
Abstract
Phosphatidylinositol 3-kinase (PI3K) plays several important roles in neuronal survival. Activation of the pathway is essential for the neuroprotective mechanisms of materials that shield neuronal cells from many stressful conditions. However, there have been no reports to date about the effect of the direct activation of the pathway in hypoxic injury of neuronal cells. We investigated whether the direct activation of the PI3K pathway inhibits neuronal cell death induced by hypoxia. Primary cultured cortical neurons (PCCNs) were exposed to hypoxic conditions (less than 1 mol% O2) and/or treated with PI3K activator. Hypoxia reduced the viability of PCCNs in a time-dependent manner, but treatment with PI3K significantly restored viability in a concentration-dependent manner. Among the signaling proteins involved in the PI3K pathway, those associated with survival, including Akt and glycogen synthase kinase-3β, were decreased shortly after exposure to hypoxia and those associated with cell death, including BAX, apoptosis-induced factor, cytochrome c, caspase-9, caspase-3, and poly(ADP-ribose) polymerase (PARP), were increased. However, treatment with PI3K activator normalized the expression levels of those signaling proteins. PARP activity and levels of ATP and NAD+ altered by hypoxia were also normalized with direct PI3K activation. All these findings suggest that direct and early activation is important for protecting neuronal cells from hypoxic injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebral infarction is one of the most common causes of death and permanent disability and is the result of a disturbance in blood flow to the brain, which causes hypoxic or anoxic conditions in neurons. Cerebral hypoxia is a pathological condition caused by a reduced supply of oxygen to the brain. Prolonged hypoxia has been shown to induce apoptosis in neuronal cells, resulting in hypoxic brain injury [1, 2]. Although hypoxia-induced cell death mechanisms still remain unclear, they include oxidative stress, excitotoxicity, inflammation, and the activation of several different cell death pathways [3]. However, recent studies have shown that transient hypoxia might enhance neuronal cell survival via the activation of the phosphatidylinositol 3-kinase (PI3K) pathway [4, 5].
The PI3K pathway is one of the most important survival pathways in neuronal cells [6–8]. In the PI3K pathway, activated PI3K phosphorylates Akt (protein kinase B) which is the direct effector of PI3K [9]. Phosphorylated Akt affects numerous downstream signaling proteins and then promotes neuronal cell survival [10]. There have been many reports showing that activation of the PI3K pathway increases neuronal cell survival in various stressful conditions [11–13]. Therefore, many studies have been performed to develop and/or to identify materials activating PI3K for the prevention of neuronal cell death [14–16].
Although several papers confirm that transient hypoxia activates the PI3K pathway [4, 5], there have been no reports showing the expression patterns of phosphorylated Akt in neuronal cells after more prolonged exposure to hypoxia. Considering the above findings about the role of the PI3K pathway in neuroprotective mechanisms of diverse materials, which might also have other protective mechanisms, we wondered whether early and direct activation of the PI3K pathway in neuronal cells in the beginning of a hypoxic insult could enhance the resistance of neuronal cells against prolonged hypoxia. Therefore, in this study, we confirmed the serial expression pattern of phosphorylated Akt in neuronal cells after exposure to hypoxia and investigated whether direct activation of PI3K at the beginning of hypoxia could protect neuronal cells against prolonged hypoxia.
Materials and Methods
Materials
To induce hypoxia, we used an anaerobic chamber (Anaerobic System Model 1025, Thermo Forma, Marietta, OH, USA). Protein protease inhibitor cocktail, tryptan blue solution, insulin, and DNase I were obtained from Sigma-Aldrich (St. Louis, MO, USA). A PI3K activator was purchased from Santa Cruz Biotech (Santa Cruz, CA, USA). It is a peptide with the sequence KKHTDDGYMPMSPGVA and a molecular weight of 1,732.8 Da. The tyrosine phosphorylated version of this peptide binds to the PI3Kinase SH2 domain activating the enzyme [17–19]. Before use in the experiments, the drugs were dissolved in distilled water and further diluted with culture medium to yield the desired final concentrations. LY294002, a PI3K inhibitor, was purchased from Sigma (St. Louis, MO, USA) to directly block PI3K.
Primary Cultures and Treatment of Cortical Neurons
All procedures using animals were consistent with Hanyang University’s guidelines for the care and use of laboratory animals. We made every effort to minimize the number of animals used and all animal suffering. Each animal was utilized only once.
Primary cultures of cortical neurons were acquired from the cerebral cortices of fetal Sprague–Dawley rats (16 days gestation) [20]. Briefly, rat embryos were decapitated, and their brains were isolated and put in a Petri dish half-filled with ice-cold Hank’s balanced salt solution (137 mM NaCl, 5.4 mM KCl, 0.3 mM Na2HPO4, 0.4 mM KH2PO4, 5.6 mM glucose, and 2.5 mM HEPES; Gibco BRL, NY, USA). Single cells separated from whole cerebral cortices were seeded on 100 mm Corning dishes (5 × 106 cells/cm2) coated with poly-l-lysine (Sigma, Saint Louis, MO, USA) or glass cover slips placed in 6- or 24-well Nunc plates (5 × 105 and 2.5 × 106 cells/cm2) and were suspended in 10 % fetal bovine serum/modified Eagle’s medium. After 24 h, the medium was changed to serum-free, neurobasal medium (NBM) supplemented with B27. Cultures were kept at 37 °C under a humidified 5 % CO2 atmosphere. After being incubated for 6 days, the cells were fixed with 4 % paraformaldehyde/PBS for anti-MAP-2 immunohistochemical staining. The neurite outgrowths were analyzed under a microscope. Only mature cultures (7 days in vitro) were used for experiments. The percentage of neuronal cells in the primary cultures was approximately 80 % [20].
To determine the best hypoxic conditions for conducting our experiments, primary cultured cortical neurons (PCCNs) were exposed to in vitro hypoxia in an anaerobic chamber (Anaerobic System Model 1025, Forma Scientific, Marietta, OH, USA) equipped with a humidified, temperature-controlled incubator. A gas mixture containing CO2 (5 mol%), O2 (1 mol%), and N2 (94 mol%) was flushed through the chamber. This procedure maintained a non-fluctuating hypoxic environment below 1 mol% O2 [21]. Cell viability was assessed after several hours of hypoxia using the lactate dehydrogenase (LDH) and MTT assays [22].
