial cells. Being highly vulnerable to ischemia-like conditions, it would be more efficient to target pericytes in order to restore vascular function in vascular-based disorders instead of directly targeting endothelial cells. Our findings are the first to demonstrate that expression of the VEGFR-1 is regulated by the hypoxic condition of OGD in brain pericytes and not in brain endothelial cells. This is in line with previous reports demonstrating that expression of the VEGFR-1 is hypoxia-inducible in some cell types [33]. Furthermore, OGD induced expression of sVEGFR-1 that is generated by differential splicing of the mRNA of VEGFR-1, which acts as a negative regulator of VEGF-A availability [18]. This result suggests that pericytes exposed to ischemic conditions negatively affect post-stroke angiogenesis, thus compromising ischemic tissue blood perfusion. The fact that OGD rapidly increased expression of the VEGFR-1 in pericytes, but not in brain endothelial cells, and that OGD did not affect levels of VEGFR-2 that is highly expressed in brain endothelial cells, but not in pericytes, delineated the important biological role of VEGFR-1 in the function of pericytes exposed to ischemia-like conditions. Collectively, these findings suggest that VEGFR-1 have distinct roles in brain pericytes compared to brain endothelial cells. In line with these results, VEGFR-1 seems to act as a signaling receptor regulating major functions in some cell types, such as monocytes, whereas it appears not to be required as a signaling receptor in endothelial cells [49, 50].
Additionally, cell-based investigations showed that VEGF-B enhanced the survival of brain pericytes exposed to OGD, partly by stabilizing expression of the anti-apoptotic protein Bcl-2. Importantly, VEGF-B enhanced the activation of AMPKα, a master key regulator of cell energy [51]. AMPKα regulates homeostasis of cell energy and metabolism by inhibiting adenosine triphosphate (ATP)-consuming pathways, such as gluconeogenesis, lipid, and protein synthesis, and activating ATP-generating pathways, such as glucose uptake and fatty acid oxidation [51]. These results indicate that VEGF-B signaling through VEGFR-1 ameliorated the capacity of brain pericytes to survive under ischemia-like conditions by restoring the activity of AMPKα. Future studies are warranted to investigate how VEGF-B/VEGFR-1 mediated AMPKα activation. It is noteworthy to mention that hypoxia alone is a potent triggered of AMPKα [52]. However, in our experimental settings, OGD induction was followed by a period of re-oxygenation. Importantly, in line with our results, re-oxygenation after hypoxia has been shown to decrease activation of the AMPKα [53]. Brain pericytes play a major role in regulating angiogenesis and inducing brain microvasculature stability via soluble and insoluble signals present at the perivascular space [5, 54, 55]. Here, we show that VEGF-B potently restored and potentiated expressions of the IL-8, PAI-1, SDF-1, and MIF, which are involved in angiogenesis, endothelial survival, and microvasculature stability, by brain pericytes exposed to ischemia-like conditions. IL-8 has been reported to regulate angiogenesis by enhancing proliferation and survival of CXC receptor-1 (CXCR-1)- and CXCR2-expressing endothelial cells [36]. PAI-1 controls proteolytic activity and cell migration during angiogenesis; thereby, it prevents uncontrolled excessive angiogenesis that may increase vascular permeability and bleeding [56]. SDF-1 plays a key role in the recruitment and retention of bone marrow-derived CXCR4+ vascular and hematopoietic cells to the neo-angiogenic niches, thereby accelerating revascularization of ischemic tissue [38]. MIF has been shown to potently stimulate pro-angiogenic responses and recombinant MIF decreased focal infarct volume in rats subjected to MCAo by promoting angiogenesis [57]. This data indicate that VEGF-B might have promoted a reparative angiogenesis by stimulating the expression and release of these signals by pericytes within the ischemic brain microvasculature. Indeed, VEGF-B increased the number of CD105+ proliferating, yet stable, brain microvasculature within the ischemic tissue. CD105 is a homodimeric membrane protein that binds transforming growth factor-β1 and 3 (TGF-β1/3) [58]. The expression of CD105 is induced in proliferating endothelial cells under physiological and pathological conditions [59]. This protein is required for vascular integrity, as mice lacking CD105 have been shown to exhibit defective vascular smooth muscle development and endothelial remodeling [60]. Importantly, CD105 has been shown to be required for endothelial survival, and thereby vascular stability, under hypoxic conditions [61].
Collectively, our findings unraveled for the first time a hitherto unknown role of VEGF-B/VEGFR-1 signaling in regulating the function of pericytes, thereby mediating neurovascular repair. These findings suggest that VEGF-B - mediated reparative angiogenesis constitutes a promising and safe therapeutic approach for ischemic stroke.
