
Abstract
Sox2 is a component of the core transcriptional regulatory network which maintains the totipotency of the cells during embryonic preimplantation period, the pluripotency of embryonic stem cells, and the multipotency of neural stem cells. This maintenance is controlled by internal loops between Sox2 and other transcription factors of the core such as Oct4, Nanog, Dax1, and Klf4, downstream proteins of extracellular ligands, epigenetic modifiers, and miRNAs. As Sox2 plays an important role in the balance between stem cells maintenance and commitment to differentiated lineages throughout the lifetime, it is supposed that Sox2 could regulate stem cells aging processes. In this review, we provide an update concerning the involvement of Sox2 in neurogenesis during normal aging and discuss its possible role in Alzheimer’s disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sox2 (sex-determining region Y (SRY)-box 2) is a transcriptional factor that is essential for maintaining self-renewal/proliferation/pluripotency of undifferentiated embryonic stem cells (ESCs) and multipotency of neural stem cells (NSCs). The Sox family of protein was identified in 1990 after the seminal discovery of the mammalian testis-determining factor SRY that carries a characteristic high-mobility group (HMG) domain that binds DNA in a sequence-specific manner [1, 2]. Four years later, Sox2 was inadvertently discovered when Dailey and colleagues identified that fibroblast growth factor 4 (FGF4) activity was regulated by an embryonically expressed factor (then called Fx) in embryonic carcinoma F9 cells [3].
Sox2 is a well-established and crucial regulator of cell fate decisions during development (Figs. 1a and 2) but also plays an important role in adult tissue homeostasis and regeneration. As a matter of fact, Sox2 is required for the totipotency of cells during embryonic preimplantation period [4], the pluripotency of ESCs [5] and the multipotency of NSCs [6, 7] (Fig. 2). Moreover, Sox2 displays the remarkable property, when co-expressed with other synergistic factors, to reprogram somatic cells into induced pluripotent stem cells (iPSCs) [8, 9] (Fig. 1b). This makes Sox2 a key factor for the control of stem cells fate and more generally neurogenesis.

Sox2 as a key component of stem cells fate and iPSCs reprogramming. a NSCs can self-renew (pink circle) or differentiate into neurons in the neurogenic niches (blue circle). Sox2 plays a central role in the fate of embryonic and neural stem cells through the control of the balance (activation/inhibition) of several self-renewal and differentiation genes. b When added, alone or in combination with Oct4, Klf4, and c-Myc, Sox2 initiate the reprogramming of somatic cells into iPSCs that can be then forced to become neurons by means of specific treatments

Roles of Sox2 in the central nervous system throughout the lifetime. Sox2 is expressed throughout the life from oocyte stage to adult neural stem cells to maintain neural stem cells potency. Sox2, together with some transcription factors such as Oct4, Nanog, Klf4, Dax1, and others constitute a core network. This network is under the control of autoregulation mechanisms, epigenetic effects, post-translational modulations by miRNAs, and some signaling pathways initiated by extracellular ligands such as LIF (JAK/STAT), FGFs (Akt), Wnt (β-catenin), Delta/Jagged (NICD) and BMP (R-SMAD). According to the time, Sox2 makes toti-, pluri- or multi-potency under the effect of these ligands. LIF leukemia inhibitory factor, FGFs fibroblast growth factors, BMP bone morphogenetic protein, TE trophectoderm, ICM inner cell mass, SVZ subventricular zone, SGZ subgranular zone, NICD Notch intracellular domain, Akt protein kinase B, STST3 signal transducer and activator of transcription 3, JAK Janus-associated tyrosine kinase, INS insulin IGF insulin growth factor, FZ frizzled, R-SMAD receptor-activated Smad proteins
Sox2 is part of the core of the pluripotency network that operates together with other transcription factors, such as Nanog and Oct4, to promote potency of stem cells (for review, see [10]). Interestingly, this network is under the control of some intracellular signaling pathways initiated by extracellular ligands such as Wnt, Notch, FGF, leukemia inhibitory factor (LIF), and bone morphogenetic protein (BMP). It can also be regulated by epigenetic modifiers and several microRNAs (miRNAs) (Fig. 2).
Thus, as Sox2 maintains stem cell potency and self-renewal throughout the lifetime, it is tempting to envision that it may be involved in stem cell aging processes. This possibility gains support when we consider that, although aging mechanisms are different in mitotic and non-mitotic cells in some aspects, there exist some correlations between Sox2 and some age-related factors.
The Roles of Sox2 in Neuroectoderm Development and Adult Neurogenesis
The expression of Sox2 has been observed in cytoplasm and nuclei of oocyte, two-cell stage to morula cells, in some trophectoderm (TE) cells, and all nuclei of inner cell mass (ICM) [11] as well as presumptive neuroectoderm (Fig. 2). In 9.5-day postcoitum mice, Sox2 RNA is seen throughout the brain, neural tube, sensory placodes, and branchial arches [12]. Knockdown and overexpression studies have shown the important role of Sox2 in preimplantation processes in mouse. Although the maternal Sox2 protein has been supposed to compensate for the loss of the function of Sox2 transcripts in mouse Sox2 homozygous mutant embryos [12], Sox2 knockdown by small interfering RNAs (siRNAs) in two-cell embryo mostly leads to a decrease in Sox2 level at morulae stage, developmental arrest at the morulae/blastocyst transition, and inability to form TE [11]. In addition, Sox2 overexpression in 1-cell embryo results in developmental arrest at two-cell stage and changes the reprogramming gene expression [4]. Moreover, Sox2 is clearly involved in stage-transition gene regulatory networks prior to implantation and then in the fate decision of three lineages of blastocyst to address the inner cells to epiblast [13]. Finally, in mice inner cell mass, Sox2 expression maintains pluripotency of ESCs and downregulation of Sox2 promotes them to trophectoderm-like cells fate [14].
ESCs maintain their pluripotency until deciding to differentiate into the progenitors of mesendoderm or neural ectoderm according to the levels of Sox2 and Oct4 expression. For instance, high Sox2 and low Oct4 levels drive ESCs to neural ectoderm fate [15]. Sox2 is expressed in chick embryo throughout the neural tube and is then restricted to the medial ventricular zone and along the entire dorsoventral axis of the developing spinal cord to maintain neural characteristics of nervous system progenitors by prevention of terminal differentiation [16]. As a matter of fact, Sox2 is expressed in proliferating central nervous system (CNS) progenitors and downregulated during their final cell cycle, and its expression inhibits neuronal differentiation and results in the maintenance of progenitor characteristics. As a corollary, inhibition of Sox2 signaling results in a loss of progenitor markers and the onset of early neuronal differentiation markers [12].
