
Abstract
Carnosic acid (CA; C20H28O4) is a phenolic diterpene found in rosemary (Rosmarinus officinalis L.) and exhibits protective properties, e.g., antioxidant, anti-inflammatory, antitumor, and antimicrobial activities. In this context, CA has been viewed as a neuroprotective agent due to its ability in rescuing neuronal cells from pro-oxidant and pro-apoptotic challenges. In the present work, we found that CA pretreatment at 1 µM for 12 h suppressed the mitochondria-related pro-oxidant and mitochondria-dependent pro-apoptotic effects of chlorpyrifos (CPF) in human neuroblastoma SH-SY5Y cells. CA prevented mitochondrial membrane potential disruption and decreased the levels of oxidative stress markers in mitochondrial membranes obtained from cells exposed to CPF. CA also inhibited cytochrome c release and activation of the caspases-9 and -3, as well as decreased DNA fragmentation, in CPF-treated cells. CA upregulated the content of glutathione (GSH) in mitochondria by a mechanism involving the activation of the phosphoinositide-3-kinase (PI3K)/Akt/nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway, since inhibition of PI3K/Akt or silencing of Nrf2 using siRNA strategy abolished the protection exerted by CA in SH-SY5Y cells. Therefore, CA protected mitochondria of SH-SY5Y cells through the activation of the PI3K/Akt/Nrf2 axis, causing upregulation of the mitochondrial GSH content and consequent antioxidant and anti-apoptotic effects.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Carnosic acid (CA; C20H28O4) is a phenolic diterpene isolated mainly from rosemary (Rosmarinus officinalis L.) and exhibits antioxidant, anti-inflammatory, antitumor, and antimicrobial activities (Yanagitai et al. 2012; Satoh et al. 2013; Birtić et al. 2015; González-Vallinas et al. 2015). Furthermore, CA is a neuroprotective agent, as recently reviewed by our research group (de Oliveira 2015). CA crosses the blood–brain barrier and may reach concentrations ranging from 1.5 to 1.9 µg/g in rat brain after subchronic exposure to a 40 % CA enriched diet, as demonstrated by Romo Vaquero et al. (2013). In this context, Satoh et al. (2008) found CA in mice brain in concentrations between 2.4 and 4.8 µmol/kg tissue after oral administration of CA. Furthermore, CA administration protected mice brain against KCN (Zhang et al. 2015) and traumatic brain injury (Miller et al. 2015).
CA is a pro-electrophilic molecule, i.e., CA becomes electrophilic after reacting with oxidants, generating a quinone form that is able to activates the nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor, the redox master regulator in mammalian cells (Satoh et al. 2008). Nrf2 activation leads to upregulation of antioxidant enzymes, such as γ-glutamyl cysteine ligase (γ-GCL), heme oxygenase-1 (HO-1), NAD(P)H:quinone oxidoreductase 1 (NQO1), glutathione peroxidase (GPx), glutathione reductase (GR), thioredoxin (Trx), and Mn-superoxide dismutase (Mn-SOD) (de Oliveira 2015; O’Connell and Hayes 2015; Espinosa-Diez et al. 2015). Nrf2 is also involved with the maintenance of the bioenergetics status in mammalian cells (Dinkova-Kostova et al. 2015; Dinkova-Kostova and Abramov 2015). Recently, we reported that CA activated Nrf2 in SH-SY5Y cells, causing upregulation of both catalytic (GCLC) and regulatory (GCLM) subunits of γ-GCL (de Oliveira et al. 2015, 2016), the rate-limiting enzyme in glutathione (GSH) synthesis (Lu 2013). CA exhibited the ability to protect SH-SY5Y cells against paraquat (de Oliveira et al. 2016) and methylglyoxal (de Oliveira et al. 2015) through the activation of the phosphoinositide-3-kinase (PI3K)/Akt/Nrf2 axis. Other research groups have previously demonstrated that CA mediated cytoprotective effects by exerting an antioxidant action using other experimental models (Chen et al. 2012; Lin et al. 2014; Wu et al. 2015). Nonetheless, whether CA would prevent mitochondrial dysfunction induced by other pro-oxidant agents remains to be determined.
Mitochondria are the major site of ATP production in mammalian cells and also coordinate cell survival and death-associated signaling pathways (Green et al. 2014; Galluzzi et al. 2014). Mitochondrial dysfunction takes a central role in several pathologies, including cardiovascular and neurodegenerative diseases (Nicholls and Budd 1998; Naoi et al. 2005; Brown and Bal-Price 2003; Bernardi et al. 1998; Rosca and Hoppel 2010; Griffiths 2012). Furthermore, mitochondrial disturbances may be consequence of exposure to toxic agents, such as drugs and pollutants (Chen et al. 2015; de Oliveira and Jardim 2016; Varga et al. 2015; Abdolghaffari et al. 2015; Yang et al. 2015). In this context, there is increasing interest in natural and synthetic agents that would be useful in preventing mitochondrial dysfunction.
Chlorpyrifos (CPF) is an organophosphate pesticide that inhibits acetylcholinesterase, causing neurotoxicity in humans (Whitney et al. 1995; Moser 2000). Due to its chemical structure, CPF is lipophilic and easily enters cells by crossing the plasma membrane (Ki et al. 2013). CPF induces redox impairment by increasing the production of reactive species in animal cells, as evidenced in several in vitro and in vivo experimental models (Ma et al. 2013; Nishi and Hundal 2013; Jasna et al. 2014; Lee et al. 2014a; Jang et al. 2015; Khalil 2015; Deng et al. 2016). Additionally, CPF elicits lipid peroxidation and DNA damage in neuronal cells, causing cell death through apoptosis (Geter et al. 2008; Saulsbury et al. 2009; Yu et al. 2008). CPF also causes mitochondrial dysfunction by interfering with the electron flux between the components of the respiratory chain (Salama et al. 2014) and by altering mitochondrial dynamics and the movement of these organelles in neuronal cells (Middlemore-Risher et al. 2011).
Therefore, based on previously findings from our research group and others, we examined here whether and how CA would prevent mitochondrial dysfunction induced by CPF in human neuroblastoma SH-SY5Y cells.