References
Iadecola C (2013) The pathobiology of vascular dementia. Neuron 80(4):844–866. doi:10.1016/j.neuron.2013.10.008
Daneman R, Prat A (2015) The blood-brain barrier. Cold Spring Harb Perspect Biol 7(1):a020412. doi:10.1101/cshperspect.a020412
Hermann DM, ElAli A (2012) The abluminal endothelial membrane in neurovascular remodeling in health and disease. Sci Signal 5(236):re4. doi:10.1126/scisignal.2002886
Stanimirovic DB, Friedman A (2012) Pathophysiology of the neurovascular unit: disease cause or consequence? Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 32(7):1207–1221. doi:10.1038/jcbfm.2012.25
ElAli A, Theriault P, Rivest S (2014) The role of pericytes in neurovascular unit remodeling in brain disorders. Int J Mol Sci 15(4):6453–6474. doi:10.3390/ijms15046453
Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions. Circ Res 97(6):512–523. doi:10.1161/01.RES.0000182903.16652.d7
Dirnagl U (2012) Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci 1268:21–25. doi:10.1111/j.1749-6632.2012.06691.x
Liu S, Agalliu D, Yu C, Fisher M (2012) The role of pericytes in blood-brain barrier function and stroke. Curr Pharm Des 18(25):3653–3662
Zechariah A, ElAli A, Doeppner TR, Jin F, Hasan MR, Helfrich I, Mies G, Hermann DM (2013) Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke; J Cereb Circ 44(6):1690–1697. doi:10.1161/STROKEAHA.111.000240
Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A (2004) Targets for vascular protection after acute ischemic stroke. Stroke; J Cereb Circ 35(9):2220–2225. doi:10.1161/01.STR.0000138023.60272.9e
Lo EH (2008) A new penumbra: transitioning from injury into repair after stroke. Nat Med 14(5):497–500. doi:10.1038/nm1735
Hayashi T, Abe K, Suzuki H, Itoyama Y (1997) Rapid induction of vascular endothelial growth factor gene expression after transient middle cerebral artery occlusion in rats. Stroke; J Cereb Circ 28(10):2039–2044
Navaratna D, Guo S, Arai K, Lo EH (2009) Mechanisms and targets for angiogenic therapy after stroke. Cell Adhes Migr 3(2):216–223
Hermann DM, Zechariah A (2009) Implications of vascular endothelial growth factor for postischemic neurovascular remodeling. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 29(10):1620–1643. doi:10.1038/jcbfm.2009.100
Lange C, Storkebaum E, de Almodovar CR, Dewerchin M, Carmeliet P (2016) Vascular endothelial growth factor: a neurovascular target in neurological diseases. Nat Rev Neurol 12(8):439–454. doi:10.1038/nrneurol.2016.88
Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, Bruggen N, Chopp M (2000) VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest 106(7):829–838. doi:10.1172/JCI9369
Zhang F, Tang Z, Hou X, Lennartsson J, Li Y, Koch AW, Scotney P, Lee C et al (2009) VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc Natl Acad Sci U S A 106(15):6152–6157. doi:10.1073/pnas.0813061106
Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L (2006) VEGF receptor signalling—in control of vascular function. Nat Rev Mol Cell Biol 7(5):359–371. doi:10.1038/nrm1911
Xie L, Mao X, Jin K, Greenberg DA (2013) Vascular endothelial growth factor-B expression in postischemic rat brain. Vascular cell 5:8. doi:10.1186/2045-824X-5-8
Sun Y, Jin K, Childs JT, Xie L, Mao XO, Greenberg DA (2004) Increased severity of cerebral ischemic injury in vascular endothelial growth factor-B-deficient mice. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 24(10):1146–1152. doi:10.1097/01.WCB.0000134477.38980.38
Li Y, Zhang F, Nagai N, Tang Z, Zhang S, Scotney P, Lennartsson J, Zhu C et al (2008) VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest 118(3):913–923. doi:10.1172/JCI33673
ElAli A, Hermann DM (2010) Apolipoprotein E controls ATP-binding cassette transporters in the ischemic brain. Sci Signal 3(142):ra72. doi:10.1126/scisignal.2001213
Kaya D, Gursoy-Ozdemir Y, Yemisci M, Tuncer N, Aktan S, Dalkara T (2005) VEGF protects brain against focal ischemia without increasing blood–brain permeability when administered intracerebroventricularly. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 25(9):1111–1118. doi:10.1038/sj.jcbfm.9600109
