
Abstract
Newly generated olfactory receptor axons grow from the peripheral to the central nervous system aided by olfactory ensheathing cells (OECs). Thus, OEC transplantation has emerged as a promising therapy for spinal cord injuries and for other neural diseases. However, these cells do not present a uniform population, but instead a functionally heterogeneous population that exhibits a variety of responses including adhesion, repulsion, and crossover during cell–cell and cell–matrix interactions. Some studies report that the migratory properties of OECs are compromised by inhibitory molecules and potentiated by chemical gradients. Here, we demonstrated that rodent OECs express all the components of the Nogo receptor complex and that their migration is blocked by myelin. Next, we used cell tracking and traction force microscopy to analyze OEC migration and its mechanical properties over myelin. Our data relate the decrease of traction force of OEC with lower migratory capacity over myelin, which correlates with changes in the F-actin cytoskeleton and focal adhesion distribution. Lastly, OEC traction force and migratory capacity is enhanced after cell incubation with the Nogo receptor inhibitor NEP1-40.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The olfactory system in the adult nervous system is renewed throughout life with the help of olfactory ensheathing cells (OECs). The elongation of newly generated olfactory receptor axons between the peripheral and central nervous system may be largely attributed to the properties of OECs. They ensheath and guide the axons of olfactory neurons that extend from the olfactory epithelium to the olfactory bulb [1–3]. Given their axon growth-promoting properties, natural or genetically modified OECs have been studied extensively, and they have been transplanted into the injured spinal cord to promote axonal regeneration [4–12]. Thus, OEC transplantation has emerged as a promising therapy for spinal cord injuries and other neural diseases [13–17]. Some OEC subpopulations have been identified in the olfactory system on the basis of their topographical distribution, intracellular cytoskeletal distribution, regenerative properties, and antigenic or gene expression profiles [18–24]. In fact, from a morphological point of view, two populations of OECs have been described in vitro and in vivo: Schwann cell-like OECs (sOECs), with fusiform bipolar form; and astrocyte-like OECs (aOECs), with a more flattened structure [25, 26]. However, the morphology of cultured OECs is strongly dependent on the culture conditions [21, 27, 28]. They transform from a fusiform to a flat-sheet shape spontaneously [29], and both sOECs and aOECs are likely to represent different morphologies of this motile and mitotic cell (see [30] for example).
Although they are useful for spinal cord repair, different OECs migratory properties have been reported after transplantation in lesioned central nervous system (CNS) [31, 32]. Indeed, Lee et al. determined by magnetic resonance tracking that OECs showed limited migration in injured spinal cord [33]. This was also reported in other studies using different techniques (e.g., [12], [34–38] among others). These results suggest that migration of OECs is modulated by specific interactions with the inhibitory substrate [39]. Indeed, in the injured spinal cord, lesioned axons and transplanted cells are confronted with a changing environment with a huge variety of growth inhibitory molecules located in the meningo-glial scar and adjacent spinal cord regions [40, 41]. Among others, the role of myelin-associated inhibitors (MAIs): Nogo-A, the myelin-associated glycoprotein (MAG) and the oligodendrocyte-myelin glycoprotein (OMgp) have been extensively studied after spinal cord lesions [42, 43]. These molecules block axon regeneration by acting thorough a common receptor: the Nogo receptor complex. This membrane receptor is formed by the GPI-anchored protein NgR1 and three putative co-receptors (p75, TROY, and Lingo-1) [42, 43]. In addition, new ligands have recently been reported to bind to the Nogo receptor complex (e.g., leucine-rich, glioma-inactivated (LGI) gene product [44], the B lymphocyte stimulator (BLyS) [45], members of the fibroblast growth factor FGF1 and FGF2 [46] and sulfatide [47]). Moreover, new receptors for MAIs have also been described: gangliosides [48], integrins [49], an NgR1 isoform: the NgR2 [50], G protein-coupled receptor 50 (GPR50) [51] and PirB [52].
OEC migration in vitro is modulated by neurotrophins, such as the glial cell-derived neurotrophic factor (GDNF), or chemicals such as lysophosphatidic acid (LPA) [53–55]. In addition, other secreted molecules involved in cell and axon migration, such as Slit-2, negatively modulate OEC migration [56]. However, although further studies are needed, it seems that Slit-2 is not over-expressed after spinal cord lesion [57], in contrast to Slit-1 and Slit-3 [57], semaphorins [58], Netrin-1 [59], chondroitin sulphate proteoglycans [60] and MAIs [61]. OECs show changes in their intracellular signaling mechanisms (Cofilin phosphorylation, RhoK activity and changes in Ca2+ stores) after Slit-2 treatment [56] that are shown by other inhibitory molecules [41]. In fact, these observations raised the notion that OECs may respond to a wide range of molecules. For MAIs, it has been reported that Nogo-66 fusion proteins (GST-Nogo-66 and His-Nogo-66) and Nogo-A enhance the adhesion of OECs, thus decreasing their migration in vitro [62]. Moreover, OECs increased its migration after PI-PLC treatment (to remove GPI-anchored NgR1) and α-NgR antibody treatment in vitro; or when NgR1-mediated signaling in a spinal cord lesion model is neutralized by α-NgR antibody infusion [62]. Taken together, these results indicate that Nogo-A/NgR signaling impairs OECs migration in vitro as well as in vivo. However, as indicated above, lesioned regions of the spinal cord may contain myelin debris as well as other inhibitors for long periods (more than 2 months in rodents [63] and years in humans [64]). Thus, in the present study, we aimed to explore the behavior and the migratory properties of OECs in the presence of myelin extracts by using single-cell tracking imaging on micropatterned substrates. On the other hand, directed cell migration requires the spatial and temporal coordination of cell adhesion and protrusion with the generation of traction forces at the cell/extra-cellular matrix (ECM) interface [65]. These traction forces are mainly generated by the acto-myosin cytoskeleton and transmitted to the ECM through focal adhesions (FAs) [66–68]. Thus, we also examined the effects of myelin on rodent OEC migration by traction force microscopy (TFM). First, we found that OECs express all the molecular machinery (Nogo receptor complex) required to transduce the inhibitory effects of MAIs, and that the migratory properties of OECs were blocked by myelin. Moreover, using TFM, we quantitatively demonstrate that OECs strongly decrease their traction stress over myelin, a finding that correlates with decreased focal contacts and redistribution of the F-actin cytoskeleton. Finally, we show that incubation of OEC cultures with the Nogo receptor 1 (NgR1) blocking peptide NEP1-40 [69] partially overcomes myelin-mediated migratory inhibition, thereby restoring the traction forces of the cells that correlate with cytoskeletal re-organization and re-appearance of focal contacts.