Importantly, beside its preponderant role in neuroectoderm development, Sox2 is also required for maintaining multipotency and self-renewal of adult NSCs (Fig. 2). In the CNS, NSCs are mostly located in the subgranular zone (SGZ) of the dentate gyrus and the subventricular zone (SVZ) although NSCs have been also isolated from dorsal root ganglia (DRG) [17], trigeminal ganglia [18], enteric nervous system (ENS) [19], and spiral ganglion in the peripheral nervous system (PNS) [20].
In the dentate gyrus, multipotent type-1 neural progenitor cells (NPCs) express Sox2 with other specific proteins such as glial fibrillary acidic protein (GFAP) [21], nestin and brain lipid-binding protein (BLBP) [22]. These cells, which divide at slow rate, display strong “stemness” properties and express Sox2 at high level. Type 2a hippocampal progenitor cells express high Sox2/low doublecortin (DCX) level, while type 2b show low Sox2/high DCX expression [22]. Type 2a and 2b progenitor cells then give rise to type 3 cells expressing DCX [23].
In SVZ, radial glia-like cells (type B cells) that express Sox2 and GFAP exhibit glial properties of both astrocytes and prenatal radial glia [6, 24] and act as NSCs. Transient amplifying cells (type C) are found in the rostral migratory stream (RMS) as bipotent cells, express oligodendrocyte transcription factor 2 (Olig2), achaete-scute homolog 1 (Ascl1), Dlx2, and paired box gene 2 (Pax2) [25, 26], and give rise to DCX-expressing type A neuroblasts [27].
Finally, immature neurons migrate toward olfactory bulb in rostral migratory chains surrounded by astrocytes to be neurons [28]. Sox2 expression has been reported in the undifferentiated proliferating Ki67- and BrdU-positive population cells in the SVZ and SGZ in adults [6, 7]. Neurosphere analysis of the properties of Sox2-positive and -negative transgenic models shows that Sox2-expressing cells have the capability of self-renewal and conversion to secondary neurospheres [29]. Although the differences in transcription factor levels between pluripotency and multipotency have not been clarified so far, adult Sox2-positive NSCs mostly stay in quiescence and, when activated, choose different fates: astrogliogenic asymmetric cell division, symmetric self-renewal to expand stem cells pool, or neurogenic fate [30].
In the PNS, Sox2 is also expressed in sensory progenitor cells [31]. Embryonic ENS neurogenesis starts with the migration of vagal and sacral neural crest cells toward the gut [32]. Uncommitted ENS progenitor cells that express Sox2 and Sox10 [33] and respond to Notch [34] and endothelin 3 [35] give rise to committed Phox2b-positive cells and migrate by downregulating Sox2 and expressing endothelin receptor type B (EDNRB) and RET receptor tyrosine kinase in an environment rich in glial cell line-derived neurotrophic factor (GDNF) and endothelin 3. Embryonic vagal and sacral neural crest cells migrate and colonize the entire length of the gut, and multipotent ENS progenitors remain, postnatally to adulthood, able to differentiate into neuronal and glial lineages [36].
In addition to ENS, NPCs have been also isolated from the embryonic [37] and adult DRG [17]. First, embryonic DRG stem cells are positive for Sox2 and Sox10 (which downregulate Sox2 expression during their migration) [31] and express neurogenins (NGNs) [38] and brain-specific homeobox/POU domain protein 3a (Brn3a) during their three-wave migration [39]. They then express TrkA, TrkB, TrkC, Runx1, Runx3, and Ret to differentiate into various sensory neurons [40]. Second, adult DRG progenitor cells that express Sox2, Pax6, Notch1, MASH1 together with glial GFAP/Olig1 migrate, produce neurospheres, and finally differentiate into neurons by increasing the expression of NeuroD, neurogenin1, and other neuronal markers [38].
Pluripotency maintenance is controlled by a network of genes and transcription factors, mostly Sox2, Oct4, Nanog [5], and lineage specifiers of ESCs [41]. Sox2 and its partner, Oct4, bind with transcriptional cis-regulatory Octamer/Sox element in the promoter of Nanog and control its expression. Nanog also regulates both Oct4 and Sox2 expression. These transcription factors bind with some gene promoters and control self-renewal and pluripotency of ESCs via internal negative/positive feedback circuits and autoregulatory loops [42, 43]. In addition to these transcription factors, other proteins such as Dax1 and Klf4 also play a role as main pluripotency regulatory factors with a potential of autoregulatory loops. Some ChIP experiments, together with other methods, show the occupancy of promoter/enhancer regions of these transcription factors by protein complexes or cis-regulatory elements [44, 45]. Sox2, Nanog, Oct4, Dax1, and Klf4 repress or activate their targets in association with cofactor complexes or histone modifiers. Interestingly, these transcription factors can share common targets. Although Sox2 regulates the expression of many target genes during the maintenance of stem cells, a biphasic effect has been evidenced for Sox2-dependent self-renewal/maintenance and differentiation. At endogenous levels, Sox2 associates with Oct3/4 and activates the genes responsible for cell maintenance. However, elevated Sox2 levels induce a decrease of these genes because of an activation of protein kinase B (Akt) signaling, an inactivation of forkhead box O1 (FoxO1) and consequently a decrease of endogenous Sox2 [46, 47]. Moreover, a concomitant augmentation of Sox2 and Oct4 disrupts the self-renewal of ESCs and induces their differentiation. In contrast, elevating Sox2 along with Oct4, Klf4, and c-Myc does not disrupt self-renewal and does not promote differentiation [48]. Finally, Sox2 has been shown to maintain retinal NPCs in an undifferentiated state in a dose-dependent manner [49].
Altogether, because Sox2 can trigger opposite effects according to its level of expression, it is of utmost importance to know how this transcription factor interacts with other partners. Indeed, it has been established that Nanog-Oct4-Sox2 clusters recruit other proteins such as P300, a histone acetyltransferase [44], and some of the interactive proteins that enhance genomic stability such as stem cell co-activator (SCC)/XPC-RAD23B-CETN2 (XPC) complex (SCC/XPC) [50]. But, the most important aspect of Sox2 regulation recently emerged with the demonstration that Sox2 was tightly linked with some so-called pluripotency extracellular ligands. The most common extracellular factors that influence stem cells pluripotency include LIF [51], FGF [52], WNT/β-catenin [53], and BMP [54]. As an example, in LIF downstream signaling, Sox2 activation has been shown to occur through Jak/Stat3 and Klf4 activation, whereas Nanog is upregulated via PI3k/Akt which affects the core transcriptional network of pluripotency in mouse ESCs [55]. Moreover, Klf4 in association with Oct3/4 and Sox2 upregulates Lefty1 expression to maintain self-renewal [56].