Yang JP, Liu HJ, Wang ZL, Cheng SM, Cheng X, Xu GL, Liu XF (2009a) The dose-effectiveness of intranasal VEGF in treatment of experimental stroke. Neurosci Lett 461(3):212–216. doi:10.1016/j.neulet.2009.06.060
Kilic U, Kilic E, Dietz GP, Bahr M (2003) Intravenous TAT-GDNF is protective after focal cerebral ischemia in mice. Stroke; J Cereb Circ 34(5):1304–1310. doi:10.1161/01.STR.0000066869.45310.50
ElAli A, Doeppner TR, Zechariah A, Hermann DM (2011) Increased blood-brain barrier permeability and brain edema after focal cerebral ischemia induced by hyperlipidemia: role of lipid peroxidation and calpain-1/2, matrix metalloproteinase-2/9, and RhoA overactivation. Stroke; J Cereb Circ 42(11):3238–3244. doi:10.1161/STROKEAHA.111.615559
Bordeleau M, ElAli A, Rivest S (2016) Severe chronic cerebral hypoperfusion induces microglial dysfunction leading to memory loss in APPswe/PS1 mice. Oncotarget 7(11):11864–11880. doi:10.18632/oncotarget.7689
Brown RC, Morris AP, O’Neil RG (2007) Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res 1130(1):17–30. doi:10.1016/j.brainres.2006.10.083
ElAli A, Bordeleau M, Theriault P, Filali M, Lampron A, Rivest S (2016) Tissue-plasminogen activator attenuates Alzheimer’s disease-related pathology development in APPswe/PS1 mice. Neuropsychopharmacol : Off Publ Am Coll Neuropsychopharmacol 41(5):1297–1307. doi:10.1038/npp.2015.279
Herz J, Reitmeir R, Hagen SI, Reinboth BS, Guo Z, Zechariah A, ElAli A, Doeppner TR et al (2012) Intracerebroventricularly delivered VEGF promotes contralesional corticorubral plasticity after focal cerebral ischemia via mechanisms involving anti-inflammatory actions. Neurobiol Dis 45(3):1077–1085. doi:10.1016/j.nbd.2011.12.026
Yen P, Finley SD, Engel-Stefanini MO, Popel AS (2011) A two-compartment model of VEGF distribution in the mouse. PLoS One 6(11):e27514. doi:10.1371/journal.pone.0027514
Tilton RG, Chang KC, LeJeune WS, Stephan CC, Brock TA, Williamson JR (1999) Role for nitric oxide in the hyperpermeability and hemodynamic changes induced by intravenous VEGF. Invest Ophthalmol Vis Sci 40(3):689–696
Okuyama H, Krishnamachary B, Zhou YF, Nagasawa H, Bosch-Marce M, Semenza GL (2006) Expression of vascular endothelial growth factor receptor 1 in bone marrow-derived mesenchymal cells is dependent on hypoxia-inducible factor 1. J Biol Chem 281(22):15554–15563. doi:10.1074/jbc.M602003200
Ozen I, Deierborg T, Miharada K, Padel T, Englund E, Genove G, Paul G (2014) Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol 128(3):381–396. doi:10.1007/s00401-014-1295-x
Sakuma R, Kawahara M, Nakano-Doi A, Takahashi A, Tanaka Y, Narita A, Kuwahara-Otani S, Hayakawa T et al (2016) Brain pericytes serve as microglia-generating multipotent vascular stem cells following ischemic stroke. J Neuroinflammation 13(1):57. doi:10.1186/s12974-016-0523-9
Li A, Dubey S, Varney ML, Dave BJ, Singh RK (2003) IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol 170(6):3369–3376
Basu A, Menicucci G, Maestas J, Das A, McGuire P (2009) Plasminogen activator inhibitor-1 (PAI-1) facilitates retinal angiogenesis in a model of oxygen-induced retinopathy. Invest Ophthalmol Vis Sci 50(10):4974–4981. doi:10.1167/iovs.09-3619
Petit I, Jin D, Rafii S (2007) The SDF-1-CXCR4 signaling pathway: a molecular hub modulating neo-angiogenesis. Trends Immunol 28(7):299–307. doi:10.1016/j.it.2007.05.007
Asare Y, Schmitt M, Bernhagen J (2013) The vascular biology of macrophage migration inhibitory factor (MIF). Expression and effects in inflammation, atherogenesis and angiogenesis. Thromb Haemost 109(3):391–398. doi:10.1160/TH12-11-0831
Simons D, Grieb G, Hristov M, Pallua N, Weber C, Bernhagen J, Steffens G (2011) Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. J Cell Mol Med 15(3):668–678. doi:10.1111/j.1582-4934.2010.01041.x
Zhang S, Zis O, Ly PT, Wu Y, Zhang S, Zhang M, Cai F, Bucala R et al (2014) Down-regulation of MIF by NFkappaB under hypoxia accelerated neuronal loss during stroke. FASEB J: Off Publ Fed Am Soc Exp Biol 28(10):4394–4407. doi:10.1096/fj.14-253625