Materials and Methods
Materials
Plastic materials utilized in cell culture were obtained from Corning, Inc. (NY, USA) and Beckton Dickson (NJ, USA). Culture analytical grade reagents and CPF were purchased from Sigma (MO, USA). All other chemicals and assay kits utilized here were obtained as described below.
Cell Culture and Chemical Treatment
Human dopaminergic neuroblastoma cell line (SH-SY5Y) was purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 HAM nutrient medium (1:1 mixture) supplemented with 10 % fetal bovine serum (FBS) and 2 mM l-glutamine in a 5 % CO2 humidified incubator at 37 °C. The cells were plated at an appropriate density according to the different experimental protocols utilized in this work. All data were obtained by performing three or five independent experiments each done in triplicate.
CPF (dissolved in DMSO) was utilized at 25–100 µM, depending on the experiment, in order to induce cytotoxicity, as previously described (Ki et al. 2013; Lee et al. 2014a; Raszewski et al. 2015). CA was utilized at 1 µM, since this concentration elicited cytoprotection efficiently against several chemical stressors in previous works, as reported by our research group (de Oliveira et al. 2015, 2016) and others (Chen et al. 2012; Lin et al. 2014; Wu et al. 2015). CA was added to the medium 12 h before CPF, which was used for additional 6 or 24 h, depending on the experiment. LY294002 (5 µM) was added to the culture medium 1 h before CA whenever necessary. Specific concentrations and periods of treatment are indicated in figure legends.
Analyses of Cellular Viability, Cytotoxicity, and Apoptosis-Related Parameters
Cellular viability was assayed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Mosmann 1983). The cells were plated onto 96-well plates, and when the culture reached 70-80 % confluence, the medium was removed and the treatments were applied. After the treatment of the cells with each chemical, 20 µL of MTT solution (5 mg/mL sterile stock solution) was added to each well (final concentration of 0.5 mg/mL MTT). The cells were left for 4 h at 37 °C in a humidified atmosphere of 5 % CO2. Then, the medium was removed and 100 µL DMSO for 30 min was utilized to dissolve the formazan crystals. The optical density of each well was measured at 570 nm in a plate reader (Molecular Devices, CA, USA). Lactate dehydrogenase (LDH) leakage assay was performed based on the manufacturer instructions (CytoTox 96-NonRadioactive Cytotoxicity Assay, Promega).
Caspase-9 enzyme activity was measured using a fluorimetric assay kit (Abcam, MA, USA; EX: 400 nm, EM: 505 nm). Caspase-3 enzyme activity was quantified using an assay kit (Sigma, MO, USA; EX: 360 nm, EM: 460 nm). We utilized a commercial ELISA kit that detects DNA fragmentation in cell lysates (Roche, Germany). The cells were incubated with 5′-bromo-2′-deoxy-uridine (BrdU) to label nuclear DNA. The BrdU-labeled DNA fragments are released from the cells to the cytoplasm during apoptosis and into cell culture during necrosis. We measured here the levels of cytoplasmic BrdU-labeled DNA fragments according to the instructions of the manufacturer. After each incubation, the samples were read at 450 nm (reference wavelength 690 nm) in a plate reader (Molecular Devices, CA, USA).
Determination of Intracellular Reactive Oxygen Species (ROS) Production
Intracellular ROS production was measured using the nonpolar compound 2′-7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (LeBel et al. 1992). In the final 30 min of each incubation time with specific chemicals (CPF and/or CA, with or without LY294002 co-treatment), the cells were loaded with 100 µM DCFH-DA dissolved in medium and were incubated for 30 min at 37 °C to allow cellular incorporation. After this period, the medium was removed and fresh medium was added to the cells, and the oxidation of intracellular DCFH was monitored during 30 min with 1 min intervals at 37 °C in a fluorescence plate reader (Molecular Devices, USA; EX: 485 nm, EM: 535 nm) (Wang and Joseph 1999).
Mitochondrial Isolation
After the incubation with specific treatments, the cells were washed and resuspended in buffer (250 mM sucrose, 20 mM HEPES—pH 7.4—10 mM KCl, 1 mM EGTA, 1 mM EDTA, 1 mM MgCl2, 1 mM dithiothreitol, 1 mM phenylmethylsulphonyl fluoride, 1 mM benzamidine, 1 mM pepstatin A, 10 mg/mL leupeptin, and 2 mg/mL aprotinin) (Wang et al. 2014). After this, cells were homogenized and nuclei, unbroken cells, and cell debris were obtained after centrifugation (1000×g, 10 min, 4 °C). To isolate cytosolic fraction, the supernatant resulting from the previous centrifugation was centrifuged again (13,000×g, 20 min, 4 °C). The final supernatant was used as cytosolic fraction and the final pellet contains the mitochondrial fraction.
Lipid Peroxidation and Protein Carbonyl Measurement
The levels of malondialdehyde (MDA, a lipid peroxidation marker) and protein carbonyl were quantified in the samples at the end of each incubation using commercial kits (Abcam, MA, USA). After reaction with the samples, DNP hydrazones (for protein carbonyl detection) and MDA were read in a plate reader (Molecular Devices, CA, USA) at 375 and 532 nm, respectively. The same procedures were applied in order to verify the levels of MDA and protein carbonyl in mitochondrial membranes after isolation of mitochondria.
Mitochondrial Membrane Potential
Mitochondrial membrane potential (MMP) was examined using a commercial kit that uses tetraethylbenzimidazolylcarbocyanide iodine (JC-1), which is a lipophilic cationic dye that accumulates in mitochondria depending on its membrane potential. JC-1 is predominantly found in a monomer form that yields green fluorescence at low MMP (emission of 530 ± 15 nm). On the contrary, JC-1 aggregates at high MMP yielding a red to orange fluorescence (emission of 590 ± 17.5 nm). The cells (1.5 × 104) were stained with JC-1 (20 µM) in dilution buffer for 10 min (37 °C) in the dark. The cells were washed twice with dilution buffer. The treatments were applied and incubated for desired period of time. The samples were read at EX: 485 nm, EM: 540, and 590 nm and cut-off at 530 nm in a fluorescence plate reader (Molecular Devices, USA).