Materials and methods
Antibodies and biochemical reagents
The following antibodies were used at a dilution of 1:1,000 for Western blotting and/or 1:500 for immunohistochemical staining, unless otherwise indicated. Lingo-1 and actin (dilution 1:10,000) were from Millipore (Billerica, MA, USA). S100β was purchased from Abcam (Cambridge, MA, USA) and TROY from R&D System (Minneapolis, MN, USA). GFAP was from DAKO (Glostrup, Denmark), p75 was from Promega (Madison, WI, USA). NgR1 was a gift from Prof. B.L. Tang (Singapore). Tubulin (1:5,000), vinculin (1:400), phalloidin-alexa594 and DAPI were purchased from Sigma (St. Louis, MO, USA). ERK phospho-threonine 202/phospho-tyrosine 204 (pERK1-2) was from Cell Signaling Technology (Beverly, MA, USA). Total ERK antibody was from Transduction Laboratories (Lexington, KY, USA). Alexa Fluor 488 goat anti-mouse and Alexa Fluor 568 goat anti-rabbit immunoglobulins were purchased from Molecular Probes (Leiden, Netherlands). The goat anti-mouse horseradish peroxidase (HRP) and rabbit anti-goat-HRP secondary antibodies used in the Western blots were from DAKO. Goat anti-rabbit-HRP was supplied by Sigma. Green fluorescent FITC-beads (0.75 μm ∅) used to bind myelin were kindly provided by Dr. A. Homs and Prof. J. Samitier (IBEC, Barcelona). In addition, the Nogo-66 (1–40) antagonist peptide (NEP1-40) was purchased from Alpha Diagnostic International (San Antonio, TX, USA). Myelin was purified from adult Sprague–Dawley rat CNS, as described [70]. All animal procedures were performed in accordance with the guidelines established by the Spanish Ministry of Science and Technology and the European Community Council Directive 86/609 EEC.
TEG3 cultures
The immortalized clonal cell line TEG3, which contains the SV40 large T antigen stable transfectant of OEG primary cultures, was prepared from adult rat olfactory bulbs [71]. Cells were maintained in ME10: DMEM–F12 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% bovine calf serum (SAFC Biosciences, Lanexa, VA, USA), 20 μg/ml pituitary extract (Invitrogen), 2 μM forskolin (Sigma), 1% penicillin–streptomycin and 1% fungizone (Invitrogen). TEG3 cells between passages 4–8 were used for the experiments.
Western-blot techniques
Cultured TEG3 cells were collected with a scraper and homogenized on ice in a buffer containing 150 mM NaCl, 50 mM HEPES, 1 mM ethylene–glycol-tetraacetic acid, 10% glycerol, 1% Triton X-100, and 13 protease inhibitor cocktail (Roche, Basel, Switzerland). The lysate was clarified by centrifugation at 12,000 × g for 15 min, and the protein content of soluble fractions was determined using the Bio-Rad detergent-compatible assay (BCA) (Bio-Rad, Hercules, CA, USA). Cell extracts (20 μg) were boiled in Laemmli sample buffer at 100°C for 10 min, subjected to 8 or 15% SDS-PAGE, and electrotransferred to nitrocellulose membranes (Amersham Biosciences, United Kingdom). Extracts of mouse brain at postnatal day five were prepared and used as controls (see [70], for technical details). After transfer, membranes were incubated overnight with primary antibodies at 4°C. The following day, they were subsequently incubated with peroxidase-tagged secondary antibodies, and peroxidase activity was detected using the ECL-plus kit (Amersham Biosciences). Active RhoA was determined with a pull-down technique using the GST-Rhotekin-binding domain, as described previously [72], following the manufacturer’s instructions (Rho Activation assay kit BK036, Tebu-Bio Barcelona, Spain).
In vitro experiments and immunocytochemical methods
Glass coverslips (12 mm ∅) were coated essentially as described [73]. Briefly, coverslips were precoated with poly-l-lysine 10 μg/ml dissolved in 0.1 M PBS (pH 7.3) and then washed. They were then incubated with myelin (0.01–0.02 μg/μl), heat-inactivated myelin (0.02 μg/μl heated at 96°C for 1 h) or 0.1 M PBS alone, and then allowed to dry. Next, the coverslips were washed and coated with laminin (2 μg/ml, dissolved in 0.1 M PBS) and washed again with 0.1 M PBS. TEG3 counted cells were seeded onto substrate-coated coverslips in ME10 medium. Some of the cells seeded on coverslips containing myelin were treated with 1 μM NEP1-40 by adding the peptide directly to the culture media. Cells were cultured for 20 h and then the coverslips were fixed in 4% buffered paraformaldehyde, permeabilized with 0.1% Triton X-100 in 0.1 M PBS, and blocked with 10% normal serum in 0.1 M PBS. Cells were sequentially incubated overnight with primary antibodies at 4°C and then with Alexa Fluor-tagged secondary antibodies for 1 h. Cells were rinsed in 0.1 M PBS, stained with 0.1 μM DAPI diluted in 0.1 M PBS for 10 min, rinsed in 0.1 M PBS, and mounted on Fluoromount™ (Vector Labs, Burlingame, CA, USA). Time course of RhoA activation and ERK1-2 phosphorylation in TEG3 cells after myelin treatment was carried out adding myelin (0.02 μg/μl) directly to the cellular media (ME10) at different times.
Adhesion stripe assays
Stripe assays were carried out as described previously [74–76]. Briefly, acid-washed coverslips were incubated overnight with poly-l-lysine (10 μg/ml; Sigma) at 37°C, and after were rinsed several times with water and then air-dried. They were then inverted onto a silicon matrix provided by Dr. J. Jung (Max Planck Institute, Tübingen, Germany). Myelin (0.02 μg/μl) or laminin (2 μg/ml; Sigma) were mixed in 0.1 M PBS and the stripes were performed. The first protein was injected into the matrix channels and incubated for 2 h at 37°C. A solution of fluorescent Alexa 594-conjugated bovine serum albumin (2% in laminin; Molecular Probes) was then injected into the channels. After 2 h at 37°C, the channels were rinsed four times by injecting 0.1 M PBS. TEG3 cells were placed on the stripes and cultured for 24 h as indicated above. Stripe-functionalized coverslips were then fixed with 4% buffered paraformaldehyde for 10 min before DAPI staining, mounted in mounted on Fluoromount™ and photo-documented in an Olympus BX61 fluorescence microscope.
Time-lapse analysis of TEG3 migration
Fluorodish cell culture dishes (World Precision Instruments, Sarasota, FL, USA) were coated with laminin or myelin as described above. We seeded 5 × 104 TEG3 cells on the coated dishes and 20–24 h later we performed the time-lapse analysis. To study cell migration, we transferred the culture dishes to an LCI system (Live Cell Instruments, Seoul, Korea) for 20 h. The multi-tracking analysis was performed with the ImageJ™ software using the plugin mTrackJ (Biomedical Imaging Group Rotterdam of the Erasmus MC-University Medical Center Rotterdam, Netherlands). Tracking was performed in an inverted Olympus microscope IX-71 (20× or 63× objectives) and the images (5 megapixels) were captured by an Olympus XC-50 camera (150 frames, one frame every 8 min). Cell tracking allows the analysis of the scrolling speed and position frame (Xt, Yt). The cell position for each frame (position pixel) was determined with Matlab™ (Math Works, Natick, MA, USA), which provides a 3D graphic (x–y–z, z = time) and the MSD. The same experiments were run over fluorodishes covered with ~70–100-μm-thick PAA gels prepared as previously described [77]. Briefly, to obtain a stiff gel of 12-kPa Young's modulus (PAA), 265 μl of an acrylamide/bis-acrylamide mixture (15% acrylamide and 6.5% bis-acrylamide, Bio-Rad) was dissolved in ultrapure water containing 0.4% of 0.2 μm diameter red fluorescent beads (Invitrogen), 0.5% ammonia persulfate and 0.05% TEMED (Bio-Rad). For multi-tracking, 3D plot, and MSD of the TEG3 migration, the mixture was added to the center of the dish, which was then coated and stored overnight at 4°C. Previous to this procedure, OEC adhesion analyses were performed using 12-, 1.4-, and 0.15-kPa PAA gels. The 12-kPa PAA gels were selected because OECs show greater adherence and migration in these gels.