To examine the effects of PI3K activation on the viability of PCCNs, we treated cortical neurons for 24 h with several concentrations of the PI3K activator (0, 0.001, 0.01, 0.1, 1, and 10 μM) and washed them several times with phosphate-buffered saline (PBS). On the basis of our data indicating the effects of PI3K activation and hypoxia on neuronal cell viability, cortical neurons were treated with several concentrations of PI3K activator (0, 0.001, 0.01, 0.1, 1, and 10 μM) and simultaneously exposed to hypoxic conditions for 6, 24, and 48 h, respectively. The cells were then gently washed, and cell viability was evaluated immediately. Cells harvested 2 h after hypoxia were used for immunodetection of hypoxia-inducible factor-alpha (HIF-1α), Akt, glycogen synthase kinase (GSK)-3β, BAX, cytoplasmic apoptosis-induced factor (AIF), and cytoplasmic cytochrome c and those 6 h after hypoxia, caspase-9, cleaved caspase-9, caspase-3, cleaved caspase-3, poly(ADP-ribose) polymerase (PARP), and cleaved PARP [22].
Finally, to confirm the direct neuroprotective effects of PI3K activation against hypoxia, we also treated cortical neurons with 10 μM LY294002, a PI3K inhibitor, prior to treatment with the PI3K activator and hypoxia. The cortical neurons were divided into the following six groups: control (group 1), hypoxia for 24 h (group 2), hypoxia + 10 μM PI3K activator for 24 h (group 3), 10 μM LY294002 for 24 h + hypoxia and 10 μM PI3K activator for 24 hrs (group 4), 10 μM LY294002 for 24 h + hypoxia for 24 h (group 5), and 10 μM LY294002 for 24 h (group 6). Cell viability was assessed by the LDH and MTT assays as described previously [22].
MTT and LDH Assays to Measure Cell Viability
Cells were plated at a density of 1 × 104 cells/well in a 96-well plate, cultured, differentiated, and treated according to the methods described above. A total of 50 μl of MTT (Sigma) was added at a concentration of 2 mg/ml after medium (200 μl) was added to each well. MTT is absorbed into the cell, and formazan is then made by the action of mitochondrial succinate dehydrogenase. Accumulation of formazan directly reflects the activity of the mitochondria and indirectly reflects cell viability. An aliquot (220 μl) of solution was removed from each well, and 150 μl of dimethyl sulfoxide (DMSO) was then added to each well. The optical density (OD) at 540 nm was measured on the enzyme-linked immunosorbent assay plate reader after all precipitate in the wells was dissolved on the microplate mixer for 10 min. All results were calibrated for an OD measured in the same conditioned well without cell culture [22]. An LDH assay kit (Promega, Madison, WI, USA) was used to evaluate the viability of PCCNs, and the assay was performed according to the manufacturer’s protocol.
TUNEL and DAPI Staining to Evaluate Apoptosis
To evaluate apoptosis, air-dried cells were fixed with 4 % paraformaldehyde in PBS for 1 h at room temperature, and apoptotic cell death was assessed by terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) according to the manufacturer’s protocol (Roche Boehringer–Mannheim, IN, USA). To stain all nuclei and to visualize intact, condensed, and fragmented nuclei, TUNEL-stained cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Sigma, Saint Louis, MO, USA). The percentage of TUNEL-positive cells was determined relative to the total number of cells [22].
Western Blot Analysis
For the western blot analyses, five groups were treated (1) without hypoxia or PI3K activator for 2 or 6 h, (2) with hypoxia for 2 or 6 h, (3) with hypoxia and 0.1 μM PI3K activator for 2 or 6 h, (4) with hypoxia and 1 μM PI3K activator for 2 or 6 h, and (5) with hypoxia and 10 μM PI3K activator for 2 or 6 h. We used western blot analysis to assay for pAkt (Ser473), Akt, pGSK-3β (Ser9), GSK-3β, BAX, cytosolic AIF, cytosolic cytochrome c, caspase-9, cleaved caspase-9, caspase-3, cleaved caspase-3, PARP, and cleaved PARP in each group. Briefly, 5 × 106 cells were washed twice in cold PBS and incubated for 10 min on ice in lysis buffer [50 mM Tris (pH 8.0), 150 mM NaCl, 0.02 % sodium azide, 0.2 % SDS, 100 μg/ml phenylmethylsulfonylfluoride (PMSF), 50 μl/ml aprotinin, 1 % Igepal 630, 100 mM NaF, 0.5 % sodium deoxy choate, 0.5 mM EDTA, 0.1 mM EGTA]. We centrifuged the cell lysates at 10,000×g and then evaluated the levels of pAkt (Ser473), Akt, pGSK-3β (Ser9), GSK-3β, BAX, caspase-9, cleaved caspase-9, caspase-3, cleaved caspase-3, PARP, and cleaved PARP in each cell lysate. To evaluate cytoplasmic AIF and cytosolic cytochrome c levels, cells were suspended in sucrose-supplemented cell extract buffer (300 mM sucrose, 10 mM HEPES at pH 7.4, 50 mM KCl, 5 mM EGTA, 5 mM MgCl2, 1 mM DTT, 10 μM cytochalasin B, and 1 mM PMSF) after washing, left on ice for 30 min, and then homogenized with 50 strokes in an ice-cold Dounce homogenizer. Unbroken cells and nuclei were pelleted by centrifugation for 10 min at 2,000×g. Mitochondria were collected by centrifugation of the resulting supernatant at 13,000×g for 10 min. The postmitochondrial fraction, namely the supernatant in the above step, was immunoblotted for AIF and cytochrome c. Protein concentrations of cell lysates and postmitochondrial fractions were determined using a Bio-Rad protein assay kit (Hercules, CA, USA). Samples containing equal amounts (20 μg) of protein were resolved by 10 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK). The membranes were blocked with 5 % skim milk and then incubated with specific primary antibodies. We used antibodies against HIF-1 (1:1,000, Cell Signaling, Beverly, MA, USA), pAkt (Ser473) (1:1,000, Cell Signaling), Akt (1:1,000, Cell Signaling), pGSK-3β (Ser9) (1:1,000, Santa Cruz Biotech, Santa Cruz, CA, USA), GSK-3β (1:1,000, Santa Cruz Biotech), BAX (1:1,000, Santa Cruz Biotech), cytosolic AIF (1:500, Cell Signaling), cytosolic cytochrome c (1:500, Santa Cruz Biotech), caspase-9 (1:1,000, Cell Signaling), cleaved caspase-9 (1:1,000, Cell Signaling), caspase-3 (1:1,000, Cell Signaling), cleaved caspase-3 (Asp 175) (1:1000, Cell Signaling), PARP (1:500, Cell Signaling), and cleaved PARP (Asp 214) (1:500, Cell Signaling). The membranes were washed with Tris-buffered saline containing 0.05 % Tween-20 (TBST) and then processed using an HRP-conjugated anti-rabbit antibody or anti-mouse antibody (Amersham Pharmacia Biotech, Piscataway, NJ, USA) followed by ECL detection (Amersham Pharmacia Biotech, Piscataway, NJ, USA) [22]. The western blot results were quantified with an image analyzer (Bio-Rad, Quantity One-4,2,0) (Bio-Rad, Hercules, CA) and were normalized to GAPDH immunostaining [22].