Zis O, Zhang S, Dorovini-Zis K, Wang L, Song W (2015) Hypoxia signaling regulates macrophage migration inhibitory factor (MIF) expression in stroke. Mol Neurobiol 51(1):155–167. doi:10.1007/s12035-014-8727-4
Yang JP, Liu HJ, Cheng SM, Wang ZL, Cheng X, Yu HX, Liu XF (2009b) Direct transport of VEGF from the nasal cavity to brain. Neurosci Lett 449(2):108–111. doi:10.1016/j.neulet.2008.10.090
Nair AB, Jacob S (2016) A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7(2):27–31. doi:10.4103/0976-0105.177703
Arai K, Jin G, Navaratna D, Lo EH (2009) Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. FEBS J 276(17):4644–4652. doi:10.1111/j.1742-4658.2009.07176.x
Winkler EA, Bell RD, Zlokovic BV (2011) Central nervous system pericytes in health and disease. Nat Neurosci 14(11):1398–1405. doi:10.1038/nn.2946
Witmer AN, Dai J, Weich HA, Vrensen GF, Schlingemann RO (2002) Expression of vascular endothelial growth factor receptors 1, 2, and 3 in quiescent endothelia. J Histochem Cytochem: Off J Histochem Soc 50(6):767–777
Suzuma K, Naruse K, Suzuma I, Takahara N, Ueki K, Aiello LP, King GL (2000) Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR, Flt1, and phosphatidylinositol 3-kinase-akt-dependent pathways in retinal vascular cells. J Biol Chem 275(52):40725–40731. doi:10.1074/jbc.M006509200
Tchaikovski V, Fellbrich G, Waltenberger J (2008) The molecular basis of VEGFR-1 signal transduction pathways in primary human monocytes. Arterioscler Thromb Vasc Biol 28(2):322–328. doi:10.1161/ATVBAHA.107.158022
Koch S, Claesson-Welsh L (2012) Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harbor Perspect Med 2(7):a006502. doi:10.1101/cshperspect.a006502
Mihaylova MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13(9):1016–1023. doi:10.1038/ncb2329
Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT (2011) Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol 31(17):3531–3545. doi:10.1128/MCB.05124-11
Liang D, Han D, Fan W, Zhang R, Qiao H, Fan M, Su T, Ma S et al (2016) Therapeutic efficacy of apelin on transplanted mesenchymal stem cells in hindlimb ischemic mice via regulation of autophagy. Sci Rep 6:21914. doi:10.1038/srep21914
Ribatti D, Nico B, Crivellato E (2011) The role of pericytes in angiogenesis. Int J Dev Biol 55(3):261–268. doi:10.1387/ijdb.103167dr
von Tell D, Armulik A, Betsholtz C (2006) Pericytes and vascular stability. Exp Cell Res 312(5):623–629. doi:10.1016/j.yexcr.2005.10.019
Stefansson S, McMahon GA, Petitclerc E, Lawrence DA (2003) Plasminogen activator inhibitor-1 in tumor growth, angiogenesis and vascular remodeling. Curr Pharm Des 9(19):1545–1564
Li Q, He Q, Baral S, Mao L, Li Y, Jin H, Chen S, An T et al (2016) MicroRNA-493 regulates angiogenesis in a rat model of ischemic stroke by targeting MIF. FEBS J 283(9):1720–1733. doi:10.1111/febs.13697
Barbara NP, Wrana JL, Letarte M (1999) Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem 274(2):584–594
Zhu Y, Sun Y, Xie L, Jin K, Sheibani N, Greenberg DA (2003) Hypoxic induction of endoglin via mitogen-activated protein kinases in mouse brain microvascular endothelial cells. Stroke; J Cereb Circ 34(10):2483–2488. doi:10.1161/01.STR.0000088644.60368.ED
Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP (1999) Defective angiogenesis in mice lacking endoglin. Science 284(5419):1534–1537
Li C, Issa R, Kumar P, Hampson IN, Lopez-Novoa JM, Bernabeu C, Kumar S (2003) CD105 prevents apoptosis in hypoxic endothelial cells. J Cell Sci 116(Pt 13):2677–2685. doi:10.1242/jcs.00470
Acknowledgments
This work was supported by grants from the Fondation du CHU de Québec (2331) and Merck-Faculty of Medicine, Laval University. NJL is supported by a scholarship from the Fondation du CHU de Québec and the Faculty of Medicine, Laval University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Jean LeBlanc, N., Guruswamy, R. & ElAli, A. Vascular Endothelial Growth Factor Isoform-B Stimulates Neurovascular Repair After Ischemic Stroke by Promoting the Function of Pericytes via Vascular Endothelial Growth Factor Receptor-1. Mol Neurobiol 55, 3611–3626 (2018). https://doi.org/10.1007/s12035-017-0478-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0478-6