The Connection Between Sox2 and Extracellular Signals
Several Sox2 regulatory mechanisms involving receptor-mediated signaling pathways have been evidenced (Fig. 2). This is the case for Akt, a downstream protein of insulin signaling pathway that is involved in neurogenesis. Mammalian insulin, insulin-like growth factor 1 (IGF1), and insulin-like growth factor 2 (IGF2) promote the MAPK/ERK pathway and activate Akt via PI(3,4) P2 and PI(3,4,5)P3. Activation of Akt modulates protein synthesis and autophagy in mammalian target of rapamycin (mTOR)-dependent and -independent manner [57]. The activity of this pathway must be maintained at optimal level for cellular homeostasis [58]. Akt-overexpressing adult hippocampal NPCs show slight increase in Sox2 expression during proliferation and loss of Sox2 expression in differentiation. It has been proposed that Akt modulates Sox2 mRNA levels rather than Sox2 protein stability [59]. Akt phosphorylates Sox2 at Thr118 and enhances self-renewal and pluripotency in mouse ESCs without phosphorylation of Oct4 [60]. As mentioned, Akt activation due to Sox2 overexpression has been shown to decrease endogenous Sox2 levels because of the phosphorylation of FoxO1 and its sequestration in cytoplasm. Nuclear FoxO1 binds to Sox2 promoter and leads to increased endogenous Sox2 expression, while phosphorylation of FoxO1 by Akt localizes it into the cytoplasm and prevents Sox2 expression [47]. FoxO1 knockdown results in the downregulation of Sox2, Oct4 and Nanog expression, and spontaneous differentiation in human ESCs, and this effect is enhanced by direct binding of FoxO1 to regulatory regions of these pluripotency markers [61]. In nervous system, FoxO-null brains reveal a decrease in Sox2-positive NSCs in SVZ [62]. Because autophagy is a downstream step of Akt signaling pathway, it has been shown that Sox2 overexpression induces autophagy by targeting autophagy-related protein 10 (ATG10) and Lc3 (ATG8b) expression, downregulation of total protein levels of Akt, p70S6K, and mTOR, and increasing phosphorylation of phosphatase and tensin homolog (PTEN). Sox2-induced autophagy promotes cellular senescence in cancer cells, and insulin treatment reverses this effect [63].
Wnt is a secreted lipid-modified protein that binds to frizzled (FZ) receptor and low-density lipoprotein receptor-related proteins and activates dishevelled (DVL) which promotes nuclear accumulation of β-catenin in canonical pathway. In the absence of Wnt, phosphorylated β-catenin makes a complex with glycogen synthase kinase 3β (GSK3β), adenomatosis polyposis coli (APC), and AXIN and is ubiquitinated for proteosome-mediated degradation. Wnt stimulation inhibits β-catenin phosphorylation by GSK3β which results in its translocation into the nucleus and interaction with T-cell factor/lymphoid enhancer factor (TCF/LEF) element in DNA [53]. In mouse ESCs, Wnt/β-catenin pathway upregulates Stat3 via interaction of β-catenin with TCF/LEF and promotes LIF downstream signaling to prevent differentiation and maintain pluripotency [64] and increases the efficacy of LIF signaling pathway. However, in adult NSCs niche, Wnt signaling increases neurogenesis through activation of β-catenin in SVZ [65] and SGZ [66]. Sox2-positive neurospheres are the major stem/progenitor cells which are stimulated by Wnt3a to increase proliferation and neurogenesis in postnatal mice olfactory epithelium [67]. The interaction between Wnt and Sox2 has been reported in mouse neural crest-derived osteoblast lineages [68], human and mouse osteosarcoma cell lines [69], xenopus retinal progenitor cells [70], as well as in postnatal and adult mouse NSCs [67, 71]. In the osteoblast lineage, Sox2 inhibits Wnt signaling pathway through the upregulation of APC and GSK3β, downregulation of Fzd receptor, and interaction with β-catenin to inhibit differentiation [68]. In xenopus retina, Sox2 inhibits Wnt/β-catenin and blocks neural differentiation, whereas its downregulation by proneural proteins results in neural differentiation [70]. The antagonizing effect of Sox2 and Wnt signaling pathway has been proposed to occur via the competition of β-catenin and Sox2 for the binding to Sox2 and TCF/LEF-binding sites (Sox/LEF) of target genes. Thus, while β-catenin displays a positive effect, Sox2 associated with histone deacetylase HDAC1 represses Sox/LEF element in the promoters of differentiation-specific genes in adult neural stem cells [71]. However, in neural crest-derived osteoblasts, Sox2 directly inhibits Wnt signaling via its c-terminal domain [72].
Notch signaling has been proposed to be a mediator of the Wnt-Sox2 cross talk in neuroepithelial cells [70]. Notch is a transmembrane receptor which is classified into four types (Notch1–4) with many ligands including Jagged (Jag1 and Jag2) and delta-like proteins in mammals. Binding of these ligands to the extracellular domain of Notch triggers an intra membrane cleavage by γ-secretase and the release of the Notch intracellular domain (NICD). NICD then translocates into the nucleus and controls the transcription of a certain number of genes. Although Notch signaling plays an important role in lateral inhibition during the development of the nervous system [73], it is also expressed in adult NSCs with an effect on their maintenance [74]. Indeed, Notch1 expression has been observed in neural precursor cells and astrocytes in SVZ and neuroblasts within the RMS, and Jagged1 and Delta1 are expressed in SVZ of adult mouse brain [75]. Importantly, Notch determines neural progenitor fate in SVZ of adult rat brains [76] and controls proliferation and differentiation through maintaining neural progenitor characteristics in a dose-dependent manner. Low levels of the active form of Notch1 promote proliferation, whereas high levels lead to growth arrest [77]. Sox2 and Jag1 are coexpressed in neurosensory development, and Jag1 maintains Sox2 expression within restricted domains of the optic epithelium through a Notch-mediated lateral induction [78]. The demonstration that activated Wnt signaling induces proneural genes in retina and leads, through Sox2 and Notch, to progenitor maintenance [70] that illustrates the fact that NPCs undergo self-renewal and differentiation through a functional interaction between Wnt, Sox2, and Notch.