Traction force measurement
Cell tractions were evaluated using constrained Fourier-transform traction microscopy (FTTM) [78]. Briefly, the displacement field was calculated by comparing fluorescent microbead images obtained during the experiment with a reference image taken at the end of the experiment after the trypsinization and the consequent detachment of OECs from the underlying substrate. The projected cell area was calculated with Matlab™, based on the manual tracing of the OEC contours determined by a phase contrast image obtained at the start of the experiment. A particle imaging velocimetry algorithm [79] was used to determine the deformation of the substrate caused by the traction forces.
Statistical analysis
Summary data are expressed as mean ± SEM (standard error of the mean) of at least three independent experiments. Means were compared by one-way ANOVA test. A value of p ≤ 0.05 was considered statistically significant.
Results
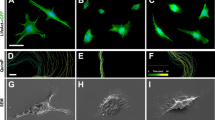
TEG3 is a clonal OEC line that shows similar growth-promoting capacity to non-modified OECs [71]. In addition, this cell line shows all the types of morphology described for primary OECs in culture [11, 80]. In a first set of experiments, we characterized the TEG3 cell line in our culture conditions (Fig. 1). In these conditions, fusiform forms were predominant over flat-shaped ones (Fig. 1a–d). Specific markers of OECs (p75, S100β, or GFAP) were present in our cultured cells (Fig. 1a–d). Next, using Western blotting, we examined whether TEG3 cells express members of the common receptor machinery for MAIs (Nogo receptor complex) [81–83]. Indeed, the revealed blots demonstrated that p75, NgR1, TROY, and Lingo1 were expressed by cultured TEG3 cells (Fig. 1e). This complex was functional since incubation with myelin increased activated RhoA as well as ERK1-2 phosphorylation (supplementary material Fig. S1). In a second set of experiments, we cultured these cells in 35-mm ∅ culture dishes patterned with dots of myelin tagged with 0.75 μm ∅ FITC beads. After Phalloidin-Alexa594 incubation and DAPI counterstaining, cells were exclusively located in regions not containing myelin-FITC (Fig. 1f). To avoid unspecific effects produced by FITC-tagged microbeads on OEC adhesion, we performed a stripe assay using brain myelin extract as substrate (Fig. 1f, h). DAPI-stained TEG3 cells were observed homogenously distributed in stripes containing poly-l-lysine and either laminin or BSA-Alexa594-laminin (Fig. 1g). In contrast, cells did not adhere to stripes containing poly-l-lysine/myelin compared to poly-l-lysine/BSA-Alexa594-laminin (Fig. 1h). Next, we performed a multiple cell migration analysis of TGE3 cells growing over poly-l-lysine + laminin- (Fig. 2a) or poly-l-lysine + myelin-coated substrates (Fig. 2b). Low-density TEG3 cultures were monitored for 20 h (20× objective; one frame every 8 min) in an inverted Olympus IX-71 microscope equipped with a cooled fluorescence camera and a cell culture incubation chamber (see the section Materials and methods for details). The migration speed of OECs grown on myelin-coated substrates (0.01 μg/μl and 0.02 μg/μl) showed a decrease of 46.7% and 57.1%, respectively, compared to those on laminin-coated substrates (Fig. 2c) (0.77 ± 0.01 μm/min (laminin); 0.41 ± 0.01 μm/min (myelin; 0.01 μg/μl); 0.33 ± 0.01 μm/min (myelin; 0.02 μg/μl) (Fig. 2c). In contrast, cell migratory capacity was restored (up to 92.2%, 0.71 ± 0.04 of the laminin migration value) by culture cells in heat-denatured myelin-coated substrate (Fig. 2c, green bar). A three-axis plot [x/y position vs. time (h)] revealed higher motile persistence on laminin-coated than on myelin-coated substrate (Fig. 2d).

Expression of Nogo receptor complex elements and effects of myelin in TEG3 cell adhesion. a–d Photomicrographs illustrating the expression of p75 (a), NgR1 (b), GFAP (c), and S100β (d) in cultured TEG3 cells. e Western blotting detected the expression of p75, NgR1, Lingo 1, and TROY in cultured TEG3 cells and brain extracts (see the section Materials and methods for details). Actin was blotted as internal control. f Photomicrographs illustrating an example of the distribution of TEG3 cells cultured on glass substrates containing spots of Alexa-tagged myelin. Note that TEG3 cells (Phalloidin-positive, red) are not localized on myelin containing regions (green). g, h Examples of stripe assays using TEG3 cells. In both cases, a coating of poly-l-lysine was uniformly distributed in all the culture area. Alternative stripes containing Alexa594-tagged BSA + laminin and laminin were generated (g) or Alexa594-tagged BSA + laminin and myelin (h). DAPI-stained TEG3 cells do not adhere in myelin-containing stripes (h) (see also Results). Scale bars: a = 25 μm pertains to b–d; f = 50 μm; g and h = 50 μm

Analysis of the migratory properties of TEG3 cells on coated glass substrates. a, b Examples of TEG3 cell migration on laminin (a) or myelin (b) coated glass substrates. Each cell trajectory is labeled with a different color line after the software analysis (ImageJ™). c Histogram showing the speed (Y axis) of cultured TEG3 cells on laminin, two different myelin concentrations and heat-denatured myelin. d Examples of three-axis representation of the trajectories of identified TEG3 cells in the focal plane (x,y) along time (z-axis, h). Note that identified TEG3 cells do not modify its (x,y) position over time (arrows) with few of them showing small displacements (arrowhead) compared to laminin. e Plot analysis of the MSD of TEG3 cells growing on laminin (red) or myelin (black). f Photomicrographs illustrating images of TEG3 cells behavior when cultured on laminin (upper panels) or myelin (lower panels). Each picture was obtained after 8 min. Note the small displacement and nuclear translocation of TEG3 cells on myelin compared to the clear nuclear translocation of parallel cultures on laminin. g Example of the NEP1-40 effects of individual TEG3 migration on myelin-coated glass substrate. The trajectory of selected TEG3 cells without treatment is labeled in yellow. Red lines show the changes observed after the incubation with NEP1-40 in the selected cells. h Histogram showing the results of the time lapse analysis and the effect of the NEP1-40 incubation. Data are represented by mean ± SEM of three different experiments and ten identified cells per experiment. Scale bars: a = 200 μm pertains to b; g = 200 μm. Asterisks in c and h indicate statistical differences (p < 0.05, ANOVA test)
To systematically analyze cell migration, we computed the mean square displacement (MSD) of cell trajectories. We then fitted the MSD to a power law expression: MSD = D × ∆t β, where D is a scaling parameter and β indicates the degree of persistence. When β = 1 the motion is random, when β < 1 the motion is subdiffusive (anti-persistent), and when β > 1 the motion is superdiffusive (persistent) [84]. For each time interval studied, cells on laminin-coated substrates migrated further than those on myelin-coated substrates (Fig. 2e). To assess the degree of persistence as a function of the time interval, we calculated the local value of β as: β = ∂ log(MSD)/∂ log(∆t) [84]. Cells growing on myelin-coated substrates exhibited a slightly subdiffuse behavior regardless of the time interval studied. In contrast, those on laminin-coated substrates moved in a slightly superdiffusive manner at short time intervals but their trajectories became progressively subdiffusive with increasing time (Fig. 2e). Taken together, these data indicate that while cells seeded on laminin-coated substrates were more motile than those on myelin-coated, they oscillated around their initial position over long intervals (Fig. 2d).