Quantification of ATP Levels
We detected ATP levels in the samples after the end of each treatment using a commercial kit following the instructions of the manufacturer (Abcam, MA, USA). After the reaction of the samples with ATP probe, the samples were read in a fluorescence plate reader (Molecular Devices, USA; EX: 535 nm, EM: 590 nm).
Detection of the Immunocontent of Mitochondrial and Cytosolic Cytochrome c
Mitochondrial and cytosolic cytochrome c immunocontents were detected by utilizing ELISA assay kits following the instructions of the manufacturer (Abcam, MA, USA).
Measurement of cleaved PARP
The levels of cleaved PARP were analyzed using a commercial kit following the instructions of the manufacturer (Cell Signaling, MA, USA).
Measurement of reduced glutathione
After each experiment, the cells were washed and collected and GSH was quantified following the protocol of a commercial kit (Abcam, MA, USA). GSH was detected after reaction with Thiol Green Indicator and the samples were read in a fluorescence plate reader (Molecular Devices, USA; EX: 490 nm, EM: 520 nm).
Nuclear Isolation
Nuclear isolation was performed by utilizing the Nuclear Extraction Kit, which was purchased from Cayman Chemical (MI, USA). Briefly, ≅1 × 107 cells (80–90 % confluence) were collected in phosphate buffered saline (PBS, ice-cold). After centrifugation (300×g, 5 min, 4 °C), cells were pelleted and then resuspended in ice-cold hypotonic buffer, which is responsible for inducing swelling of the cells. The addition of 10 % Nonidet P-40 dissolved the cell membranes, then allowing access to the cytoplasmic fraction without causing nuclear membrane damage. Centrifugation (13,000×g for 30 s, 4 °C) was performed in order to separate cytoplasmic fraction from nuclei. The pelleted nuclei were lysed in ice-cold extraction buffer. Finally, another centrifugation (14,000×g for 10 min, 4 °C) was performed to isolate nuclear extracts, which were used in the quantification of Nrf2 in the cell nucleus.
Measurement of Nrf2 Immunocontent in Nuclear Samples
After nuclear extraction, quantification of the nuclear immunocontent of Nrf2 was performed by utilizing an ELISA assay kit following the instructions of the manufacturer (Active Motif, CA, USA). Briefly, samples (30 µg protein) were added into the wells containing a monoclonal capture antibody specific for Nrf2. Then, a detection antibody specific for Nrf2 was added to the wells. A horseradish peroxidase labeled anti-rabbit IgG was pipetted into the wells after washing. Finally, a substrate solution (TMB) was added, generating color that was detected in a microplate reader (Molecular Devices, CA, USA) at 450 nm.
Nrf2 Knockdown by siRNA Transfection
Nrf2 silencing was obtained by the transfection of the SH-SY5Y cells transiently with Nrf2 siRNA (5′-CCCATTGATGTTTCTGATCTA-3′) or All Stars Negative Control (NC) siRNA by applying the Lipofectamine RNAiMAX reagent (Life Technologies, CA, USA) (de Oliveira et al. 2015, 2016).
Statistical Analyses
Statistical analyses were performed with GraphPad 5.0 software. The results are presented as the mean ± standard deviation (SD) of three or five independent experiments each done in triplicate; p values were considered significant when p < 0.05. Differences in experimental groups were determined by one-way ANOVA followed by the post hoc Tukey’s test.
Results
Cell Viability and Cellular Redox Parameters
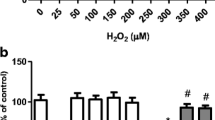
According to Fig. 1a, CPF decreased cell viability in a dose-dependent manner. Moreover, CPF exposure for 24 h induced cytotoxicity in SH-SY5Y cells (Fig. 1b). Based on these data, we utilized CPF at 100 µM for the other experiments, we performed here.

The effects of chlorpyrifos at different concentrations for 24 h on SH-SY5Y cell viability (a) and cytotoxicity (b). Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group, **p < 0.001 versus the control group
In order to verify whether CA would protect SH-SY5Y cells against CPF, we evaluated cell viability and redox-related parameters. CA pretreatment (1 µM for 12 h) prevented loss of viability (Fig. 2a) and cytotoxicity (Fig. 2b) in CPF-treated cells. CA also abrogated ROS production (Fig. 2c), lipid peroxidation (Fig. 2d), and protein carbonylation (Fig. 2e) in cells exposed to CPF. We utilized CA at 1 µM because this concentration elicited better results when compared to CA at lower or higher concentrations (Fig. S1).

The effects of a pretreatment with carnosic acid at 1 µM for 12 h on cell viability (a), cytotoxicity (b), ROS production (c), total lipid peroxidation (d), and total protein carbonylation (e). Chlorpyrifos was utilized at 100 µM for 24 h. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group, a different from chlorpyrifos-treated group; a p < 0.05 versus chlorpyrifos-treated cells
Mitochondrial Function
CA efficiently reduced lipid peroxidation (Fig. 3a) and protein carbonylation (Fig. 3b) in the membranes of mitochondria isolated from CPF-treated cells. CA also prevented MMP disruption (Fig. 3c) and ATP production impairment (Fig. 3d) in this experimental model.

The effects of a pretreatment with carnosic acid at 1 µM for 12 h on mitochondrial membrane a MDA content, b protein carbonyl content, c mitochondrial membrane potential (MMP), and d ATP levels. Chlorpyrifos was utilized at 100 µM for 24 h. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group; a p < 0.05 versus chlorpyrifos-treated cells
Apoptosis-Related Parameters
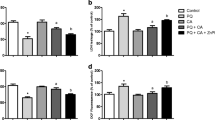
To determine whether CA pretreatment would prevent CPF-triggered apoptosis, we examined apoptotic parameters associated with the intrinsic apoptotic pathway (i.e., which is dependent on mitochondria). CA pretreatment prevented release of cytochrome c to the cytosol (Fig. 4a) and loss of mitochondrial cytochrome c (Fig. 4b), consequently decreasing caspase-9 (Fig. 4c) and caspase-3 (Fig. 4d) activities. As expected, CA also suppressed cleavage of PARP (Fig. 4e) and fragmentation of DNA (Fig. 4f) in CPF-administrated cells. Therefore, CA efficiently prevented mitochondria-dependent cell death in SH-SY5Y cells exposed to CPF.