The Connection Between Sox2, Epigenetic Modifications, and miRNAs
The self-renewal, maintenance, and differentiation of NSCs are regulated by epigenetic mechanisms that modulate DNA without altering genomic sequences. DNA methylation and histone acetylation play an important role in activating and silencing Sox2 expression in undifferentiated NSCs and differentiated neurons. DNA methylation mostly occurs in the cytosine residues of CpG islands by DNA methyltransferases (DNMT1, DNMT2a, and DNMT2b in mammals), while histone acetyltransferases (HATs) and histone deacetylases (HDACs) catalyze histone acetylation and deacetylation. Sox2 regulatory region 1 (SRR1) and SRR2 enhancers, located 4 kb upstream and downstream from transcription start site (TSS) of Sox2 gene respectively, are involved in the epigenetic regulation of Sox2 expression. SRR1 includes POU transcription factor motif, and SRR2 contains Octamer/Sox2-binding sequence in ESCs [79], and these enhancers are also present in NSCs or NPCs [80]. In one hand, in differentiated neurons, CpGs are highly methylated at SRR1 and SRR2, whereas in NPCs, these enhancers have undergone H3 histone acetylation and demethylation [81], and it has been shown that P27kip1 decreases Sox2 expression via its binding to Sox2-SRR2 enhancer [82]. In the other hand, Sox2 binds to distal enhancers of many genes in ESCs and NPCs, and mapping of the binding sites within 1 kb from TSS has shown that the most occupied sites in ESCs and NPCs are different [83].
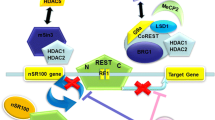
The association of Sox2 with the histone deacetylase HDAC1 has been evidenced in ESCs and NSCs. HDAC1 is one of the class I HDAC family which is found in repressive complexes such as Sin3, NuRD, CoREST, and PRC2 [84]. In addition to HDAC1, Sox2 also binds to HDAC2 and has been shown to be part of protein complexes by using multiple domains to interact with its partners [85]. Embryonic and trophoblast stem cells express high level of Sox2 and HDAC1. In ESCs, HDAC1 occupies the promoters of most of the pluripotency-related genes including Sox2 and supports self-renewal and pluripotency [86]. Sox2/HDAC represses Sox/LEF element in the promoters of differentiation-specific genes such as Neuro1 in adult NSCs [71]. Sox2 has been found to be associated with the mSin3A-HDAC complex [87], which consists of HDAC1/2, Sin3A/3B, RbAp46/48, and SAP18/30, and interacts with Nanog in mammals. Nanog maintains self-renewal of NSCs by activating specific target genes under the control of the binding of Sox2 and Oct4 to its Octamer/Sox element [88], while during differentiation, Nanog is downregulated [89]. The mSin3A-HDAC complex binds to the Nanog promoter of ESCs and interacts with Sox2 to stimulate Nanog expression during proliferation, whereas this interaction is destroyed during differentiation [87].
The maintenance of stem cells is mostly controlled by polycomb group (PcG) protein-mediated histone modifications and DNA methylation, whereas trithorax group (TrxG)-mediated histone modification plays a role in differentiation [90] via acetylation of H3K27 (H3K27ac), H3K4me3, dimethylation of Lys36 on histone H3 (H3K36me2), and/or nucleosome-remodelling activities [91]. The repressive function of Sox2 on target genes is addressed by polycomb repressive complex 2 (PRC2), one of the PcG proteins. In the epigenetic control of repression, enhancer of zeste homologue 2 (EZH2), a component of PRC2, methylates lysine 27 of histone H3 (H3K27me), and PRC2 then recruits polycomb repressive complex 1 (PRC1).
The PRC1 complex contains Bmi-1 and also Ring1A proteins which are ubiquitin ligases for H2AK119, and these chromatin modifications results in the repressive function of Sox2, whereas these modifications are lost via some activators during differentiation [92, 93]. Bmi-1, as a member of PRC1, binds to repressive tri-methyl lysine 27 of histone H3 (H3K27me3) during the epigenetic control of stem cell self-renewal [94], and overexpression/knockdown of Bmi-1 has demonstrated its role in self-renewal of NSCs [95, 96]. Bmi-1 suppresses P16INK4a and P19Arf expression, promotes stem cell self-renewal in the central and peripheral nervous system [97], and affects aging process via Ink4a/Arf locus [98]. Importantly, Sox2 increases the expression of Bmi-1 in osteoblast progenitor cells [68].
In addition to the epigenetic control of Sox2 in the nucleus, it can also be modulated by miRNAs at a post-transcriptional level. MicroRNAs, which are transcribed as primary transcripts (pri-miRNAs) by RNA polymerase II, are processed by a microprocessor complex containing Drosha and DGCR8 in the nucleus and matured by Dicer and transactivation response (TAR) RNA-binding protein (TRBP). Mature miRNAs are then incorporated into the RNA-induced silencing complex (RISC) including argonaute proteins and the single-stranded miRNA and recognize target mRNAs through the seed match sequences. It inhibits the expression of the target mRNA through deadenylation followed by mRNA degradation and blockade of translation at the initiation step or at the elongation step [99]. Several connections between Sox2 and some miRNAs have been illustrated recently. First, endogenous miR-145 represses the Oct4, Sox2, and Klf4 3′-UTR reporters in human ESCs under self-renewal conditions [100]. Second, Oct4, Sox2, and Nanog bind to the promoter region of the miR-302 cluster of miRNAs in human ESCs and regulate transcription of miR-302 which leads to the translational repression of targets such as cyclin D1 and provides a link between the transcription factors of pluripotency and cell cycle regulators in pluripotent cells [101]. Third, Nanog, Oct3/4, Rex1, and Sox2 have been identified as regulators of the miR302-367 cluster in ESCs. The comparison of transcriptional activity between the 525-bp (PROM-525) and 974-bp (PROM-974) fragments in the promoter of this cluster shows that Oct3/4 and Nanog inhibition has a negative effect on PROM-974 activity and that they act as transcriptional activators of the miR302-367 gene. In contrast, repression of Sox2 is associated with a significant increase in PROM-974 activity, thereby suggesting a potential negative regulation on the miR302-367 promoter [102]. Fourth, Sox2 expression is inhibited by miR-126 via targeting the Sox2 3′-UTR through two binding sites in gastric cancer cells [103]. Fifth, miR-296, miR-470, and miR-134 are significantly upregulated during self-renewal because of their effects on Nanog, Oct4, and Sox2, while miR-134 can silence Sox2 in ESCs [104]. Finally, miR-137 is a direct target of Sox2 and methyl CpG binding protein 2 (MeCP2) that inhibit neuronal differentiation and maturation in adult SGZ NSCs [105].