High-power microscopic observation of cells growing on laminin and myelin showed the specific details of their migration (Fig. 2f). Cells growing on laminin-coated substrate migrated in a similar manner to a fibroblast-like cell with dynamic lamellipodia and further nuclear translocation to the cell leading edge, leaving a tail process (Fig. 2f, upper panels). In fact, the cells translocated the nuclei between caudal to rostral cell locations following the largest lamellipodia. However, cells growing on myelin displayed decreased lamellipodia dynamics and motile small lamellipodia were observed in several cell domains (Fig. 2f, lower panels). This finding contrasts with the behavior of cells growing on laminin, where two main lamellipodia were located on the opposite sides of the fusiform cell, with few transient lamellipodia that displaced laterally along the cell (see also Fig. 3d, e).

Analysis of the migratory properties of TEG3 cells on coated PAA gels. a, b Examples of TEG3 cell migration on PAA-laminin (a) or PAA-Myelin (b) gels. Each cell trajectory is labeled with a different color line after the software analysis (ImageJ™). c Histograms illustrating the quantitative results of the migration assays. Notice that TEG3 cells, irrespective of the coating (laminin or myelin), showed increased speed on PAA gels than glass substrates. d, e High-power photomicrographs illustrating examples of TEG3 cells growing on PAA-laminin (d) and PAA-myelin (e). Notice the strong reduction of cell protrusions over PAA-myelin gels. f MSD plot of TEG3 cells growing on PAA (laminin and myelin gel, black and green lines) or in glass-coated substrates (laminin and myelin, red and blue lines). See Results for details. g Three-axis plots illustrating the migratory trajectories of cultured TEG3 cells in PAA-laminin and PAA-myelin. Notice the decreased number of trajectory changes of identified TEG3 cells on PAA-myelin (arrows). Plot scales are of Fig. 2. Scale bars: a = 200 μm; d = 20 μm. Data in (c) is represented as mean ± SEM of a three different experiments. Asterisks in c indicate statistical differences (p < 0.05, ANOVA test)
We next analyzed the migration distance of identified cells growing on myelin in the absence or presence of the NgR1 inhibitor peptide NEP1-40, to block MAIs effects (Fig. 2g, h). Non-treated cells migrated shorter distances, showing a decrease of 48% compared to those on laminin-coated substrates (0.39 ± 0.03 vs. 0.75 ± 0.02 μm/min, respectively). These distances increased by around 33% after the addition of the NgR1 blocking peptide (0.52 ± 0.03 μm/min) (Fig. 2h).
Cells generate traction forces against their substrate during adhesion and migration, and traction forces are used, in part, by cells to sense the substrate. Thus, using TFM, we aimed to determine the distribution of traction forces in OECs cultured on a well-characterized polyacrylamide (PAA) gel, which is linearly elastic, optically clear, and amenable to protein conjugation [85] (Fig. 3). First we cultured OECs in PAA gels containing laminin (2 μg/ml) or myelin (0.02 μg/μl) and performed time-lapse analysis as above for 10 h (Fig. 3a, b). TEG3 cells growing on PAA coated with laminin or myelin displayed slightly higher migration speeds compared to those cultured on coated glass culture plates (Fig. 3c). Cultured cells over PAA containing laminin or myelin showed similar morphologies than those cultured on glass (Fig. 3d, e). MSD analysis indicates that cells on laminin-coated substrates (glass or PAA) showed similar motion properties being more motile than those on myelin-coated substrates (Fig. 3f, g).
Using TFM, we observed that TEG3 cells seeded on laminin-coated substrates transferred higher strain energy (0.037 ± 0.0061 pJ) to their underlying substrate than those seeded on myelin-coated substrates (0.0033 ± 0.0007 pJ) (Fig. 4). Treatment with NEP1-40 induced a significant increase (2.75-fold increase) in strain energy (0.0091 ± 0.0016 pJ) (Fig. 4b). These findings suggest that increased traction force generation provides OECs with greater migratory capacity. To study the mechanisms underlying changes in traction force generation in laminin versus myelin substrates as well as the effect of Nogo receptor complex blockage by NEP1-40, we examined the intracellular distribution of F-actin (Fig. 5a, b) and FAs (vinculin-positive) (Fig. 5c–f). Results indicate the negligible presence of Phalloidin-Alexa594-positive stress fibers (Fig. 5b) and FAs (Fig. 5e) in TEG3 cells cultured on myelin substrates in contrast to laminin (Fig. 5a (F-actin) and Fig. 5c, d (FAs)). In addition, the distribution of FAs in cells incubated with NEP1-40 was partially recovered as determined after vinculin labeling (Fig. 5f).

TFM analysis of cultured TEG3 cells. a Quantitation of cellular traction forces of cultured TEG3 cells in PAA-laminin, PAA-myelin, and PAA-myelin + NEP1-40. A phase-contrast image of examples of cultured cells is shown on the left side. The scale in μm is also displayed in the lower and the left side of each image. In addition, the force map of cultured cells is shown on the right column. Arrows indicate the direction of the bead displacement. The color scale indicates the magnitude of the cellular traction forces (measured in Pa). b Histogram illustrating the quantitative results of the TFM analysis. Data in b is represented as mean ± SEM of the TFM analysis of 61 cells (PAA-laminin), 83 cells (PAA-myelin) and 62 cells (PAA-myelin + NEP1-40). Asterisks in b indicate statistical differences (p < 0.05, ANOVA test)

Changes in stress fibers and FA distribution in cultured TEG3 cells. a, b Photomicrographs illustrating the distribution of F-actin in cultured TEG3 cells growing on laminin (a) or myelin (b). Stress fibers are clearly identified in a and poorly defined in b. Arrows in a and b point to cellular protrusions. c–f Distribution of FA (arrows) in cultured TEG3 cells on laminin (c, d), myelin (e), and myelin + NEP1-40. Notice the loss of FA and the diffuse vinculin-staining in TEG3 cells cultured over myelin (arrowheads in e) and the recovery of the FA (vinculin-positive) after incubation with NEP1-40 (arrows in f). Scale bars: a = 20 μm pertains to b–f
Discussion
As a result of their intrinsic properties, OECs and genetically modified OECs have been extensively transplanted into the injured spinal cord to promote axonal regeneration [4–11]. However, several studies failed to find unique migratory properties of OECs when implanted into damaged brain or spinal cord lesion (see Introduction for references). Although several authors examined OEC interaction with growing axons [86, 87], a number of parallel studies began to analyze the intrinsic migratory properties of OECs in order to improve their effectiveness after transplantation. Thus, research into OEC migration and proliferation has recently been performed on over glass surfaces with artificially generated gradients [53, 54, 56, 88], in collagen scaffolds [89, 90], in nanofibers [91], and in biomaterial-coated substrates [92, 93]. It has been demonstrated that TROY [94] and NgR1 are expressed in OECs [62, 95]. However, the present study is the first to report that cultured rodent OECs express all the elements of the Nogo receptor complex. In addition, we demonstrate that this receptor complex is active since OECs activate RhoK and increase ERK1-2 phosphorylation in response to myelin. Indeed, myelin inhibits OEC migration over glass surfaces but also in linearly elastic PAA gels.