Immunocytochemistry for the Evaluation of the Location of AIF
Immunocytochemistry was used to assess the translocation of AIF into the nucleus during apoptosis, as described previously [23]. Briefly, the cells were fixed on slides using 4 % paraformaldehyde in PBS, pH 7.4, for 15 min at room temperature. The specimens were washed twice with ice-cold PBS, incubated for 10 min with PBS containing 0.25 % Triton X-100 (or 100 μM digitonin or 0.5 % saponin), and then washed in PBS three times for 5 min. Then, the samples were blocked with 1 % BSA in PBST for 30 min and incubated with an anti-AIF polyclonal antibody (1:100, Cell Signaling) in PBST containing 1 % BSA in a humidified chamber overnight at 4 °C. After the cells were washed, an Alexa 488 anti-rabbit secondary antibody (1:100, Molecular Probes, Eugene, OR) was applied for 1 h at room temperature in the dark. The medium containing the secondary antibody was removed, and then the cells were washed three times with PBS for 5 min each in the dark. A coverslip was mounted with a drop of mounting medium containing DAPI. Cells without the anti-AIF antibody served as negative controls.
PARP-1 Activity
PARP activity was assessed using an enzymatic activity assay (Universal Colorimetric PARP assay kit, Trevigen Inc, Gaithersburg, MD, USA). The assay measures the incorporation of biotinylated poly(ADP-ribose) onto histone proteins in cell homogenates according to manufacturer instructions. The amount of PARP-1 activity assayed on cellular samples (50 μg) was determined by comparison to a standard curve.
Determination of Intracellular NAD+ and ATP Levels
The level of intracellular ATP was measured by using a colorimetric assay kit (Abcam, Cambridge, MA, USA). Briefly, cells were lysed in ATP assay buffer and centrifuged at 15,000×g for 2 min to pellet insoluble materials. The supernatant was added to a 96-well plate and followed by addition of ATP assay buffer to a final volume of 50 μl/well. The absorbance was read at 550 nm using a micro-plate reader from Molecular Devices. The NAD+ and NADH levels were determined using a NAD/NADH assay kit (Abcam, Cambridge, MA, USA). Briefly, cells were lysed in NADH/NAD extraction buffer and centrifuged at 14,000×g for 5 min to pellet insoluble materials. The supernatant was added to the NAD Cycling Mixture. The absorbance was read at 450 nm using a microplate reader from Molecular Devices. The ATP content and NAD+ were calculated based on a standard curve. Both ATP and NAD+ levels were normalized to total protein concentrations of cell lysates determined using Bio-Rad protein assay kit.
Statistical Analysis
All data are presented as the mean ± SD of five or more independent experiments. Statistical comparisons between different treatment groups were performed with Tukey’s test after one-way ANOVA. P values less than 0.05 were considered statistically significant.
Results
Expression of HIF-1α and phosphorylated Akt (Ser473) under hypoxic conditions
To confirm that primary cultured cortical neurons (PCCNs) were affected by hypoxic injury, we measured levels of HIF-1α, a key regulator of the hypoxic response, and phosphorylated Akt (Ser473) by western blotting. HIF-1α expression rapidly increased in response to hypoxia after 2 h (Fig. 1a). However, phosphorylated Akt at Ser473 slightly decreased 2 h after the exposure to hypoxia and then significantly increased after that (Fig. 1b).

Expression of HIF-1α and phosphorylated Akt (Ser473) in PCCNs in response to hypoxia. Expression of HIF-1α in PCCNs dramatically increases from just after the exposure to hypoxia, but it becomes normalized over time (a). However, expression of phosphorylated Akt (Ser473) slightly decreases just after the exposure to hypoxia and then significantly increases in a time-dependent manner (b). *p < 0.05 by Tukey’s test after one-way ANOVA (compared with the control group [no hypoxia]) (n = 5)
Effect of Hypoxia and PI3K Activation on the Viability of Primary Cultured Cortical Neurons
To confirm the effect of the hypoxic condition used in the present study on PCCN viability, we incubated PCCNs in the anaerobic chamber with different exposure times. Cell viability was measured with MTT assay and LDH assay. Under hypoxic conditions, cell viability was significantly reduced in a time-dependent manner (Fig. 2a).

Effects of hypoxia and PI3K activation on PCCN viability. Prolonged hypoxia markedly induces neuronal cell death (a). PI3K activator does not affect viability of PCCNs (b). Treatment of PI3K activator reduces neuronal cell death induced by hypoxia in a concentration-dependent manner (c). Pretreatment of a specific PI3K inhibitor blocks the neuroprotective effect of PI3K activator (d). *p < 0.05 (when compared with the control group), #p < 0.05 (compared with PCCNs under hypoxic condition without PI3K activator), and $p < 0.05 (compared with PCCNs under hypoxic condition and 10 μM PI3K activator) by Tukey’s test after one-way ANOVA (n = 5)
To evaluate the effect of PI3K activator itself on PCCNs, we treated PCCNs for 24 h with several different concentrations of PI3K activator. Cell viability was decreased with more than 100 μM PI3K activator treatment in MTT and LDH assays (Fig. 2b).