Sox2 in the Aging Nervous System
Neurogenesis in the Adult Brain and During Aging
Although the occurrence of adult neurogenesis along the SVZ-olfactory bulb axis of the brain has been evidenced more than 50 years ago [106, 107], it long remained controversial, and the cells responsible for continued neuronal and glial production were not identified. The two main properties of NSCs are their multipotency and their dual capacity of self-renewal and differentiation. During the past years, many intrinsic factors (growth factors, morphogens, neurotransmitters, and others) have been shown to regulate the decision of NSCs to proliferate or differentiate (see [108] for review). Thus, NSCs appear as a long-life source of neurons and glia (for recent review see [109]), a concept that makes obsolete the dogma that the CNS lacks regenerative power.
Very interesting is the fact that extrinsic factors such as aging, stress, and inflammation can also modulate the fate of NSCs. Although most of the studies demonstrate a decrease in proliferative activity of neural precursors with aging [110–112], some show a constant proliferation capacity throughout life [113]. Several processes have been considered to be involved in the aging of NSCs such as reduced commitment and fate changes, increased cell death, an imbalance between symmetric and asymmetric division or gliogenesis versus neurogenesis, increased quiescence, failure of the stem cells self-renewal, senescence, telomeres shortening, and telomerase deficits [114–117]. However, validation of such molecular mechanisms is still under discussion.
Aging, Oxidative Stress, and Sox2
Reactive oxygen species (ROS) producing oxidative stress are considered as the main cause of aging in non-mitotic cells, whereas the reactivity of stem cells is mostly different. For instance, it has been documented that hematopoietic stem cells (HSCs) self-renewal increases under low level ROS condition [118]. However, NSCs, when compared with HSCs, are mostly in a quiescent state, and their self-renewal, proliferation, and multipotency increase in response to elevated endogenous ROS levels. High level of ROS has been observed in DCX- positive cells rather than in Sox2- or GFAP-positive cells in SVZ [119]. In SGZ, ROS is transiently produced and reaches its highest level in intermediate precursor cells that exhibit the highest rate of proliferation, while inhibition of neurogenesis leads to a decline of oxidative stress markers [120]. The high level of endogenous ROS which is associated with higher neurosphere formation, proliferation, and multipotency of NSCs has been evidenced to be dependent on NADPH oxidase (NOX) enzymes and PI3/Akt pathway [119]. However, the effects of free radicals on the markers of NSCs may demonstrate the role of multipotency transcription factors in aging.
As far as Sox2 is concerned, Sox2-positive cells mostly observed in low ROS conditions in SVZ [119] and low levels of O2, when compared with higher levels, enhance the expression of Sox2 and Oct4 via hypoxia-inducible factor 2α (HIF-2α) in human glioblastoma cells [121]. Thus, Sox2-positive cells, which mostly include NSCs, have been suggested to maintain low levels of ROS in quiescent state to keep their stemness property. In these cells, oxidative response is mediated by FoxO3 [122], and the balance between Akt/FoxO and JNk/FoxO has been supposed to address cells to quiescence or senescence [123]. Because a direct interaction between FoxO1 and Sox2 has been documented [47], it is proposed that the role of Sox2 in NSCs maintenance under low level ROS is correlated to FoxO proteins.
Aging, Senescence Processes, and Sox2
Senescence, a hayflick limitation and exhaustion of cell division, has been considered as an aging mechanism in mitotic cells. The number of senescent cells increases during aging, and senescence is considered as a cell cycle arrest. Indeed, cell exhaustion and senescence mechanisms include inhibition of cyclin-dependent protein kinases (CDKs)/cyclins and modulation of the mechanisms in charge of chromosome cycle (DNA replication, nuclear envelope breakdown, chromosome condensation, and spindle assembly) [124]. Moreover, telomere shortening through p53 pathway, stress-induced senescence through p16-pRB pathway, oncogenes or loss of tumor suppressor genes, oxidative stress, loss of enriched environment and cellular contacts are considered as senescence mechanisms [125, 126]. It has been evidenced that telomeres shortening and telomerase deficits mediated by p53 regulation and Notch signaling pathways impair neurogenesis and neuritogenesis in NSCs [115]. Moreover, the plasticity of the histone modification marks, H3K9m3 and H4K30m, shows more similarity at telomeric regions of iPSCs and ESCs compared with differentiated mouse embryonic fibroblasts (MEFs). Interestingly, telomerase activity increases in human iPSCs induced by Oct3/4, Sox2, Klf4, and c-Myc in human dermal fibroblasts [9]. However, the role of Sox2 in telomere elongation remains poorly defined.
In senescence-induced p19ARF/p53/p21 pathway, the tumor suppressor protein p53 is activated downstream to DNA damage through some checkpoint kinase pathways such as DNA-dependent protein kinase (DNA-PK), ataxia telangectasia mutated (ATM), ATM rad-3 related (ATR), checkpoint kinase 1 (CHK1), checkpoint kinase 2 (CHK2), and MAPK-activated protein kinase 2 (MK2) to induce senescence or transient cell cycle arrest as a tumor suppression [127]. P53 knockdown increases self-renewal, proliferation, and apoptosis mostly via p21 in mice NSCs [128]. Moreover, p53 downregulates E2f target genes and activates cellular senescence via p19ARF, p16INK4a, or p21 [125]. It has been documented that p21 inhibits Sox2 expression since p21-deficient mice demonstrate high population of GFAP/Sox2-positive B-type NSCs with higher amount of Sox2, p53, and p19ARF proteins compared to wild-type NSCs. In these cells, a decrease of γH2AX-positive cells under Sox2 knockdown condition has been determined, and DNA damage occurs concomitantly with Sox2 overexpression in p21-null mice NSCs. Finally, elevated Sox2 levels trigger growth arrest and impairment of self-renewal, DNA damage, and senescence in a p53-dependent manner [129].
In p27Kip1-mediated senescence pathway, overexpression of PTEN leads to the upregulation of the CDK inhibitor p27Kip1 and to the negative regulation of PI3K/Akt signaling pathway [130]. Interestingly, p27Kip1 has been considered as a Sox2 repressor, and p27Kip1 null MEFs can be reprogrammed to iPSCs cells. In p27Kip1-null iPSCs cells, H3K9me3 and H3K27me3 that are both repressive modifications, increase at Sox2-SRR2 enhancer similarly to retinoic acid-induced differentiation condition of MEFs. Additional experiments showed that p27Kip1 directly binds to the Sox2-SRR2 enhancer sequence and decreases Sox2 expression [82].