From a biophysical point of view, our MSD data analysis indicates that cultured OECs migrate in an anti-persistent (subdiffusive) manner both over laminin and myelin on glass and PAA. In addition, our results may explain, to some extent, the findings of Gudiño-Cabrera and coworkers, who reported that transplanted OECs migrate preferentially in the opposite direction to the regenerating axon target and thus fail to promote axon regeneration [31]. Furthermore, OECs transplanted into the lesioned spinal cord must be placed on both sides of the lesion and, close to it, in order to be effective (e.g., [35]). On the basis of our MSD data, we propose that additional factors are required to generate a persistent and directed movement of OECs in vivo [96], as reported in vitro [29]. Indeed, a study by Su et al. indicates that TNF-α released by reactive astrocytes in the meningo-glial scar attracts OECs [97]. This result may explain why OECs invade the lesion cavity containing reactive glia when transplanted at shorter distances [35]. In fact, OECs interact and intermingle with astroglial cells in contrast to other cell types (e.g., Schwann cells) [98]. Moreover, OECs transplanted into the lesion cavity only migrate over short distances in adjacent regions [12]. Indeed, these surrounding areas are rich in myelin debris as well as other inhibitory molecules (e.g., [41]).
OECs show lower migratory potential in lesioned spinal cord than controls [99]. In this regard, anti-NgR-treated OECs show greater migration in spinal cord in white matter tracts than controls [62]. In addition, in the same study, OECs cultured on Nogo-66- or Nogo-His-coated coverslips showed increased adhesion and reduced migration as a result of enhanced formation of paxillin-positive FAs and increased cell spreading, which was overcome by incubation with Y-27632 (a RhoK inhibitor) [62]. In our study, OECs cultured on myelin showed decreased spreading and fewer cell protrusions, stress fibers and FAs (vinculin-positive), and these effects were partially reversed by the NEP1-40 peptide. These data also paralleled those obtained from PAA gels. The differences in cell spreading between our data and those reported by Su and coworkers could be attributed to their use of a recombinant peptide or Nogo-A protein instead of complete myelin extract (containing several inhibitory proteins and lipids; see Introduction for details). The studies show different cell spreading of OECs growing over myelin or Nogo-66 peptides. Although the cell spreading of OECs cultured over Nogo-A is not illustrated by Su et al. we cannot completely rule out that the cell spreading mechanism in OECs could be different from that of other cell types. A study by Oertle et al. reported that Nogo-66, Nogo-C, and Nogo-66 peptide 4 (shown to be the inhibitory regions of Nogo-66 by GrandPre and coworkers [100]) are not inhibitory for fibroblasts spreading in culture (see Fig. 2 of [101]). In fact, two Nogo-A regions (59–172 aa and 544–725 aa) showed the increased inhibition of cell spreading in cultured fibroblasts. Thus, we conclude that specific Nogo-A domains may have a different effect on OECs spreading in a balanced way. On the other hand, in the same study, the authors report that higher concentrations of the cell-spreading inhibitory peptides became anti-adhesive. It has been reported that cell adhesion and migration showed a biphasic distribution and an optimal balance between adhesion and migration must be achieved by cells before they can migrate [102]. Indeed, if adhesion is lower or higher than an optimal level, migration may be decreased.
On the other hand, molecules other than Nogo-A may interact with Nogo receptor complex (e.g., OMgp, MAG, sulfatide) as well as with other undetermined MAIs receptors expressed by cultured OECs. Although further studies for OECs are needed, it has been described that the N-terminal domain inhibits COS-7 or HUVEC adhesion by acting through αVβ3, α5 and α4 integrins, leading to decreased activity of focal adhesion kinase (FAK) in treated cells [49]. Our data showing a moderate recovery of the migratory properties and strain energy (see below) of TEG3 after NEP1-40 incubation suggest the presence of parallel inhibitory mechanisms.
Cell migration relies on the interplay between two cycles, one involving extension and contraction of its cytoskelekon, and the other involving formation and retraction of its adhesions. Traction forces come into play in the synchronization of both cycles by causing forced de-adhesion at the trailing edge and by driving the resulting forward motion of the cell body. Therefore, the balance between traction force and adhesion strength can be understood as a direct indicator of cell migration capacity. Using TFM and single-cell tracking, we showed that even if OECs cells on laminin exhibit increased concentration of focal adhesion proteins, the increased traction force is sufficient to tilt the mechanical balance toward a pro-migratory phenotype. The contribution of FAs to cell migration has been widely debated. Su et al. (2007) reported that OECs growing on Nogo-66 resembled flat-sheet cells. This observation could be attributed to a maintenance role of FA with little migration potential, as suggested for flat-shaped OECs. However, although we cannot rule out cell-specific factors for OECs, FA distribution and FA-mediated signaling in several cell types correlates with traction force parameters, nuclear translocation and cell migration (e.g., keratinocytes [103], invasive tumor cells [104], or fibroblasts [105, 106]).
In addition, our data show that OECs cultured on PAA substrates, the mechanical properties of which clearly differ from substrates coated on glass slides, increase their migratory potential. In this regard, recent data report increased proliferation and migration of OECs growing on several scaffolds (see above). Thus, it is feasible that a nanostructured scaffold would serve as a mechanical guide for OEC migration, thereby preventing the interaction of these cells with a putative inhibitory ECM. In this respect, migrating cells sense and respond to external stimuli. In fact, mechanical inputs can also be powerful regulators of cell behavior [103]. In physiological conditions, the migration of most cells occurs in an adhesion-dependent manner and involves the formation of FA at the membrane and the generation of mechanical forces via the actin-myosin network [103]. MAIs and receptors modulate the establishment of this network as well as microtubule dynamics [81, 107]. Indeed, MAI-induced arrest of CNS axon growth is either induced after MAI receptor, the Ca2+-dependent activation of the epidermal growth factor receptor (EGFr), or induced by sequential RhoA/RhoK/LIM-kinase/cofilin phosphorylation, leading to actin depolymerization [108]. In parallel, microtubule stabilization is also compromised after exposure to MAIs [109]. Our results reinforce the notion that molecules present in myelin extracts act on Nogo receptor complex as well as on other receptors, and may modulate OEC cytoskeleton and FAs distribution. In conclusion, our data suggest that a cell-based strategy using OECs to overcome the inhibitory action of myelin is required in order to enhance the migratory properties and the persistence of OECs in axon re-growth and their functional recovery in the lesioned CNS.