To evaluate the effect of PI3K activation on PCCNs under hypoxic conditions, PCCNs were exposed to hypoxia for several hours and were treated simultaneously with several different concentrations of PI3K activator. Cell viability was measured by LDH assay. Compared to PCCNs under hypoxic conditions without treatment, the viability of PCCNs treated with PI3K activator under hypoxic condition gradually increased in a concentration-dependent manner up to 1 μM (Fig. 2c).
To confirm the role of PI3K activation in neurons exposed to hypoxic injury, PCCNs were separated into six groups: control (group 1), hypoxia for 24 h (group 2), hypoxia + 10 μM PI3K activator for 24 h (group 3), 10 μM LY294002 for 24 h + hypoxia and 10 μM PI3K activator for 24 h (group 4), 10 μM LY294002 for 24 h + hypoxia for 24 h (group 5), and 10 μM LY294002 for 24 h (group 6). In this study, pretreatment with the PI3K inhibitor in group 4 resulted in an approximately 13 % decrease in cell viability as compared with group 3 (Fig. 2d).
Anti-apoptotic Effects of PI3K Activation Under Hypoxic Conditions
TUNEL and DAPI staining showed that the percentage of cells undergoing apoptosis under hypoxic conditions was markedly increased after 24 h of hypoxia (p < 0.01) but significantly decreased when the cells were treated with a PI3K activator (1 and 10 μM) (p < 0.05) (Fig. 3). Treatment with 1 or 10 μM PI3K activator resulted in the greatest decrease in the percentage of apoptotic cells.

Anti-apoptotic effect of PI3K activation on PCCNs during exposure to hypoxic conditions for 24 hrs. PCCNs were analyzed using TUNEL and DAPI staining. The percentage of TUNEL-positive cells induced by hypoxia decreased when the cells were treated with a PI3K activator. The data are presented as the percentage of TUNEL-positive cells ± SD. Each treatment group was compared with the other groups using Tukey’s test after one-way ANOVA (n = 5). *p < 0.05 (compared with the control group) and #p < 0.05 (compared with PCCNs exposed to hypoxia alone). The scale bar is 50 μm, and the magnification is 200×
Effects of Hypoxia and PI3K Activation on the Levels of Intracellular Signaling Proteins
To confirm the effects of PI3K activation on several intracellular signaling proteins, we measured the expression levels of Akt, phosphorylated Akt, GSK3-β, phosphorylated GSK3-β, BAX, cytosolic AIF, cytosolic cytochrome c, caspase-9, cleaved caspase-9, caspase-3, cleaved caspase-3, PARP, and cleaved PARP.
The results of western blotting showed that the immunoreactivities (IRs) of phosphorylated Akt (Ser473) and phosphorylated GSK-3β (Ser9), which are survival-related proteins, in PCCNs were significantly increased with the treatment of PI3K activator in a concentration-dependent manner up to 1 μM when compared with PCCNs treated only with hypoxia (Fig. 4). In contrast, treatment with PI3K activator significantly decreased the expression of BAX, AIF, cytochrome c, cleaved caspase-9, cleaved caspase-3, and cleaved PARP, which are pathways associated with cell death, in a concentration-dependent manner up to 10 μM (Fig. 4).

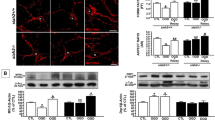
Immunoreactivities (IRs) of pAkt (Ser473), pGSK-3β (Ser9), BAX, AIF, cytochrome c, caspase-9, caspase-3, and PARP in PCCNs. Immunoreactivities (IRs) were assessed by western blotting. Data were expressed as a ratio of the simultaneously assayed control group’s value and were compared using Tukey’s test after a one-way ANOVA (n = 5). Representative ECL radiographs of the immunoblots demonstrate that combined treatment with 10 μM PI3K activator increased the IRs of pAkt (Ser473) (a) and pGSK-3β (Ser9) (b), and decreased the IRs of BAX (c), cytosolic AIF (d), cytosolic cytochrome c (e), cleaved caspase-9 (f), cleaved caspase-3 (g), and cleaved PARP (h) when compared with PCCNs under hypoxia alone. *p < 0.05 (when compared with control group) and #p < 0.05 (when compared with PCCNs under hypoxia without treatment)
In addition, immunocytochemistry was performed to evaluate the location of AIF. This assay showed that the level of nuclear AIF increased after hypoxia for 2 h but that treatment with the PI3K activator decreased the level of nuclear AIF in a concentration-dependent manner (Fig. 5).

Location of AIF in PCCNs after hypoxia and/or PI3K activator treatment. Immunocytochemistry was performed to evaluate the location of AIF after hypoxia and/or PI3K activator treatment. The nuclear AIF level increased after hypoxia for 2 h, but treatment with the PI3K activator led to a decrease in the nuclear AIF level in a concentration-dependent manner. The scale bar is 50 μm, and the magnification is ×400
Effects of Hypoxia and PI3K Activation on Poly(ADP-ribose) Polymerase Activity
To confirm the effect of PI3K activation on cells under hypoxic conditions, we used the poly(ADP-ribose) polymerase assay kit. The results showed that PARP activity increased in PCCNs under hypoxic conditions. In contrast, treatment with PI3K activator significantly decreased the PARP activity (Fig. 6a).

Effects of hypoxia and PI3K activation on PARP activity and levels of ATP and NAD+ in PCCNs. PARP activity in PCCNs is increased by hypoxia but is normalized with PI3K activation (a). Treatment of PI3K activator increases intracellular ATP level in PCCNs which is decreased by hypoxia (b). Intracellular NAD+ level in PCCNs which is decreased by hypoxia is restored with the treatment of PI3K activator (c). *p < 0.05 (when compared with the control group) and #p < 0.05 (compared with PCCNs under hypoxic condition without PI3K activator) by Tukey’s test after one-way ANOVA, (n = 5).