The effects of a pretreatment with carnosic acid at 1 µM for 12 h on a cytosolic cytochrome c content, b mitochondrial cytochrome c content, c caspase-9 activity, d caspase-3 activity, e cleaved PARP levels, and f DNA fragmentation. Chlorpyrifos was utilized at 100 µM for additional 24 h. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group; a p < 0.05 versus chlorpyrifos-treated cells
Mitochondrial Parameters and Mechanism of Mitochondrial Protection
To verify by which mechanism CA exerted mitochondrial protection against CPF, we analyzed the levels of GSH in the mitochondria of SH-SY5Y cells. As depicted in Fig. 5a, CA upregulated GSH levels in a time-dependent manner. We also investigated the mechanism involved in the CA-elicited increase in mitochondrial GSH levels, as demonstrated in Fig. 5b. CA induced an increase in the levels of GSH by a PI3K/Akt-associated mechanism, since LY294002 (specific inhibitor of PI3K) suppressed the effects of CA on mitochondrial GSH levels. LY294002 alone did not change the mitochondrial levels of GSH in this experimental model. We also examined whether Nrf2 activation would be involved in the regulation of GSH synthesis in this experimental model. As may be observed in Fig. 5c, Nrf2 knockdown blocked the CA-induced increase in the mitochondrial GSH.

The effects of a treatment with carnosic acid at 1 µM for different periods on a mitochondrial GSH content. In b, the effects of a pretreatment with LY294002 (5 µM for 1 h prior exposure to CA) are shown regarding the mitochondrial levels of GSH. The effects of Nrf2 knockdown (48 h) on the mitochondrial GSH levels in CA-treated SH-SY5Y cells are shown in c. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group; a p < 0.05 versus CA-treated cells; # p < 0.05 versus the NC siRNA-treated cells
To evaluate whether the PI3K/Akt/Nrf2 signaling pathway was also involved in the modulation of redox parameters associated with mitochondria, we utilized LY294002 as a co-treatment in SH-SY5Y cells. We found that PI3K/Akt inhibition suppressed the antioxidant effects elicited by CA on mitochondrial membranes, namely inhibition of lipid peroxidation (Fig. 6a) and protein carbonylation (Fig. 6b). Furthermore, LY294002 blocked MMP improvement caused by CA pretreatment in CPF-treated cells (Fig. 6c). LY294002 co-treatment also abrogated the effects of CA on cytochrome c release (Fig. 7a) and on the levels of cytochrome c in mitochondria (Fig. 7b).

The effects of LY294002 (5 µM; cells were exposed to LY294002 1 h prior CA treatment for 12 h) on a lipid peroxidation in mitochondrial membranes, b protein carbonylation in mitochondrial membranes, and c mitochondrial membrane potential (MMP). d cytosolic cytochrome c levels, and e mitochondrial cytochrome c levels. Cells were exposed to CPF for additional 24 h. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group; a p < 0.05 versus chlorpyrifos-treated cells; b p < 0.05 versus CPF + CA-treated cells

The effects of (5 µM; cells were exposed to LY294002 1 h prior CA treatment for 12 h) on a cytosolic cytochrome c levels and b mitochondrial cytochrome c levels. Cells were exposed to CPF for additional 24 h. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group; a p < 0.05 versus chlorpyrifos-treated cells; b p < 0.05 versus CPF + CA-treated cells
Involvement of PI3K/Akt Signaling Pathway in the Cytoprotective Effects Elicited by CA
Using LY294002 as a co-treatment, we investigated whether the PI3K/Akt signaling pathway would be participating in the cytoprotective effects induced by CA. According to Fig. 8a, LY294002 abrogated the protective effect of CA regarding cell viability. Also, LY294002 blocked the inhibitory effect of CA on cytotoxicity (Fig. 8b). Consequently, inhibition of PI3K/Akt signaling pathway suppressed the effect of CA on DNA fragmentation in cells exposed to CPF (Fig. 8c).

The effects of LY294002 (5 µM; cells were exposed to LY294002 1 h prior CA treatment for 12 h) on a cell viability, b LDH leakage, and c DNA fragmentation. Cells were exposed to CPF for additional 24 h. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group; a p < 0.05 versus chlorpyrifos-treated cells; b p < 0.05 versus CPF + CA-treated cells
Involvement of Nrf2 in the Mitochondrial Protection Exerted by CA
Nrf2 knockdown abrogated the CA-induced improvement of MMP in CPF-treated SH-SY5Y cells (Fig. 9a). Additionally, CA elicited anti-apoptotic effect through a Nrf2-dependent mechanism, since Nrf2 knockdown prevented the CA-triggered inhibition on DNA fragmentation in cells exposed to CPF (Fig. 9b).

The effects of Nrf2 siRNA (48 h) on mitochondrial membrane potential (a) and DNA fragmentation (b) of chlorpyrifos (CPF) and/or carnosic acid-treated cells. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group; **p < 0.05 versus the CPF-treated cells transfected with negative control (NC) siRNA; a p < 0.05 versus the CPF-treated cells transfected with NC siRNA; b p < 0.05 versus the CPF and CA-treated cells transfected with NC siRNA
To analyze whether Nrf2 would be involved in the cytoprotective effects elicited by CA on CPF-treated cells, we utilized Nrf2 knockdown strategy, as depicted in Fig. 10. CA failed to prevent loss of cell viability (Fig. 10a) and CPF-induced cytotoxicity (Fig. 10b) in SH-SY5Y cells. Data demonstrating the efficacy of Nrf2 knockdown are shown in Fig. S2.