Ink4a/Arf locus encodes p16INK4a and p19Arf, which are mediators of cellular senescence, and their expression increases during aging [131]. In p16INK4a deficient mice, the number of newborn neurons increases, and p16INK4a upregulation causes a decline of NPCs in SVZ during aging [132]. Bmi-1, which suppresses p16INK4a and p19Arf expression, is downregulated during aging, which results in an increase of p53 positive effect on the promoter regions of antioxidant response genes, promotes oxidative stress in brain cortical neurons, and triggers aging processes [133]. Interestingly, it has been identified as a critical regulator of Sox2-dependent self-renewal in osteoblasts [68].
Aging, iPSCs Reprogramming, and Sox2
During reprograming, Oct4, Klf4, and Sox2 repress Ink4a/Arf locus whereas aging upregulates it. Thus, Ink4/Arf locus expression is considered to be responsible for the decreased reprogramming associated with aging [134]. Induction of reprogramming factors (Sox2, Oct4, Klf4, and c-Myc) triggers senescence via upregulation of p16INK4a, p21cip1, and p53 which results in DNA damage. Because inducing telomerase activity results in declined senescence, the cost of this rejuvenation is an increased tumorigenesis. For this reason, tumor formation is a barrier for reprogramming to pluripotent stem cells. In this context, Sox2 has been evidenced to modulate the expression of tumor progression genes and plays a role as an oncogene in esophageal and lung squamous cell carcinoma [135, 136]. Among reprogramming factors, increased Sox2 has been shown to correlate with an accumulation of p53 [129], and Sox2 has been reported as a single factor for the reprogramming of mouse and human fibroblasts into NSCS without tumor formation [137].
Aging, Intracellular Signaling, and Sox2
Reduction of PI3/Akt pathway enhances translation of anti-stress proteins and autophagy-dependent clearance of misfolded proteins (via a reduction of FoxO1 and an augmentation of mTOR downstream signaling) and extends lifespan [63] (Fig. 3). Moreover, lysosomal autophagy protects cells against oxidative stress and enhances the degradation of dysfunctional mitochondria [138]. It has been evidenced that autophagy is promoted by Sox2 via the upregulation of ATG10 and LC3, which triggers senescence by increasing p16INK4a, p21, and p53 in cancer cells [139]. Although Sox2 overexpression has been reported to upregulate Akt in ESCs [47], Sox2-induced autophagy has been shown to promote senescence in cancer cells through downregulation of Akt but not class III PI3K signaling [139].

The functional cross-talk between Sox2 and some signaling pathways that are involved in the aging processes. Wnt/β-catenin signaling pathway efficacy which promotes differentiation genes through the binding of β-catenin with TCF/LEF decreases in hippocampal astrocytes during aging. Sox2 inhibits Wnt signaling pathway by upregulation of APC and GSK3β, downregulation of FZ receptor, interaction with β-catenin to inhibit differentiation in the osteoblast lineage and repression of Sox/LEF element associated with HDAC1 in the promoters of differentiation-specific genes in adult neural stem cells. In mammalian insulin/IGF signaling pathway, Akt is upregulated during aging, which increases mTORC1 and authophagy and protein synthesis. It phosphorylates Sox2 at Thr118 to modulate Sox2 protein stability and inhibits FoxO1 which increases endogenous Sox2 expression. Sox2 induces autophagy via upregulation of ATG10 and LC3 and results in senescence in cancer cells. Sox2 suppresses LINE-1 expression which has upregulation during aging. In senescence pathways, Sox2 increases the expression of Bmi-1 which suppresses P16INK4a and P19Arf expression. On the other hand, p21 and p27kip1 which mediate senescence pathways inhibit Sox2 expression. Elevated Sox2 levels trigger impairment of self-renewal and senescence in a p53-dependent manner. Notch signaling efficacy also declines in aging via decrease in Notch, Jagged1, and NICD in neuroblasts and astrocytes in SVZ. TCF/LEF T-cell factor/lymphoid enhancer factor, APC adenomatosis polysis coli, GSK3β glycogen synthase kinase 3β, FZ frizzled, β-cat β-catenin, HDAC1 histone deacetylase 1, IGF insulin-like growth factor, IIS insulin/IGF-like signaling, IRS insulin receptor substrates, PDK1 phosphatidylinositol-dependent protein kinase 1, mTORC1 mammalian target of rapamycin complex 1, FoxO1 Forkhead box O1, CC cancer cells, OC osteoblast cells, NSC neural stem cells, ESC embryonic stem cells, NICD Notch intracellular domain, p phosphorylated, ROS reactive oxygen species, LINE1 long interspersed nucleotide element-1
Wnt/β-catenin signaling pathway promotes the expression of differentiation genes, and Wnt expression is decreased in hippocampal astrocytes during aging, thereby driving NPCs to quiescence [140]. Interestingly, Sox2 reduces Wnt signaling through the upregulation of APC and GSK3β and the downregulation of Fzd in osteoblast lineage [68] and the binding to Sox/LEF element via Sox2/HDAC1 in adult NSCs [71]. This Sox2-dependent pathway may thus be considered as a cause of the reduction of Wnt signaling during aging. In addition, in aging HSCs, Wnt signaling shifts from canonical to non-canonical pathway through the upregulation of Wnt5a and the downregulation of Wnt3a [141]. In the nervous system, Wnt3a expression, as well as the number of Wnt3a-secreting astrocytes, decreases in adult hippocampal NSCs, thereby affecting the expression of some pro-neural genes such as NeuroD1 and long interspersed nucleotide element 1 (LINE-1) and leading to a decline of neurogenesis during aging [142].
Notch1, which is expressed in neuroblasts and astrocytes in SVZ, as well as Jagged1 and NICD that regulate progenitor fate, all decreases in SVZ of aged rat brain when compared with young adult [76]. For this reason, it has been hypothesized that Notch1 signaling could play an important role in the aging-dependent declined of neurogenesis in SVZ [76] although a direct relationship between Sox2 and Notch signaling pathway remains to be firmly established.
Aging, LINE-1 Transposable Element, and Sox2
LINE-1 is a transposable element, the gene of which includes two open reading frames (ORF1 and ORF2) encoding proteins that bind to nucleic acids as well as other elements presenting reverse transcriptase and endonuclease properties. After transcription, LINE-1 mRNA is translocated into the cytoplasm for translation of ORF1 and ORF2 proteins which reintegrate LINE-1 into the genome via target primed reverse transcription (TPRT) process [143]. LINE-1 activity in the brain is greater than in other parts of the body and is mostly expressed in the spinal cord and the dentate gyrus [144]. It creates DNA double-strand breaks [145], point mutations, rearrangements, damaged chromatin, and retrotransposition, which lead to genome instability [146] and an imbalance between damage and repair during aging [147]. It has been proposed that in contrast with the impact of beneficial genetic variation on evolution, LINE-1 activation has a cost on longevity and causes aging [143]. Interestingly, methyl-CpG-binding protein (MECP2) and Sox2 are able to repress LINE-1 transcription through Sox/LEF-binding sites on LINE-1 promoter sequence [71]. Indeed, two Sox-specific SRY-binding sites are present in the LINE-1 5′-UTR close to the CpG islands and during differentiation, Sox2 and MECP2 expressions are lower in neural progenitor cell [144]. Indeed, it has been shown that the neuronal specificity of somatic LINE-1 retrotransposition in NPCs is partially due to the transition of a Sox2/HDAC1 repressor complex to a Wnt-mediated TCF/LEF transcriptional activation [71].