Effects of Hypoxia and PI3K Activation on ATP and NAD+ Level
To investigate the effects of hypoxia and PI3K activation on the level of intracellular ATP and NAD+ of PCCNs, we treated PCCNs with hypoxia and/or diverse concentrations of PI3K activator (0.1, 1, and 10 μM), and then ATP and NAD+ levels were measured. As shown in Figs. 6b and c, hypoxia significantly decreased the level of intracellular ATP and NAD+, respectively. However, treatment with PI3K activator increased those levels (p < 0.05).
Discussion
In the present study, PI3K activator itself did not affect the viability of PCCNs up to 10 μM (Fig. 2a), but hypoxia for more than 24 h significantly decreased viability (Figs. 2b and 3). Co-treatment with PI3K activator markedly reduced hypoxia-induced neuronal cell death in a concentration-dependent manner (Figs. 2c, d and 3). The neuroprotective effect of the PI3K activator was associated with the direct activation of PI3K as demonstrated by the fact that LY294002, a specific PI3K inhibitor, significantly antagonized the effect of the PI3K activator (Fig. 2d). Molecular studies showed that hypoxia decreased the survival-related signals including phosphorylated Akt (Ser473) and phosphorylated GSK-3β (Ser9) and increased death-associated signals including BAX, cytosolic AIF, cytosolic cytochrome c, activated capase-9, activated caspase-3, and cleaved PARP (Fig. 4). However, co-treatment with PI3K activator significantly recovered such survival signals and inhibited those associated with cell death in a concentration-dependent manner (Fig. 4). Because the main purpose of our experiment was to determine the effect of the early activation of PI3K on hypoxic cortical neurons, western blotting for upstream signaling proteins, such as PI3K, Akt, and GSK-3β, was performed after 2 h of hypoxia. However, western blotting for downstream signaling proteins, such as activated caspase-9, activated caspase-3, and cleaved PARP, was performed after 6 h of hypoxia. These time points were selected based on previous reports showing that the levels of the upstream signaling proteins are changed within a few hours of the onset of hypoxia [24–26] and that the levels of the downstream proteins are changed after a longer duration of hypoxia [27, 28]. In addition, in many in vitro hypoxic/ischemic experiments, western blotting was performed in the range of 2–8 h [29–31], which is similar to the time range used in our experiment. To confirm the location of AIF, we also performed immunocytochemistry and found that the level of nuclear AIF, which is known to increase during apoptosis [23], increased after hypoxia, but treatment with the PI3K activator decreased the level of nuclear AIF in a concentration-dependent manner (Fig. 5). It was also found that PARP activity was dramatically increased after exposure to hypoxia for 2 h, but treatment with PI3K activator significantly reduced it (Fig. 6a). Although the levels of ATP and NAD+ were markedly decreased in PCCNs exposed to hypoxia for 2 h, treatment with PI3K activator effectively recovered those levels (Fig. 6b and c).
The PI3K pathway is well known to be crucial in neuronal cell survival. It has been confirmed that the activation of the PI3K pathway is critically associated with the neuroprotective mechanisms of uncountable neurotrophic factors, chemicals, and medicines against several neurotoxic conditions [14, 16, 32, 33]. It has also been reported that the activation of the pathway is involved in the neuroprotective effects of many materials enhancing neuronal cell survival under hypoxic/ischemic conditions [34–36]. However, to the best of our knowledge, there have been no reports to date about the effect of direct activation of the PI3K pathway in neuronal cells injured by hypoxia. Therefore, we aimed to confirm the direct role of the PI3K pathway for the protection of neuronal cells from hypoxic injury and a few novel findings were demonstrated by this study.
First, we found that the direct activation of the PI3K pathway with a specific and direct PI3K activator efficiently protected neuronal cells from hypoxic injury. PI3K has been shown to be involved in cell growth, proliferation, differentiation, and motility of neural stem cells [37] and in survival of neuronal cells [6–8]. Various survival factors, including insulin-like growth factor 1 [38], vascular endothelial growth factor [39], nerve growth factor [40], epidermal growth factor [40], and erythropoietin [41], activate PI3K. Activated PI3K produces diverse 3-phosphorylated phosphoinositides. Two are phosphatidylinositol(3,4,5)P3 and phosphatidylinositol(3,4)P2 [42]. These 3-phosphorylated phosphoinositides directly bind and activate Akt [42]. Activated AKT phosphorylates a group of molecules, including GSK-3β [43] and Bax [44], thereby blocking mitochondrial cytochrome c release and caspase activity as also confirmed in the present study. This sequential process inhibits neuronal cell death, especially apoptosis. Based on many previous reports showing that the activation of the PI3K pathway plays an important role in the neuroprotective mechanism of numerous neurotrophic factors, chemicals, and medicines protecting neuronal cells from hypoxia [14, 16, 32, 33], the hypothesis that the direct activation of the PI3K pathway could protect neuronal cells against hypoxic injury seems reasonable. However, there has been no report about the effect of the direct activation of the PI3K pathway on hypoxia-induced neuronal cell injury. In addition, we also know that the above-described materials can have other neuroprotective mechanisms besides the activation of the PI3K pathway which may contribute to their neuroprotective effect. So, we studied the role of the direct activation of the pathway for the prevention of neuronal cell death induced by hypoxia. Treatment with a specific and direct PI3K activator effectively restored neuronal cell survival under hypoxic stress and pretreatment with a specific PI3K inhibitor completely blocked the effect of PI3K activator. These findings strongly support the hypothesis that the direct and specific activation of the PI3K pathway effectively prevents hypoxia-induced neuronal cell death.