References
Ramon-Cueto A, Valverde F (1995) Olfactory bulb ensheathing glia: a unique cell type with axonal growth-promoting properties. Glia 14(3):163–173
Doucette R (1995) Olfactory ensheathing cells: potential for glial cell transplantation into areas of CNS injury. Histol Histopathol 10(2):503–507
Katoh H, Shibata S, Fukuda K, Sato M, Satoh E, Nagoshi N, Minematsu T, Matsuzaki Y, Akazawa C, Toyama Y, Nakamura M, Okano H (2011) The dual origin of the peripheral olfactory system: placode and neural crest. Mol Brain 4(1):34
Ramon-Cueto A, Cordero MI, Santos-Benito FF, Avila J (2000) Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron 25(2):425–435
Li Y, Field PM, Raisman G (1997) Repair of adult rat corticospinal tract by transplants of olfactory ensheathing cells. Science 277(5334):2000–2002
Navarro X, Valero A, Gudino G, Fores J, Rodriguez FJ, Verdu E, Pascual R, Cuadras J, Nieto-Sampedro M (1999) Ensheathing glia transplants promote dorsal root regeneration and spinal reflex restitution after multiple lumbar rhizotomy. Ann Neurol 45(2):207–215
Ramon-Cueto A, Nieto-Sampedro M (1994) Regeneration into the spinal cord of transected dorsal root axons is promoted by ensheathing glia transplants. Exp Neurol 127(2):232–244
Ruitenberg MJ, Plant GW, Christensen CL, Blits B, Niclou SP, Harvey AR, Boer GJ, Verhaagen J (2002) Viral vector-mediated gene expression in olfactory ensheathing glia implants in the lesioned rat spinal cord. Gene Ther 9(2):135–146
Ramon-Cueto A, Plant GW, Avila J, Bunge MB (1998) Long-distance axonal regeneration in the transected adult rat spinal cord is promoted by olfactory ensheathing glia transplants. J Neurosci 18(10):3803–3815
Garcia-Alias G, Lopez-Vales R, Fores J, Navarro X, Verdu E (2004) Acute transplantation of olfactory ensheathing cells or Schwann cells promotes recovery after spinal cord injury in the rat. J Neurosci Res 75(5):632–641
Moreno-Flores MT, Bradbury EJ, Martin-Bermejo MJ, Agudo M, Lim F, Pastrana E, Avila J, Diaz-Nido J, McMahon SB, Wandosell F (2006) A clonal cell line from immortalized olfactory ensheathing glia promotes functional recovery in the injured spinal cord. Mol Ther 13(3):598–608
Ramer LM, Au E, Richter MW, Liu J, Tetzlaff W, Roskams AJ (2004) Peripheral olfactory ensheathing cells reduce scar and cavity formation and promote regeneration after spinal cord injury. J Comp Neurol 473(1):1–15
Raisman G (2007) Repair of spinal cord injury by transplantation of olfactory ensheathing cells. C R Biol 330(6–7):557–560
Moreno-Flores MT, Avila J (2006) The quest to repair the damaged spinal cord. Recent Pat CNS Drug Discov 1(1):55–63
Radtke C, Kocsis JD, Vogt PM (2009) Chapter 22: Transplantation of olfactory ensheathing cells for peripheral nerve regeneration. Int Rev Neurobiol 87:405–415
Ramon-Cueto A, Munoz-Quiles C (2011) Clinical application of adult olfactory bulb ensheathing glia for nervous system repair. Exp Neurol 229(1):181–194
Su Z, He C (2010) Olfactory ensheathing cells: biology in neural development and regeneration. Prog Neurobiol 92(4):517–532
Windus LC, Lineburg KE, Scott SE, Claxton C, Mackay-Sim A, Key B, St John JA (2010) Lamellipodia mediate the heterogeneity of central olfactory ensheathing cell interactions. Cell Mol Life Sci 67(10):1735–1750
Guerout N, Derambure C, Drouot L, Bon-Mardion N, Duclos C, Boyer O, Marie JP (2010) Comparative gene expression profiling of olfactory ensheathing cells from olfactory bulb and olfactory mucosa. Glia 58(13):1570–1580
Vincent AJ, West AK, Chuah MI (2005) Morphological and functional plasticity of olfactory ensheathing cells. J Neurocytol 34(1–2):65–80
Au E, Roskams AJ (2003) Olfactory ensheathing cells of the lamina propria in vivo and in vitro. Glia 41(3):224–236
Roet KC, Bossers K, Franssen EH, Ruitenberg MJ, Verhaagen J (2011) A meta-analysis of microarray-based gene expression studies of olfactory bulb-derived olfactory ensheathing cells. Exp Neurol 229(1):10–45
Richter MW, Fletcher PA, Liu J, Tetzlaff W, Roskams AJ (2005) Lamina propria and olfactory bulb ensheathing cells exhibit differential integration and migration and promote differential axon sprouting in the lesioned spinal cord. J Neurosci 25(46):10700–10711
Novikova LN, Lobov S, Wiberg M, Novikov LN (2011) Efficacy of olfactory ensheathing cells to support regeneration after spinal cord injury is influenced by method of culture preparation. Exp Neurol 229(1):132–142
Kumar R, Hayat S, Felts P, Bunting S, Wigley C (2005) Functional differences and interactions between phenotypic subpopulations of olfactory ensheathing cells in promoting CNS axonal regeneration. Glia 50(1):12–20
Franceschini IA, Barnett SC (1996) Low-affinity NGF-receptor and E-N-CAM expression define two types of olfactory nerve ensheathing cells that share a common lineage. Dev Biol 173(1):327–343
Higginson JR, Barnett SC (2011) The culture of olfactory ensheathing cells (OECs)-a distinct glial cell type. Exp Neurol 229(1):2–9
Richter M, Westendorf K, Roskams AJ (2008) Culturing olfactory ensheathing cells from the mouse olfactory epithelium. Methods Mol Biol 438:95–102
Huang ZH, Wang Y, Cao L, Su ZD, Zhu YL, Chen YZ, Yuan XB, He C (2008) Migratory properties of cultured olfactory ensheathing cells by single-cell migration assay. Cell Res 18(4):479–490
van den Pol AN, Santarelli JG (2003) Olfactory ensheathing cells: time lapse imaging of cellular interactions, axonal support, rapid morphologic shifts, and mitosis. J Comp Neurol 458(2):175–194
Gudino-Cabrera G, Pastor AM, de la Cruz RR, Delgado-Garcia JM, Nieto-Sampedro M (2000) Limits to the capacity of transplants of olfactory glia to promote axonal regrowth in the CNS. Neuroreport 11(3):467–471
Nieto-Sampedro M (2003) Central nervous system lesions that can and those that cannot be repaired with the help of olfactory bulb ensheathing cell transplants. Neurochem Res 28(11):1659–1676
Lee IH, Bulte JW, Schweinhardt P, Douglas T, Trifunovski A, Hofstetter C, Olson L, Spenger C (2004) In vivo magnetic resonance tracking of olfactory ensheathing glia grafted into the rat spinal cord. Exp Neurol 187(2):509–516
Lu P, Yang H, Culbertson M, Graham L, Roskams AJ, Tuszynski MH (2006) Olfactory ensheathing cells do not exhibit unique migratory or axonal growth-promoting properties after spinal cord injury. J Neurosci 26(43):11120–11130
Pearse DD, Sanchez AR, Pereira FC, Andrade CM, Puzis R, Pressman Y, Golden K, Kitay BM, Blits B, Wood PM, Bunge MB (2007) Transplantation of Schwann cells and/or olfactory ensheathing glia into the contused spinal cord: Survival, migration, axon association, and functional recovery. Glia 55(9):976–1000
Amemori T, Jendelova P, Ruzickova K, Arboleda D, Sykova E (2010) Co-transplantation of olfactory ensheathing glia and mesenchymal stromal cells does not have synergistic effects after spinal cord injury in the rat. Cytotherapy 12(2):212–225
Pettersson J, Lobov S, Novikova LN (2010) Labeling of olfactory ensheathing glial cells with fluorescent tracers for neurotransplantation. Brain Res Bull 81(1):125–132