The effects of Nrf2 siRNA (48 h) on cell viability (a) and LDH leakage (b) of chlorpyrifos (CPF) and/or carnosic acid-treated cells. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group; **p < 0.05 versus the CPF-treated cells transfected with negative control (NC) siRNA; a p < 0.05 versus the CPF-treated cells transfected with NC siRNA; b p < 0.05 versus the CPF and CA-treated cells transfected with NC siRNA
Discussion
CA exerts neuroprotection in both in vitro and in vivo experimental models, at least in part, due to its antioxidant property (de Oliveira 2015). CA is an Nrf2 activator and may upregulate this transcription factor more intensely than sulforaphane, as previously reported (Kraft et al. 2004; Hong et al. 2005). In the present work, CA pretreatment at 1 µM efficiently prevented cytotoxicity and mitochondrial dysfunction induced by CPF, a pro-oxidant agent with pro-apoptotic capacity (Bagchi et al. 1995; Lassiter et al. 2009; Lee et al. 2012, 2014b). CA upregulated GSH synthesis through a PI3K/Akt/Nrf2 axis, causing an increase in the mitochondrial content of GSH and preventing MMP disruption, as well as abolishing the CPF-triggered intrinsic apoptotic pathway. PI3K/Akt signaling pathway inhibition or Nrf2 silencing suppressed the protective effects of CA in cells exposed to CPF, demonstrating that part of the mechanism of cytoprotection elicited by CA was dependent on those signaling pathways.
We previously demonstrated that CA prevented cytotoxicity against paraquat (de Oliveira et al. 2016) and methylglyoxal (de Oliveira et al. 2015). Furthermore, other research groups have demonstrated that CA protected neuronal cells against 6-hydroxydopamine (Chen et al. 2012; Lin et al. 2014; Wu et al. 2015) and amyloid-β peptide (Azad et al. 2011; Rasoolijazi et al. 2013; Meng et al. 2015). CA activates Nrf2 causing an increase in the immunocontents of antioxidant enzymes, such as HO-1, γ-GCL, NQO-1, Mn-SOD, GPx, and GR (Chen et al. 2012; de Oliveira et al. 2015, 2016). Additionally, CA exhibits anti-apoptotic effects, at least in part, by preserving mitochondrial function and inhibiting cytochrome c release to the cytosol (de Oliveira 2015). Actually, we observed here that CA blocked cytochrome c release from mitochondria in CPF-treated cells (Fig. 4a). CPF is a pro-apoptotic agrochemical triggering cell death by a mechanism involving JNK and p38 protein kinases activation (Ki et al. 2013), dynamin-related protein 1 (Drp1) migration to mitochondria, and increased caspase-3 activity (Park et al. 2015), leading to DNA fragmentation, a hallmark of apoptosis (Raszewski et al. 2015). Drp1 is a protein involved in the regulation of mitochondrial fission, an event that plays an important role during mitophagy (Fukushima et al. 2001; Zhu et al. 2004). Thus, CPF induces mitochondrial dysfunction not only due to its intrinsic pro-oxidant capacity, but also by altering mitochondrial architecture and dynamics. Further research would be necessary in order to evaluate the role of CA in preventing impairments in mitochondrial dynamics resulting from exposure to CPF or other agent able to induce mitochondrial dysfunction.
GSH synthesis upregulation has been associated with neuroprotection in several experimental models (Agarwal and Shukla 1999; Bakshi et al. 2015; Mitra et al. 2015). Moreover, GSH depletion has been linked to mitochondrial dysfunction in brain cells (Heales et al. 1995). The control of GSH production is exerted by signaling pathways upstream Nrf2, such as JNK, p38, ERK1/2, and PI3K/Akt (Forman and Dickinson 2003; Lu 2009, 2013; Satoh et al. 2013). GSH is considered the main non-enzymatic antioxidant defense in mammalian cells due to its direct role as antioxidant and also because this molecule is utilized to convert hydrogen peroxide (H2O2) to water by GPx, among other functions (Morris et al. 2014). Mitochondrial uptake of GSH is a complex event and needs to be coordinated according to the mitochondrial needs for this antioxidant agent (Lash 2006). In this context, CA activates PI3K/Akt signaling pathway, but not others in SH-SY5Y cells, as previously reported by us (de Oliveira et al. 2015, 2016) and by others (Lin et al. 2014). The upregulation of the PI3K/Akt pathway was previously demonstrated to take a role in the activation of Nrf2, since PI3K/Akt inhibition suppressed Nrf2 activation in SH-SY5Y cells treated with CA (de Oliveira et al. 2015, 2016). Here, we found that PI3K/Akt signaling inhibition caused a decrease in the mitochondrial content of GSH in CA-treated cells (Fig. 5b) and abrogated the antioxidant effects of CA on mitochondrial membranes (Fig. 6a, b), as well as impaired MMP (Fig. 6c) and favored cytochrome c release to the cytosol (Fig. 6d) in CPF-treated cells. The CA-induced Nrf2-dependent increase in the content of GSH in mitochondria was part of the mechanism by which CA protected mitochondria against CPF, as demonstrated here. CA failed to prevent loss of MMP in SH-SY5Y cells in which Nrf2 was silenced (Fig. 8a), favoring apoptosis in those cells (Fig. 8b).
Overall, we demonstrated here, for the first time, that CA was effective in preventing mitochondrial dysfunction and redox impairment in CPF-treated SH-SY5Y cells by a mechanism involving the activation of the antioxidant and pro-survival PI3K/Akt/Nrf2 axis.