The second novel finding is the expression pattern of phosphorylated Akt after the exposure to hypoxia, namely expression of phosphorylated Akt (Ser473) could differ depending on the type of cell exposed to hypoxia. It is well known that phosphorylated Akt (Ser473) increases after exposure to hypoxia to enhance the resistance of neuronal cells against hypoxic injury [45]. However, the expression pattern of phosphorylated Akt (Ser473) after a hypoxic insult has not yet been well established. In the present study, we found that phosphorylated Akt decreased slightly in PCCNs for a short period after hypoxia and then significantly increased after that (Fig. 1b). This finding is contrary to the general concept that phosphorylated Akt increases after exposure to hypoxia. We propose that phosphorylated Akt (Ser473) slightly decreases in neuronal cells just after hypoxia and the dying process begins in some of the vulnerable neuronal cells during this period, and that the phosphorylation of Akt at Ser473 increases in other neuronal cells resistant to hypoxia to enhance their survival. We also propose that the early expression of HIF-1 after hypoxia might involve this increase of phosphorylated Akt considering their expression patterns after hypoxia, namely HIF-1 significantly increased 2 h after hypoxia when the amount of phosphorylated Akt (Ser473) was slightly decreased. These findings suggest that phosphorylated Akt (Ser473) does not always increase after hypoxia, but its expression differs depending on the duration of hypoxia. In addition, our previous study and a report by Zhang et al. showed that phosphorylated Akt (Ser473) increased continuously in primary cultured neural stem cells and in the RN46 cell line which is an immortalized neuronal cell line derived from E13 brainstem raphe [5] after hypoxia. Therefore, the expression of phosphorylated Akt (Ser473) might be different depending on the cell type. All of these findings are novel.
Based on these findings, we hypothesized that early treatment with a PI3K activator could increase phosphorylated Akt (Ser473) which decreases shortly after exposure to hypoxia. To test this hypothesis, we performed molecular studies including western blotting for the detection of upstream signaling proteins, such as phosphorylated GSK-3β, BAX, cytosolic AIF, and cytosolic cytochrome c, 2 h after hypoxia. Downstream signaling proteins, such as caspase-9, caspase-3, and PARP, were evaluated 6 h after hypoxia based on a previous report showing that activity of caspase-3 was maximized 6 h after hypoxia [46]. Western blotting data showed that survival-related signaling proteins were decreased and those associated with cell death were increased and that treatment with a direct and specific PI3K activator recovered all the signaling proteins to a similar level as the control PCCNs. These findings also support the hypothesis that direct and specific PI3K activation protects neuronal cells from hypoxic injury.
The third novel finding is the effect of PI3K activation on PARP activity, the ATP level, and the NAD+ level under ischemic conditions. PARP is involved in a variety of physiological and pathological events, such as DNA replication, DNA repair, and gene expression. In response to DNA damage caused by events such as hypoxia, PARP is activated after only a few minutes, and it has been reported that 3 min of hypoxia can induce a threefold increase of PARP activity after 15 min [47]. It has also been reported that increased PARP activity along with the depletion of NAD+ and ATP could occur without occult neuronal damage [48]. In addition, it has been reported that even 10-min hypoxia induced significant ATP depletion [49] and that neuronal NAD+ depletion was remarkably induced 30 min after treatment with N-methyl-N′-nitro-N-nitrosoguanidine causing PARP activation [50]. Considering all these findings, we thought that the determination of the NAD+ and ATP levels after 2 h of hypoxia was reasonable in this study, and we found that PI3K activation directly or indirectly affected PARP activity, the ATP level, and the NAD+ level in PCCNs under hypoxic stress (Fig. 6). Poly ADP-ribosylation of nuclear proteins is a post-transcriptional event that occurs in response to DNA damage. Hypoxia is known to be an inducer of PARP activity as shown in Fig. 6a. Over-activation of PARP overuses ATP, which is a substrate of PARP, which then induces neuronal cell death [51]. In addition, hyperactive PARP is involved in inflammation [52]. Therefore, inhibition of PARP activity after hypoxia is very critical for the prevention of neuronal cell death. Our novel finding that direct and specific PI3K activation reduced PARP activity to a similar level to the control PCCNs led us to think that the regulation of PARP activity by PI3K activation also contributed to the neuroprotection of PI3K activation against hypoxia. Normalized PARP activity by PI3K activation also contributed to the normalization of the levels of ATP and NAD+ (Fig. 6b, c). Although the fact that PI3K activation can increase ATP levels has been demonstrated previously [53], our finding that PI3K activation affected PARP activity and then normalized the levels of ATP and NAD+ in neuronal cells under hypoxia is novel.
An additional interesting finding is the HIF-1 expression pattern. In general, HIF-1 expression is well known to be increased after hypoxia in various cells. However, it is also well established that the HIF-1α protein is continuously synthesized and rapidly degraded in normoxic cells [54]. In a previous report, HIF-1α expression was detected in brain and muscles cells but not in liver, kidney, heart, and spleen cells under conditions without any stress [55]. In addition, the expression of HIF-1α under normoxic conditions has been reported in a few cell lines, such as primary cortical neurons [26] and mesenchymal stromal cells [56]. In our repeated experiments, we continuously found that HIF-1α was expressed in primary cortical neurons under normoxic conditions and that the expression level of this protein significantly increased under hypoxic conditions. All of these findings led us hypothesize that HIF-1α can be expressed under normoxic conditions but that its expression markedly increases under hypoxic conditions. We believe that HIF-1α could be expressed in different cell lines under different conditions.
There were some limitations in this study: (1) this study was performed under in vitro conditions, and therefore, the results could be different under in vivo conditions where more complicated factors may be involved, (2) only a single dose of PI3K inhibitor (10 μM) was used because our previous data showed that treatment with LY294002 more than 10 μM induced neurotoxicity, and (3) only a non-fluctuating hypoxic environment below 1 mol% O2 was used for the main study. Therefore, the toxicological relevance of our findings to hypoxic injury is difficult to ascertain in humans.
In conclusion, our findings suggest that direct and specific PI3K activation prevents neuronal cell death against hypoxia via the activation of downstream targets of the PI3K pathway and the normalization of the levels of PARP activity, ATP, and NAD+.