Li BC, Li Y, Chen LF, Chang JY, Duan ZX (2011) Olfactory ensheathing cells can reduce the tissue loss but not the cavity formation in contused spinal cord of rats. J Neurol Sci 303(1–2):67–74
Saxton MJ (2007) Modeling 2D and 3D diffusion. Methods Mol Biol 400:295–321
Fawcett JW (2006) Overcoming inhibition in the damaged spinal cord. J Neurotrauma 23(3–4):371–383
Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nat Rev Neurosci 5(2):146–156
Cao Z, Gao Y, Deng K, Williams G, Doherty P, Walsh FS (2010) Receptors for myelin inhibitors: Structures and therapeutic opportunities. Mol Cell Neurosci 43(1):1–14
Giger RJ, Venkatesh K, Chivatakarn O, Raiker SJ, Robak L, Hofer T, Lee H, Rader C (2008) Mechanisms of CNS myelin inhibition: evidence for distinct and neuronal cell type specific receptor systems. Restor Neurol Neurosci 26(2–3):97–115
Thomas R, Favell K, Morante-Redolat J, Pool M, Kent C, Wright M, Daignault K, Ferraro GB, Montcalm S, Durocher Y, Fournier A, Perez-Tur J, Barker PA (2010) LGI1 is a Nogo receptor 1 ligand that antagonizes myelin-based growth inhibition. J Neurosci 30(19):6607–6612
Zhang L, Zheng S, Wu H, Wu Y, Liu S, Fan M, Zhang J (2009) Identification of BLyS (B lymphocyte stimulator), a non-myelin-associated protein, as a functional ligand for Nogo-66 receptor. J Neurosci 29(19):6348–6352
Lee H, Raiker SJ, Venkatesh K, Geary R, Robak LA, Zhang Y, Yeh HH, Shrager P, Giger RJ (2008) Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci 28(11):2753–2765
Winzeler AM, Mandemakers WJ, Sun MZ, Stafford M, Phillips CT, Barres BA (2011) The lipid sulfatide is a novel myelin-associated inhibitor of CNS axon outgrowth. J Neurosci 31(17):6481–6492
Worter V, Schweigreiter R, Kinzel B, Mueller M, Barske C, Bock G, Frentzel S, Bandtlow CE (2009) Inhibitory activity of myelin-associated glycoprotein on sensory neurons is largely independent of NgR1 and NgR2 and resides within Ig-Like domains 4 and 5. PloS one 4(4):e5218
Hu F, Strittmatter SM (2008) The N-terminal domain of Nogo-A inhibits cell adhesion and axonal outgrowth by an integrin-specific mechanism. J Neurosci 28(5):1262–1269
Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ (2005) The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci 25(4):808–822
Grunewald E, Kinnell HL, Porteous DJ, Thomson PA (2009) GPR50 interacts with neuronal NOGO-A and affects neurite outgrowth. Mol Cell Neurosci 42(4):363–371
Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322(5903):967–970
Cao L, Su Z, Zhou Q, Lv B, Liu X, Jiao L, Li Z, Zhu Y, Huang Z, Huang A, He C (2006) Glial cell line-derived neurotrophic factor promotes olfactory ensheathing cells migration. Glia 54(6):536–544
Yan H, Lu D, Rivkees SA (2003) Lysophosphatidic acid regulates the proliferation and migration of olfactory ensheathing cells in vitro. Glia 44(1):26–36
Windus LC, Chehrehasa F, Lineburg KE, Claxton C, Mackay-Sim A, Key B, St John JA (2011) Stimulation of olfactory ensheathing cell motility enhances olfactory axon growth. Cell Mol Life Sci 68(19):3233–3247
Huang ZH, Wang Y, Su ZD, Geng JG, Chen YZ, Yuan XB, He C (2011) Slit-2 repels the migration of olfactory ensheathing cells by triggering Ca2+ -dependent cofilin activation and RhoA inhibition. J Cell Sci 124(Pt 2):186–197
Wehrle R, Camand E, Chedotal A, Sotelo C, Dusart I (2005) Expression of netrin-1, slit-1 and slit-3 but not of slit-2 after cerebellar and spinal cord lesions. Eur J Neurosci 22(9):2134–2144
Pasterkamp RJ, Giger RJ, Ruitenberg MJ, Holtmaat AJ, De Wit J, De Winter F, Verhaagen J (1999) Expression of the gene encoding the chemorepellent semaphorin III is induced in the fibroblast component of neural scar tissue formed following injuries of adult but not neonatal CNS. Mol Cell Neurosci 13(2):143–166
Low K, Culbertson M, Bradke F, Tessier-Lavigne M, Tuszynski MH (2008) Netrin-1 is a novel myelin-associated inhibitor to axon growth. J Neurosci 28(5):1099–1108
Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB (2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416(6881):636–640
Schwab ME (1990) Myelin-associated inhibitors of neurite growth and regeneration in the CNS. Trends Neurosci 13(11):452–456
Su Z, Cao L, Zhu Y, Liu X, Huang Z, Huang A, He C (2007) Nogo enhances the adhesion of olfactory ensheathing cells and inhibits their migration. J Cell Sci 120(Pt 11):1877–1887
Buss A, Schwab ME (2003) Sequential loss of myelin proteins during Wallerian degeneration in the rat spinal cord. Glia 42(4):424–432
Buss A, Sellhaus B, Wolmsley A, Noth J, Schwab ME, Brook GA (2005) Expression pattern of NOGO-A protein in the human nervous system. Acta Neuropathol 110(2):113–119
Lauffenburger DA, Horwitz AF (1996) Cell migration: a physically integrated molecular process. Cell 84(3):359–369
Balaban NQ, Schwarz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L, Geiger B (2001) Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol 3(5):466–472
Beningo KA, Dembo M, Kaverina I, Small JV, Wang YL (2001) Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J Cell Biol 153(4):881–888
Bershadsky AD, Balaban NQ, Geiger B (2003) Adhesion-dependent cell mechanosensitivity. Annu Rev Cell Dev Biol 19:677–695
GrandPre T, Li S, Strittmatter SM (2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417(6888):547–551
Seira O, Gavin R, Gil V, Llorens F, Rangel A, Soriano E, del Rio JA (2010) Neurites regrowth of cortical neurons by GSK3beta inhibition independently of Nogo receptor 1. J Neurochem 113(6):1644–1658
Moreno-Flores MT, Lim F, Martin-Bermejo MJ, Diaz-Nido J, Avila J, Wandosell F (2003) Immortalized olfactory ensheathing glia promote axonal regeneration of rat retinal ganglion neurons. J Neurochem 85(4):861–871
Ren XD, Kiosses WB, Schwartz MA (1999) Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J 18(3):578–585
Fournier AE, GrandPre T, Strittmatter SM (2001) Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409(6818):341–346
Nguyen-Ba-Charvet KT, Brose K, Marillat V, Sotelo C, Tessier-Lavigne M, Chedotal A (2001) Sensory axon response to substrate-bound Slit2 is modulated by laminin and cyclic GMP. Mol Cell Neurosci 17(6):1048–1058
Prestoz L, Chatzopoulou E, Lemkine G, Spassky N, Lebras B, Kagawa T, Ikenaka K, Zalc B, Thomas JL (2004) Control of axonophilic migration of oligodendrocyte precursor cells by Eph-ephrin interaction. Neuron Glia Biol 1(1):73–83
Bribian A, Esteban PF, Clemente D, Soussi-Yanicostas N, Thomas JL, Zalc B, de Castro F (2008) A novel role for anosmin-1 in the adhesion and migration of oligodendrocyte precursors. Dev Neurobiol 68(13):1503–1516
Dembo M, Wang YL (1999) Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys J 76(4):2307–2316
Butler JP, Tolic-Norrelykke IM, Fabry B, Fredberg JJ (2002) Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol Cell Physiol 282(3):C595–C605
Trepat X, Wasserman MR, Angelini TE, Millet E, Weitz DA, Butler JP, Fredberg JJ (2009) Physical forces during collective cell migration. Nat Phys 5(6):426–430