References
Abdolghaffari AH, Baghaei A, Solgi R, Gooshe M, Baeeri M, Navaei-Nigjeh M, Hassani S, Jafari A, Rezayat SM, Dehpour AR, Mehr SE, Abdollahi M (2015) Molecular and biochemical evidences on the protective effects of triiodothyronine against phosphine-induced cardiac and mitochondrial toxicity. Life Sci 139:30–39. doi:10.1016/j.lfs.2015.07.026
Agarwal R, Shukla GS (1999) Potential role of cerebral glutathione in the maintenance of blood-brain barrier integrity in rat. Neurochem Res 24:1507–1514
Azad N, Rasoolijazi H, Joghataie MT, Soleimani S (2011) Neuroprotective effects of carnosic Acid in an experimental model of Alzheimer’s disease in rats. Cell J 13:39–44
Bagchi D, Bagchi M, Hassoun EA, Stohs SJ (1995) In vitro and in vivo generation of reactive oxygen species, DNA damage and lactate dehydrogenase leakage by selected pesticides. Toxicology 104:129–140
Bakshi R, Zhang H, Logan R, Joshi I, Xu Y, Chen X, Schwarzschild MA (2015) Neuroprotective effects of urate are mediated by augmenting astrocytic glutathione synthesis and release. Neurobiol Dis 82:574–579. doi:10.1016/j.nbd.2015.08.022
Bernardi P, Colonna R, Costantini P, Eriksson O, Fontaine E, Ichas F, Massari S, Nicolli A, Petronilli V, Scorrano L (1998) The mitochondrial permeability transition. BioFactors 8:273–281
Birtić S, Dussort P, Pierre FX, Bily AC, Roller M (2015) Carnosic acid. Phytochemistry 115:9–19. doi:10.1016/j.phytochem.2014.12.026
Brown GC, Bal-Price A (2003) Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 27:325–355
Chen JH, Ou HP, Lin CY, Lin FJ, Wu CR, Chang SW, Tsai CW (2012) Carnosic acid prevents 6-hydroxydopamine-induced cell death in SH-SY5Y cells via mediation of glutathione synthesis. Chem Res Toxicol 25:1893–1901. doi:10.1021/tx300171u
Chen L, Na R, Boldt E, Ran Q (2015) NLRP3 inflammasome activation by mitochondrial reactive oxygen species plays a key role in long-term cognitive impairment induced by paraquat exposure. Neurobiol Aging 36:2533–2543. doi:10.1016/j.neurobiolaging.2015.05.018
de Oliveira MR (2015) The dietary components carnosic acid and carnosol as neuroprotective agents: a mechanistic view. Mol Neurobiol. doi:10.1007/s12035-015-9519-1
de Oliveira MR, Jardim FR (2016) Cocaine and mitochondria-related signaling in the brain: a mechanistic view and future directions. Neurochem Int 92:58–66. doi:10.1016/j.neuint.2015.12.006
de Oliveira MR, Ferreira GC, Schuck PF, Dal Bosco SM (2015) Role for the PI3K/Akt/Nrf2 signaling pathway in the protective effects of carnosic acid against methylglyoxal-induced neurotoxicity in SH-SY5Y neuroblastoma cells. Chem Biol Interact 242:396–406. doi:10.1016/j.cbi.2015.11.003
de Oliveira MR, Ferreira GC, Schuck PF (2016) Protective effect of carnosic acid against paraquat-induced redox impairment and mitochondrial dysfunction in SH-SY5Y cells: role for PI3K/Akt/Nrf2 pathway. Toxicol In Vitro 32:41–54. doi:10.1016/j.tiv.2015.12.005
Deng Y, Zhang Y, Lu Y, Zhao Y, Ren H (2016) Hepatotoxicity and nephrotoxicity induced by the chlorpyrifos and chlorpyrifos-methyl metabolite, 3,5,6-trichloro-2-pyridinol, in orally exposed mice. Sci Total Environ 544:507–514. doi:10.1016/j.scitotenv.2015.11.162
Dinkova-Kostova AT, Abramov AY (2015) The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med 88:179–188. doi:10.1016/j.freeradbiomed.2015.04.036
Dinkova-Kostova AT, Baird L, Holmström KM, Meyer CJ, Abramov AY (2015) The spatiotemporal regulation of the Keap1-Nrf2 pathway and its importance in cellular bioenergetics. Biochem Soc Trans 43:602–610. doi:10.1042/BST20150003
Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, Lamas S (2015) Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol 6:183–197. doi:10.1016/j.redox.2015.07.008
Forman HJ, Dickinson DA (2003) Oxidative signaling and glutathione synthesis. BioFactors 17:1–12
Fukushima NH, Brisch E, Keegan BR, Bleazard W, Shaw JM (2001) The GTPase effector domain sequence of the Dnm1p GTPase regulates self-assembly and controls a rate-limiting step in mitochondrial fission. Mol Biol Cell 12:2756–2766
Galluzzi L, Bravo-San Pedro JM, Kroemer G (2014) Organelle-specific initiation of cell death. Nat Cell Biol 16:728–736. doi:10.1038/ncb3005
Geter DR, Kan HL, Lowe ER, Rick DL, Charles GD, Gollapudi BB, Mattsson JL (2008) Investigations of oxidative stress, antioxidant response, and protein binding in chlorpyrifos exposed rat neuronal PC12 cells. Toxicol Mech Methods 18:17–23. doi:10.1080/15376510701389530
González-Vallinas M, Reglero G, Ramírez de Molina A (2015) Rosemary (Rosmarinus officinalis L.) extract as a potential complementary agent in anticancer therapy. Nutr Cancer 67:1221–1229. doi:10.1080/01635581.2015.1082110
Green DR, Galluzzi L, Kroemer G (2014) Metabolic control of cell death. Science 345:1250256. doi:10.1126/science.1250256
Griffiths EJ (2012) Mitochondria and heart disease. Adv Exp Med Biol 942:249–267. doi:10.1007/978-94-007-2869-1_11
Heales SJ, Davies SE, Bates TE, Clark JB (1995) Depletion of brain glutathione is accompanied by impaired mitochondrial function and decreased N-acetyl aspartate concentration. Neurochem Res 20:31–38
Hong F, Freeman ML, Liebler DC (2005) Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol 18:1917–1926. doi:10.1021/tx0502138