References
Malhotra R, Lin Z, Vincenz C, Brosius FC 3rd (2001) Hypoxia induces apoptosis via two independent pathways in Jurkat cells: differential regulation by glucose. Am J of Physiol Cell Physiol 281:C1596–603
Mattiesen WR, Tauber SC, Gerber J, Bunkowski S, Bruck W, Nau R (2009) Increased neurogenesis after hypoxic-ischemic encephalopathy in humans is age related. Acta Neuropathol 117:525–34. doi:10.1007/s00401-009-0509-0
Banasiak KJ, Xia Y, Haddad GG (2000) Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog Neurobiol 62:215–49
Alvarez-Tejado M, Naranjo-Suarez S, Jimenez C, Carrera AC, Landazuri MO, del Peso L (2001) Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells: protective role in apoptosis. J Biol Chem 276:22368–74. doi:10.1074/jbc.M011688200
Zhang SX, Gozal D, Sachleben LR, Rane M Jr, Klein JB, Gozal E (2003) Hypoxia induces an autocrine-paracrine survival pathway via platelet-derived growth factor (PDGF)-B/PDGF-beta receptor/phosphatidylinositol 3-kinase/Akt signaling in RN46A neuronal cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 17:1709–11. doi:10.1096/fj.02-1111fje
Crowder RJ, Freeman RS (1998) Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. The Journal of Neurosci: the Official J of the Soc for Neurosci 18:2933–43
Peltier J, O’Neill A, Schaffer DV (2007) PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol 67:1348–61. doi:10.1002/dneu.20506
Yao R, Cooper GM (1995) Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 267:2003–6
Franke TF, Kaplan DR, Cantley LC, Toker A (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science 275:665–8
Song G, Ouyang G, Bao S (2005) The activation of Akt/PKB signaling pathway and cell survival. Journal of cellular and molecular medicine 9:59–71
Ha KS et al (2003) Nitric oxide prevents 6-hydroxydopamine-induced apoptosis in PC12 cells through cGMP-dependent PI3 kinase/Akt activation. FASEB J: Official Publ of the Fed of Am Soc for Exp Biol 17:1036–47. doi:10.1096/fj.02-0738com
Namikawa K et al (2000) Akt/protein kinase B prevents injury-induced motoneuron death and accelerates axonal regeneration. The J of Neurosci: the Official J of the Soc for Neurosci 20:2875–86
Park HH, Lee KY, Kim SH, Lee YJ, Koh SH (2009) l-DOPA-induced neurotoxicity is reduced by the activation of the PI3K signaling pathway. Toxicology 265:80–6. doi:10.1016/j.tox.2009.09.011
Choi H et al (2012) Coenzyme Q10 protects against amyloid beta-induced neuronal cell death by inhibiting oxidative stress and activating the P13K pathway. Neurotoxicology 33:85–90. doi:10.1016/j.neuro.2011.12.005
Cui D, Wang L, Qi A, Zhou Q, Zhang X, Jiang W (2012) Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS One 7:e35324. doi:10.1371/journal.pone.0035324
Lee YJ et al (2009) Cilnidipine mediates a neuroprotective effect by scavenging free radicals and activating the phosphatidylinositol 3-kinase pathway. J Neurochem 111:90–100. doi:10.1111/j.1471-4159.2009.06297.x
Garcia P, Shoelson SE, George ST, Hinds DA, Goldberg AR, Miller WT (1993) Phosphorylation of synthetic peptides containing Tyr-Met-X-Met motifs by nonreceptor tyrosine kinases in vitro. J Biol Chem 268:25146–51
Rordorf-Nikolic T, Van Horn DJ, Chen D, White MF, Backer JM (1995) Regulation of phosphatidylinositol 3′-kinase by tyrosyl phosphoproteins. Full activation requires occupancy of both SH2 domains in the 85-kDa regulatory subunit. J Biol Chem 270:3662–6
Shoelson SE, Chatterjee S, Chaudhuri M, White MF (1992) YMXM motifs of IRS-1 define substrate specificity of the insulin receptor kinase. Proc Natl Acad Sci U S A 89:2027–31
Noh MY, Koh SH, Kim Y, Kim HY, Cho GW, Kim SH (2009) Neuroprotective effects of donepezil through inhibition of GSK-3 activity in amyloid-beta-induced neuronal cell death. J Neurochem 108:1116–25. doi:10.1111/j.1471-4159.2008.05837.x
Li B, Xu W, Luo C, Gozal D, Liu R (2003) VEGF-induced activation of the PI3-K/Akt pathway reduces mutant SOD1-mediated motor neuron cell death. Brain research Molecular brain research 111:155–64
Lee YJ, Park HH, Koh SH, Choi NY, Lee KY (2011) Amlodipine besylate and amlodipine camsylate prevent cortical neuronal cell death induced by oxidative stress. J Neurochem 119:1262–70. doi:10.1111/j.1471-4159.2011.07529.x
Zhang W, Shokeen M, Li D, Mehta JL (2003) Identification of apoptosis-inducing factor in human coronary artery endothelial cells. Biochem Biophys Res Commun 301:147–51
Li L, Qu Y, Mao M, Xiong Y, Mu D (2008) The involvement of phosphoinositid 3-kinase/Akt pathway in the activation of hypoxia-inducible factor-1alpha in the developing rat brain after hypoxia-ischemia. Brain Res 1197:152–8. doi:10.1016/j.brainres.2007.12.059
Liu C et al (2010) Neuroprotection by baicalein in ischemic brain injury involves PTEN/AKT pathway. J Neurochem 112:1500–12. doi:10.1111/j.1471-4159.2009.06561.x
Zhang L et al (2009) Signaling pathway involved in hypoxia-inducible factor-1alpha regulation in hypoxic-ischemic cortical neurons in vitro. Neurosci Lett 461:1–6. doi:10.1016/j.neulet.2009.03.091
Li L, Qu Y, Li J, Xiong Y, Mao M, Mu D (2007) Relationship between HIF-1alpha expression and neuronal apoptosis in neonatal rats with hypoxia-ischemia brain injury. Brain Res 1180:133–9. doi:10.1016/j.brainres.2007.08.059
Lin CH, Chen PS, Gean PW (2008) Glutamate preconditioning prevents neuronal death induced by combined oxygen-glucose deprivation in cultured cortical neurons. Eur J Pharmacol 589:85–93. doi:10.1016/j.ejphar.2008.05.047
Guo S et al (2008) Glucose up-regulates HIF-1 alpha expression in primary cortical neurons in response to hypoxia through maintaining cellular redox status. J Neurochem 105:1849–60. doi:10.1111/j.1471-4159.2008.05287.x