A Role for Sox2 in Alzheimer’s Disease?
Alzheimer’s disease (AD) is the most common form of neurodegenerative syndrome worldwide and is characterized by a progressive loss of memory and cognitive functions ultimately leading to dementia, vascular hemorrhage, and death.
At the brain level, post-mortem AD patients manifest a massive neuronal loss with particular damages in the regions that are responsible for memory and language. The affected brain areas display two main pathological hallmarks: (i) extracellular amyloid plaques that are mainly composed of the amyloid-β peptide and (ii) intraneuronal neurofibrillary tangles (NFT) that are due to aggregation of the hyperphosphorylated tau protein. Although they can rarely be (less than 1 %) of genetic origin (familial forms due to mutations on some genes), most of AD cases are sporadic. Nevertheless, all AD forms are characterized by the abnormal aggregation of a set of peptides called amyloid-β peptides (Aβ) that is intimately linked to the onset of the disease.
AD, the Amyloid Hypothesis, and βAPP Processing
According to the amyloid hypothesis, β-amyloid peptide (Aβ) that is produced from the β-amyloid precursor protein (βAPP) accumulates because of an imbalance between its production and clearance and initiates subsequent deleterious events (tau hyperphosphorylation, inflammation and neuronal death) that ultimately lead to memory deficits and dementia. Aβ peptides are produced through the so-called amyloidogenic pathway by the sequential cleavages by β- and γ-secretases [148] (Fig. 4). The β-secretase has been identified in 1999 by four independent research groups as a new aspartyl protease called Beta-site APP-cleaving enzyme 1 (BACE1) [149], while γ-secretase is a generic term defining an heterotetrameric complex composed of presenilin 1 or 2, anterior pharynx defective-1 (Aph-1), presenilin enhancer-2 (Pen-2), and nicastrin [150].

The connections between Sox2 and some signaling pathways that are modified in AD pathology. Since Sox2 is decreased in AD brain and because βAPP overexpression triggers an augmentation of Sox2 levels, one can reasonably envision that Sox2 may have an impact on the development of AD. Moreover, several Sox2-connected signaling pathways are affected in AD. Thus, the efficacy of Wnt, which protects hippocampal neurons from Aβ oligomers declines in AD. GSK3β, which phosphorylates both β-catenin and tau protein increases in AD and stimulates Aβ production. Sox2 combined with HDAC1 represses Sox/LEF element in the promoters of differentiation-specific genes in adult neural stem cells. Although Sox2 inhibits Wnt signaling pathway via an interaction with GSK3b in the osteoblast lineage, its effect in AD is unknown. NICD, the downstream effector of Notch signaling, is produced by γ-secretase, a shared enzyme between βAPP processing and Notch signaling, and inhibits AICD-Tip60-Fe65 complex through physical interaction. The β-secretase BACE1 also interacts with the Jag1-Notch pathway and NICD production. Activation of Akt by insulin/IGF1 increases in AD thereby leading to more GSK3β, tau phosphorylation, and mTOR levels in the cortex. However, the level of FoxO1 expression declines as a result of overexpression of Akt in AD. Although Sox2 phosphorylation by Akt and its upregulation by FoxO1 have been evidenced in cancer cells, the occurrence of such effects in AD remains to be determined. AD Alzheimer’s disease, APP amyloid precursor protein, sAPP soluble APP, AICD APP intracellular domain, Aβ amyloid-β peptide, Sec secretase, TCF/LEF T-cell factor/lymphoid enhancer factor, APC adenomatosispolysis coli, GSK3β glycogen synthase kinase 3β, FZ frizzled, β-cat β-catenin, HDAC1 histone deacetylase 1, IGF insulin-like growth factor, IIS insulin/IGF-like signaling, PDK1 phosphatidylinositol-dependent protein kinase 1, IRS insulin receptor substrates, mTORC1 mammalian target of rapamycin complex 1, CC cancer cells, OC osteoblast cells, FoxO1 Forkhead box O1, p phosphorylated, ROS reactive oxygen species, NICD Notch intracellular domain
On the other hand, there exists an alternative non-amyloidogenic α-secretase cleavage mainly performed by two enzymes (ADAM10 and ADAM17) that are members of the disintegrin family of metalloprotease and are respectively responsible for the constitutive and PKC-regulated pathways [151]. This proteolytic event occurs in the middle of the Aβ sequence, thereby precluding its production. Moreover, this cleavage leads to the secretion of the large neurotrophic and neuroprotective-secreted sAPPα fragment as well as its C-terminal counterpart C83.
AD and Stem Cells
It has been documented that proliferation and differentiation of NSCs first decrease in AD before a subsequent increase as a compensatory mechanism in SVZ and SGZ. Moreover, neurogenesis marker proteins such as DCX, PSA-NCAM, TUC-4, and NeuroD are upregulated in the subgranular zone of dentate gyrus in AD hippocampus [152]. Recently, stem cell therapy for patients with AD and iPSCs usage in AD animal models have been envisioned with a particular focus on the core transcriptional network (including Sox2) that is responsible for the maintenance of pluripotency and multipotency. Because many evidences suggest that adult neurogenesis contributes to learning and memory [153], stem cell therapy has been considered as a possible treatment for neurodegenerative disorders. In spite of the limitation in transplantation, mainly due to grafts rejection and tumor production, derived neurons or glial cells from ESCs or NSCs have been tested as an effective treatment in vivo in animal models of AD [154–157]. As a proof of concept, transplantation of ESCs-derived neurospheres into some cortical areas in nucleus basalis of Meynert lesion in AD mice model has been shown to trigger the production of ChAT-positive and serotonin-positive neurons around the graft [154]. Moreover, transplantation of NSCs with transgenic expression of human nerve growth factor (hNGF) into the brain enhances cognitive performance in rat model of AD [155], and behavioral recovery has been observed following the induction of mouse ESCs-derived NPCs into cholinergic cells and their transplantation into the mouse models of AD [156]. Finally, transplanted human NSCs line was found to differentiate in cerebral cortex, hippocampus, striatum, and septum in a rat model of AD [157].