Pastrana E, Moreno-Flores MT, Gurzov EN, Avila J, Wandosell F, Diaz-Nido J (2006) Genes associated with adult axon regeneration promoted by olfactory ensheathing cells: a new role for matrix metalloproteinase 2. J Neurosci 26(20):5347–5359
Llorens F, Gil V, del Rio JA (2011) Emerging functions of myelin-associated proteins during development, neuronal plasticity, and neurodegeneration. FASEB J 25(2):463–475
Yamashita T, Fujitani M, Yamagishi S, Hata K, Mimura F (2005) Multiple signals regulate axon regeneration through the Nogo receptor complex. Mol Neurobiol 32(2):105–111
Zhang S, Zhang Q, Zhang JH, Qin X (2008) NgR acts as an inhibitor to axonal regeneration in adults. Front Biosci 13:2030–2040
Bursac P, Lenormand G, Fabry B, Oliver M, Weitz DA, Viasnoff V, Butler JP, Fredberg JJ (2005) Cytoskeletal remodelling and slow dynamics in the living cell. Nat Mater 4(7):557–561
Califano JP, Reinhart-King CA (2010) Exogenous and endogenous force regulation of endothelial cell behavior. J Biomech 43(1):79–86
Sorensen A, Moffat K, Thomson C, Barnett SC (2008) Astrocytes, but not olfactory ensheathing cells or Schwann cells, promote myelination of CNS axons in vitro. Glia 56(7):750–763
Chuah MI, Hale DM, West AK (2011) Interaction of olfactory ensheathing cells with other cell types in vitro and after transplantation: Glial scars and inflammation. Exp Neurol 229(1):46–53
Vukovic J, Ruitenberg MJ, Roet K, Franssen E, Arulpragasam A, Sasaki T, Verhaagen J, Harvey AR, Busfield SJ, Plant GW (2009) The glycoprotein fibulin-3 regulates morphology and motility of olfactory ensheathing cells in vitro. Glia 57(4):424–443
Mollers S, Heschel I, Damink LH, Schugner F, Deumens R, Muller B, Bozkurt A, Nava JG, Noth J, Brook GA (2009) Cytocompatibility of a novel, longitudinally microstructured collagen scaffold intended for nerve tissue repair. Tissue Eng Part A 15(3):461–472
Wang B, Zhao Y, Lin H, Chen B, Zhang J, Wang X, Zhao W, Dai J (2006) Phenotypical analysis of adult rat olfactory ensheathing cells on 3-D collagen scaffolds. Neurosci Lett 401(1–2):65–70
Shen Y, Qian Y, Zhang H, Zuo B, Lu Z, Fan Z, Zhang P, Zhang F, Zhou C (2010) Guidance of olfactory ensheathing cell growth and migration on electrospun silk fibroin scaffolds. Cell Transplant 19(2):147–157
Martin-Lopez E, Nieto-Diaz M, Nieto-Sampedro M (2010) Differential adhesiveness and neurite-promoting activity for neural cells of chitosan, gelatin, and poly-l-lysine films. J Biomater Appl. PMID 20876636
Martin-Lopez E, Alonso FR, Nieto-Diaz M, Nieto-Sampedro M (2010) Chitosan, gelatin and poly(l-lysine) polyelectrolyte-based scaffolds and films for neural tissue engineering. J Biomater Sci Polym Ed. PMID 21192838
Morikawa Y, Hisaoka T, Kitamura T, Senba E (2008) TROY, a novel member of the tumor necrosis factor receptor superfamily in the central nervous system. Ann N Y Acad Sci 1126:A1–A10
Woodhall E, West AK, Vickers JC, Chuah MI (2003) Olfactory ensheathing cell phenotype following implantation in the lesioned spinal cord. Cell Mol Life Sci 60(10):2241–2253
Lankford KL, Sasaki M, Radtke C, Kocsis JD (2008) Olfactory ensheathing cells exhibit unique migratory, phagocytic, and myelinating properties in the X-irradiated spinal cord not shared by Schwann cells. Glia 56(15):1664–1678
Su Z, Yuan Y, Chen J, Cao L, Zhu Y, Gao L, Qiu Y, He C (2009) Reactive astrocytes in glial scar attract olfactory ensheathing cells migration by secreted TNF-alpha in spinal cord lesion of rat. PLoS One 4(12):e8141
Lakatos A, Franklin RJ, Barnett SC (2000) Olfactory ensheathing cells and Schwann cells differ in their in vitro interactions with astrocytes. Glia 32(3):214–225
Deng C, Gorrie C, Hayward I, Elston B, Venn M, Mackay-Sim A, Waite P (2006) Survival and migration of human and rat olfactory ensheathing cells in intact and injured spinal cord. J Neurosci Res 83(7):1201–1212
GrandPre T, Nakamura F, Vartanian T, Strittmatter SM (2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403(6768):439–444
Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, Huber AB, Simonen M, Schnell L, Brosamle C, Kaupmann K, Vallon R, Schwab ME (2003) Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. J Neurosci 23(13):5393–5406
Wang JH, Lin JS (2007) Cell traction force and measurement methods. Biomech Model Mechanobiol 6(6):361–371
Fournier MF, Sauser R, Ambrosi D, Meister JJ, Verkhovsky AB (2010) Force transmission in migrating cells. J Cell Biol 188(2):287–297
Mierke CT, Rosel D, Fabry B, Brabek J (2008) Contractile forces in tumor cell migration. Eur J Cell Biol 87(8–9):669–676
Wang HB, Dembo M, Hanks SK, Wang Y (2001) Focal adhesion kinase is involved in mechanosensing during fibroblast migration. Proc Natl Acad Sci USA 98(20):11295–11300
Li S, Guan JL, Chien S (2005) Biochemistry and biomechanics of cell motility. Annu Rev Biomed Eng 7:105–150
Schwab ME (2010) Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci 11(12):799–811
Hsieh SH, Ferraro GB, Fournier AE (2006) Myelin-associated inhibitors regulate cofilin phosphorylation and neuronal inhibition through LIM kinase and Slingshot phosphatase. J Neurosci 26(3):1006–1015
Mimura F, Yamagishi S, Arimura N, Fujitani M, Kubo T, Kaibuchi K, Yamashita T (2006) Myelin-associated glycoprotein inhibits microtubule assembly by a Rho-kinase-dependent mechanism. J Biol Chem 281(23):15970–15979
Acknowledgments
The authors thank R. Rycroft for linguistic advice and G. Tormen for technical assistance. This work was supported by FP7-PRIORITY, the MICINN (BFU2009-10848) and grants from the Instituto Carlos III (PI11/03028) and SGR2009-366 (Generalitat of Catalunya) to JADR. SN and PC were supported by the MICINN. DR is supported by a fellowship from the National Commission for Science and Technology (CONICYT, Chile). AB is a Sara Borrell postdoctoral research of the Instituto Carlos III. XT acknowledges support of the Spanish Ministry for Science and Innovation (BFU2009-07595) and the European Research Council (Grant Agreement 242993). F. Wandosell was supported by MICINN (SAF2009-12249-C02-01).
Author information
Authors and Affiliations
Corresponding authors
Additional information
S. Nocentini and D. Reginensi contribute equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
18_2011_893_MOESM1_ESM.tif
Fig. S1. Myelin treatment activates RhoA and induces ERK1-2 phosphorylation in TEG3 cells. Time course of RhoA activation (a) and ERK1-2 phosphorylation (b) in TEG3 cells after myelin treatment. See the sections Materials and methods and Results for details (TIFF 2036 kb)
Rights and permissions
About this article
Cite this article
Nocentini, S., Reginensi, D., Garcia, S. et al. Myelin-associated proteins block the migration of olfactory ensheathing cells: an in vitro study using single-cell tracking and traction force microscopy. Cell. Mol. Life Sci. 69, 1689–1703 (2012). https://doi.org/10.1007/s00018-011-0893-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0893-1