Lopez-Hernandez B, Posadas I, Podlesniy P, Abad MA, Trullas R, Cena V (2012) HIF-1alpha is neuroprotective during the early phases of mild hypoxia in rat cortical neurons. Exp Neurol 233:543–54. doi:10.1016/j.expneurol.2011.11.040
Zhang L et al (2010) PI3K/Akt signaling pathway is required for neuroprotection of thalidomide on hypoxic-ischemic cortical neurons in vitro. Brain research 1357:157–65. doi:10.1016/j.brainres.2010.08.007
Quesada A, Lee BY, Micevych PE (2008) PI3 kinase/Akt activation mediates estrogen and IGF-1 nigral DA neuronal neuroprotection against a unilateral rat model of Parkinson’s disease. Dev Neurobiol 68:632–44. doi:10.1002/dneu.20609
Yu W, Mechawar N, Krantic S, Quirion R (2011) Alpha7 nicotinic receptor activation reduces beta-amyloid-induced apoptosis by inhibiting caspase-independent death through phosphatidylinositol 3-kinase signaling. J Neurochem 119:848–58. doi:10.1111/j.1471-4159.2011.07466.x
Shao JL et al (2011) H2S protects hippocampal neurons from anoxia-reoxygenation through cAMP-mediated PI3K/Akt/p70S6K cell-survival signaling pathways. J of Mol Neurosci: MN 43:453–60. doi:10.1007/s12031-010-9464-4
Sharma S, Yang B, Xi X, Grotta JC, Aronowski J, Savitz SI (2011) IL-10 directly protects cortical neurons by activating PI-3 kinase and STAT-3 pathways. Brain Res 1373:189–94. doi:10.1016/j.brainres.2010.11.096
Sun X, Yao H, Douglas RM, Gu XQ, Wang J, Haddad GG (2010) Insulin/PI3K signaling protects dentate neurons from oxygen-glucose deprivation in organotypic slice cultures. J Neurochem 112:377–88. doi:10.1111/j.1471-4159.2009.06450.x
Sato A et al (2010) Regulation of neural stem/progenitor cell maintenance by PI3K and mTOR. Neurosci Lett 470:115–20. doi:10.1016/j.neulet.2009.12.067
Singleton JR, Dixit VM, Feldman EL (1996) Type I insulin-like growth factor receptor activation regulates apoptotic proteins. J Biol Chem 271:31791–4
Jin KL, Mao XO, Greenberg DA (2000) Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci U S A 97:10242–7
Carter AN, Downes CP (1992) Phosphatidylinositol 3-kinase is activated by nerve growth factor and epidermal growth factor in PC12 cells. J Biol Chem 267:14563–7
Ruscher K et al (2002) Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. the J of Neurosci: the Official Journal of the Soc for Neurosci 22:10291–301
Auger KR, Serunian LA, Soltoff SP, Libby P, Cantley LC (1989) PDGF-dependent tyrosine phosphorylation stimulates production of novel polyphosphoinositides in intact cells. Cell 57:167–75
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378:785–9. doi:10.1038/378785a0
Matsuzaki H et al (1999) Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J Neurochem 73:2037–46
Zhao H, Sapolsky RM, Steinberg GK (2006) Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol 34:249–70. doi:10.1385/MN:34:3:249
Wang X, Mori T, Sumii T, Lo EH (2002) Hemoglobin-induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke; a journal of cerebral circulation 33:1882–8
Strosznajder RP, Gadamski R, Czapski GA, Jesko H, Strosznajder JB (2003) Poly(ADP-ribose) polymerase during reperfusion after transient forebrain ischemia: its role in brain edema and cell death. Journal of molecular neuroscience: MN 20:61–72. doi:10.1385/JMN:20:1:61
Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB (2010) Poly(ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol 41:187–96. doi:10.1007/s12035-010-8124-6
Santos MS, Moreno AJ, Carvalho AP (1996) Relationships between ATP depletion, membrane potential, and the release of neurotransmitters in rat nerve terminals. An in vitro study under conditions that mimic anoxia, hypoglycemia, and ischemia. Stroke; a J of Cereb Circ 27:941–50
Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA (2010) NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. The J of Neurosci: the Official J of the Soc for Neurosci 30:2967–78. doi:10.1523/JNEUROSCI.5552-09.2010
Nicoletti VG, Stella AM (2003) Role of PARP under stress conditions: cell death or protection? Neurochem Res 28:187–94
Skaper SD (2003) Poly(ADP-Ribose) polymerase-1 in acute neuronal death and inflammation: a strategy for neuroprotection. Ann N Y Acad Sci 993:217–28, discussion 287–8
Huang TJ, Verkhratsky A, Fernyhough P (2005) Insulin enhances mitochondrial inner membrane potential and increases ATP levels through phosphoinositide 3-kinase in adult sensory neurons. Mol Cell Neurosci 28:42–54. doi:10.1016/j.mcn.2004.08.009
Shi H (2009) Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr Med Chem 16:4593–600
Stroka DM et al (2001) HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 15:2445–53. doi:10.1096/fj.01-0125com
Chacko SM, Ahmed S, Selvendiran K, Kuppusamy ML, Khan M, Kuppusamy P (2010) Hypoxic preconditioning induces the expression of prosurvival and proangiogenic markers in mesenchymal stem cells. Am J of Physiol Cell Physiol 299:C1562–70. doi:10.1152/ajpcell.00221.2010
Acknowledgments
This work was supported by a grant (2012R1A1B3000473) from the Korea Research Foundation (KRF).
Conflict of Interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Min Young Noh and Young Seo Kim equally contributed to this work.
Rights and permissions
About this article
Cite this article
Noh, M.Y., Kim, Y.S., Lee, KY. et al. The Early Activation of PI3K Strongly Enhances the Resistance of Cortical Neurons to Hypoxic Injury via the Activation of Downstream Targets of the PI3K Pathway and the Normalization of the Levels of PARP Activity, ATP, and NAD+ . Mol Neurobiol 47, 757–769 (2013). https://doi.org/10.1007/s12035-012-8382-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-012-8382-6