Jang Y, Lee AY, Jeong SH, Park KH, Paik MK, Cho NJ, Kim JE, Cho MH (2015) Chlorpyrifos induces NLRP3 inflammasome and pyroptosis/apoptosis via mitochondrial oxidative stress in human keratinocyte HaCaT cells. Toxicology 338:37–46. doi:10.1016/j.tox.2015.09.006
Jasna JM, Anandbabu K, Bharathi SR, Angayarkanni N (2014) Paraoxonase enzyme protects retinal pigment epithelium from chlorpyrifos insult. PLoS One 9:e101380. doi:10.1371/journal.pone.0101380
Khalil AM (2015) Toxicological effects and oxidative stress responses in freshwater snail, Lanistes carinatus, following exposure to chlorpyrifos. Ecotoxicol Environ Saf 116:137–142. doi:10.1016/j.ecoenv.2015.03.010
Ki YW, Park JH, Lee JE, Shin IC, Koh HC (2013) JNK and p38 MAPK regulate oxidative stress and the inflammatory response in chlorpyrifos-induced apoptosis. Toxicol Lett 218:235–245. doi:10.1016/j.toxlet.2013.02.003
Kraft AD, Johnson DA, Johnson JA (2004) Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci 24:1101–1112. doi:10.1523/JNEUROSCI.3817-03.2004
Lash LH (2006) Mitochondrial glutathione transport: physiological, pathological and toxicological implications. Chem Biol Interact 163:54–67. doi:10.1016/j.cbi.2006.03.001
Lassiter TL, MacKillop EA, Ryde IT, Seidler FJ, Slotkin TA (2009) Is fipronil safer than chlorpyrifos? Comparative developmental neurotoxicity modeled in PC12 cells. Brain Res Bull 78:313–322. doi:10.1016/j.brainresbull.2008.09.020
LeBel CP, Ischiropoulos H, Bondy SC (1992) Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5:227–231
Lee JE, Park JH, Shin IC, Koh HC (2012) Reactive oxygen species regulated mitochondria-mediated apoptosis in PC12 cells exposed to chlorpyrifos. Toxicol Appl Pharmacol 263:148–162. doi:10.1016/j.taap.2012.06.005
Lee JE, Park JH, Jang SJ, Koh HC (2014a) Rosiglitazone inhibits chlorpyrifos-induced apoptosis via modulation of the oxidative stress and inflammatory response in SH-SY5Y cells. Toxicol Appl Pharmacol 278:159–171. doi:10.1016/j.taap.2014.04.021
Lee JE, Lim MS, Park JH, Park CH, Koh HC (2014b) Nuclear NF-κB contributes to chlorpyrifos-induced apoptosis through p53 signaling in human neural precursor cells. Neurotoxicology 42:58–70. doi:10.1016/j.neuro.2014.04.001
Lin CY, Chen JH, Fu RH, Tsai CW (2014) Induction of Pi form of glutathione S-transferase by carnosic acid is mediated through PI3K/Akt/NF-κB pathway and protects against neurotoxicity. Chem Res Toxicol 27:1958–1966. doi:10.1021/tx5003063
Lu SC (2009) Regulation of glutathione synthesis. Mol Aspects Med 30:42–59. doi:10.1016/j.mam.2008.05.005
Lu SC (2013) Glutathione synthesis. Biochim Biophys Acta 1830:3143–3153. doi:10.1016/j.bbagen.2012.09.008
Ma P, Wu Y, Zeng Q, Gan Y, Chen J, Ye X, Yang X (2013) Oxidative damage induced by chlorpyrifos in the hepatic and renal tissue of Kunming mice and the antioxidant role of vitamin E. Food Chem Toxicol 58:177–183. doi:10.1016/j.fct.2013.04.032
Meng P, Yoshida H, Tanji K, Matsumiya T, Xing F, Hayakari R, Wang L, Tsuruga K, Tanaka H, Mimura J, Kosaka K, Itoh K, Takahashi I, Kawaguchi S, Imaizumi T (2015) Carnosic acid attenuates apoptosis induced by amyloid-β 1–42 or 1–43 in SH-SY5Y human neuroblastoma cells. Neurosci Res 94:1–9. doi:10.1016/j.neures.2014.12.003
Middlemore-Risher ML, Adam BL, Lambert NA, Terry AV Jr (2011) Effects of chlorpyrifos and chlorpyrifos-oxon on the dynamics and movement of mitochondria in rat cortical neurons. J Pharmacol Exp Ther 339:341–349. doi:10.1124/jpet.111.184762
Miller DM, Singh IN, Wang JA, Hall ED (2015) Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp Neurol 264:103–110. doi:10.1016/j.expneurol.2014.11.008
Mitra S, Siddiqui WA, Khandelwal S (2015) C-Phycocyanin protects against acute tributyltin chloride neurotoxicity by modulating glial cell activity along with its anti-oxidant and anti-inflammatory property: a comparative efficacy evaluation with N-acetyl cysteine in adult rat brain. Chem Biol Interact 238:138–150. doi:10.1016/j.cbi.2015.06.016
Morris G, Anderson G, Dean O, Berk M, Galecki P, Martin-Subero M, Maes M (2014) The glutathione system: a new drug target in neuroimmune disorders. Mol Neurobiol 50:1059–1084. doi:10.1007/s12035-014-8705-x
Moser VC (2000) Dose-response and time-course of neurobehavioral changes following oral chlorpyrifos in rats of different ages. Neurotoxicol Teratol 22:713–723
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63
Naoi M, Maruyama W, Shamoto-Nagai M, Yi H, Akao Y, Tanaka M (2005) Oxidative stress in mitochondria: decision to survival and death of neurons in neurodegenerative disorders. Mol Neurobiol 31:81–93
Nicholls DG, Budd SL (1998) Neuronal excitotoxicity: the role of mitochondria. BioFactors 8:287–299
Nishi K, Hundal SS (2013) Chlorpyrifos induced toxicity in reproductive organs of female Wistar rats. Food Chem Toxicol 62:732–738. doi:10.1016/j.fct.2013.10.006
O’Connell MA, Hayes JD (2015) The Keap1/Nrf2 pathway in health and disease: from the bench to the clinic. Biochem Soc Trans 43:687–689. doi:10.1042/BST20150069
Park JH, Ko J, Hwang J, Koh HC (2015) Dynamin-related protein 1 mediates mitochondria-dependent apoptosis in chlorpyrifos-treated SH-SY5Y cells. Neurotoxicology 51:145–157. doi:10.1016/j.neuro.2015.10.008
Rasoolijazi H, Azad N, Joghataei MT, Kerdari M, Nikbakht F, Soleimani M (2013) The protective role of carnosic acid against beta-amyloid toxicity in rats. ScientificWorldJournal 2013:917082. doi:10.1155/2013/917082
Raszewski G, Lemieszek MK, Łukawski K, Juszczak M, Rzeski W (2015) Chlorpyrifos and cypermethrin induce apoptosis in human neuroblastoma cell line SH-SY5Y. Basic Clin Pharmacol Toxicol 116:158–167. doi:10.1111/bcpt.12285
Romo Vaquero M, García Villalba R, Larrosa M, Yáñez-Gascón MJ, Fromentin E, Flanagan J, Roller M, Tomás-Barberán FA, Espín JC, García-Conesa MT (2013) Bioavailability of the major bioactive diterpenoids in a rosemary extract: metabolic profile in the intestine, liver, plasma, and brain of Zucker rats. Mol Nutr Food Res 57:1834–1846. doi:10.1002/mnfr.201300052
Rosca MG, Hoppel CL (2010) Mitochondria in heart failure. Cardiovasc Res 88:40–50. doi:10.1093/cvr/cvq240
Salama M, El-Morsy D, El-Gamal M, Shabka O, Mohamed WM (2014) Mitochondrial complex I inhibition as a possible mechanism of chlorpyrifos induced neurotoxicity. Ann Neurosci 21:85–89. doi:10.5214/ans.0972.7531.210303
Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton SA (2008) Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem 104:1116–1131. doi:10.1111/j.1471-4159.2007.05039.x
Satoh T, McKercher SR, Lipton SA (2013) Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med 65:645–657. doi:10.1016/j.freeradbiomed.2013.07.022
Saulsbury MD, Heyliger SO, Wang K, Johnson DJ (2009) Chlorpyrifos induces oxidative stress in oligodendrocyte progenitor cells. Toxicology 259:1–9. doi:10.1016/j.tox.2008.12.026
Varga ZV, Ferdinandy P, Liaudet L, Pacher P (2015) Drug-induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol 309:1453–1467. doi:10.1152/ajpheart.00554.2015
Wang H, Joseph JA (1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med 27:612–616
Wang K, Zhu L, Zhu X, Zhang K, Huang B, Zhang J, Zhang Y, Zhu L, Zhou B, Zhou F (2014) Protective effect of paeoniflorin on Aβ25-35-induced SH-SY5Y cell injury by preventing mitochondrial dysfunction. Cell Mol Neurobiol 34:227–234. doi:10.1007/s10571-013-0006-9
Whitney KD, Seidler FJ, Slotkin TA (1995) Developmental neurotoxicity of chlorpyrifos: cellular mechanisms. Toxicol Appl Pharmacol 134:53–62
Wu CR, Tsai CW, Chang SW, Lin CY, Huang LC, Tsai CW (2015) Carnosic acid protects against 6-hydroxydopamine-induced neurotoxicity in in vivo and in vitro model of Parkinson’s disease: involvement of antioxidative enzymes induction. Chem Biol Interact 225:40–46. doi:10.1016/j.cbi.2014.11.011
Yanagitai M, Itoh S, Kitagawa T, Takenouchi T, Kitani H, Satoh T (2012) Carnosic acid, a pro-electrophilic compound, inhibits LPS-induced activation of microglia. Biochem Biophys Res Commun 418:22–26. doi:10.1016/j.bbrc.2011.12.087
Yang G, Zhou Z, Cen Y, Gui X, Zeng Q, Ao Y, Li Q, Wang S, Li J, Zhang A (2015) Death receptor and mitochondria-mediated hepatocyte apoptosis underlies liver dysfunction in rats exposed to organic pollutants from drinking water. Drug Des Devel Ther 9:4719–4733. doi:10.2147/DDDT.S86843
Yu F, Wang Z, Ju B, Wang Y, Wang J, Bai D (2008) Apoptotic effect of organophosphorus insecticide chlorpyrifos on mouse retina in vivo via oxidative stress and protection of combination of vitamins C and E. Exp Toxicol Pathol 59:415–423. doi:10.1016/j.etp.2007.11.007
Zhang D, Lee B, Nutter A, Song P, Dolatabadi N, Parker J, Sanz-Blasco S, Newmeyer T, Ambasudhan R, McKercher SR, Masliah E, Lipton SA (2015) Protection from cyanide-induced brain injury by the Nrf2 transcriptional activator carnosic acid. J Neurochem 133:898–908. doi:10.1111/jnc.13074
Zhu PP, Patterson A, Stadler J, Seeburg DP, Sheng M, Blackstone C (2004) Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J Biol Chem 279:35967–35974
Acknowledgments
This work was supported by CNPq. GCF is supported by Edital APQ1/FAPERJ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None to declare.
Electronic supplementary material
Below is the link to the electronic supplementary material.
12640_2016_9620_MOESM1_ESM.pdf
The effects of a pretreatment with carnosic acid (CA) at 0.1–2 μM for 12 h on the viability of SH-SY5Y cells exposed to chlorpyrifos (CPF) for 24 h. Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group, a different from CPF-treated cells. Supplementary material 1 (PDF 96 kb)
12640_2016_9620_MOESM2_ESM.pdf
The effects of Nrf2 siRNA (48 h) on nuclear Nrf2 content in SH-SY5Y cells exposed to carnosic acid (CA). Data are presented as the mean ± SD of three or five independent experiments each done in triplicate. One-way ANOVA followed by the post hoc Tukey’s test, *p < 0.05 versus the control group, a p < 0.05 versus carnosic acid-treated cells transfected with negative control (NC) siRNA. Supplementary material 2 (PDF 97 kb)
Rights and permissions
About this article

Cite this article
de Oliveira, M.R., Peres, A., Ferreira, G.C. et al. Carnosic Acid Affords Mitochondrial Protection in Chlorpyrifos-Treated Sh-Sy5y Cells. Neurotox Res 30, 367–379 (2016). https://doi.org/10.1007/s12640-016-9620-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-016-9620-x