However, because accessibility and isolation of NSCs out of their in vivo environment are problematic, an alternative to NSCs transplantation was awaited. In 2006, a huge step forward in the field of cell biology occurred with the first description of a simple method to dedifferentiate somatic cells (fibroblasts) to embryonic-like iPSCs in mouse [8]. One protocol consists of the retroviral delivery of a cocktail of just four genes (Sox2, Oct4, Klf4, and c-Myc), and this method was shown to also work for other species including humans [9]. Interestingly, it has been demonstrated recently that iPSCs induction can be performed by the sole Sox2 transcription factor [137]. These pluripotent iPSCs can then be differentiated into any kind of cells according to the treatment applied and thereby provide a simple way to obtain cultured cells issued from a whole organism. This approach allowed, via iPSCs, the obtention of genuine neurons issued from fibroblasts of AD patients. This so-called “AD modeling” process has been successfully achieved by several laboratories during the past 3 years [158].
βAPP, βAPP-Derived Metabolites, βAPP-Cleaving Secretases and Stem Cells
Several lines of evidence have established the occurrence of a functional link between NSCs fate and βAPP biology. First, neurogenesis and the number of BrdU-positive cells decrease in SVZ and SGZ in βAPP-overexpressing transgenic mice models of AD [159–161]. Second, βAPP overexpression in embryonic stem cells has been evidenced to promote differentiation with an altered morphology of human ESCs colonies, neuronal markers, and neurite outgrowths [162]. Third, βAPP and its parent protein APLP2 can respectively upregulate neuronal migration and NSCs differentiation during mammalian cortical development [163, 164]. Altogether, this most likely means that βAPP itself, Aβ, βAPP C-terminal fragments, and possibly others play a role in the AD-dependent decrease of NPCs differentiation.
As far as βAPP-derived fragments are concerned, ESCs express the three secretases responsible for βAPP processing and, as a consequence, produce detectable levels of Aβ, C99, and sAPPα [165]. Not surprisingly, it has been established that Aβ and sAPPα treatments modulate the proliferation/differentiation of ESCs into neural progenitor cells in various manners (induction of proliferation by soluble Aβ and fibrillar Aβ, induction of differentiation by sAPPα, and inhibition of proliferation by oligomeric Aβ) [165].
Considering Aβ, although a neurogenic effect of oligomeric Aβ42 has been described [166], a correlation between the impairment of the differentiation into neurons and Aβ production/deposition has been evidenced [159, 167]. Another study established that Aβ decreases neurogenesis via apoptosic pathways and downregulation of β-catenin in newborn neurons, thereby leading to Wnt/β-catening signaling impairment in glial progenitor cells [168].
Concerning the large secreted sAPP fragments, the fact that sAPP-binding sites, which regulate proliferation of adult progenitors in response to either epidermal growth factor (EGF) or sAPP and increase the number of BrdU- and epidermal growth factor receptor (EGF-R)-positive NSCs, is present in SVZ goes in favor of a role for sAPP in neurogenesis [169]. In addition to SVZ, both sAPPα and sAPPβ regulate proliferation and differentiation of NSCs in SGZ [170] with sAPPβ being more potent than sAPPα at inducing differentiation in human ESCs [162]. Although the underlying signaling pathway is still poorly understood, it has been established that sAPPα modulates depolarization-induced neurite outgrowth via MAPK activity in NPCs-derived neurons [171].
Finally, the βAPP intracellular domain AICD impairs adult neurogenesis in AD mouse model by inducing inflammation and reducing adult hippocampal neurogenesis in an age-dependent manner [172]. Moreover, proliferation of NSCs declines in the SGZ of hippocampus of AICD transgenic mice, and AICD downregulates cell survival without specific effect on differentiation of newly generated hippocampal cells [173].
Regarding the impact of the βAPP-cleaving proteases (more commonly called “secretase” and responsible for the production of the here above mentioned metabolites) on NSCs fate, pharmacological inhibition of the amyloidogenic β-secretase BACE1 has been shown to suppress proliferation and promote NPCs formation [165], whereas another study established that BACE1 knockout increases astrogenesis and decreases neurogenesis [174]. These data appear conflicting, and further studies are now required to delineate the exact role of this enzyme in neurogenesis under normal and pathological (AD) conditions. Information concerning the involvement of presenilin 1 (PS1) (the catalytic core of the amyloidogenic γ-secretase complex) in neurogenesis under physiological conditions are still lacking at present, and the only data published so far have reported that FAD-linked PS1 mutations impair adult neurogenesis in transgenic mouse models of AD [167, 175–178]. Finally, the role of the nonamyloidogenic α-secretases ADAM10 and ADAM17 in adult neurogenesis under normal and pathological conditions is still unknown. Concerning ADAM10, it has been suggested, but not proved, that it could play a role in neuronal maturation during cortex development via the processing of Notch [179]. As far as ADAM17 is concerned, a role for this protease in the upregulation of proliferation after stroke in the SVZ has been evidenced [180], and an increase of proliferation/decrease of differentiation via Notch signaling has been shown in glioblastoma stem cells [181].
AD and Sox2
It is widely accepted that neurogenesis contributes to learning and memory. Thus, considering the strong implication of Sox2 in regulating the fate of stem cells and given the fact that neurogenesis and cognitive functions including memory are impaired in AD, one can reasonably postulate that this transcription factor could play an important role in the development of this neurodegenerative disease. Two independent studies have supported this hypothesis. First, Sox2 deficiency not only impairs neurogenesis but also induces neuronal degeneration in the adult mouse brain [6]. Second, Sox2 levels are strongly decreased in the brain of transgenic mouse model of AD as well as in the brain of AD patients [182]. Of utmost importance is the observation that the Sox2 decrease in AD cases positively correlates with the severity of the disease [182]. These complementary data strongly support the fact that any decrease of Sox2 could favor AD pathology.
Intracellular Signaling Pathways as Common Denominators to Sox2 and AD
It is striking that most of the signaling pathways that interact with the Sox2 transcription factor are also implicated in AD (Fig. 4). First, the Sox2-regulating Insulin/IGF1/Akt signaling pathway increases in AD and leads to more phosphorylation of Akt targets such as GSK3β, Tau, and Mtor, while PTEN is downregulated in cortex of AD patients [183]. In addition, some studies showed that the inhibition of PI3K/Akt pathway increases the level of the γ-secretase complex and the ubiquitination of PS1 [184], whereas the stimulation of this pathway promotes phosphorylation and inactivation of IRS-1/2 as a mechanism of insulin resistance in AD. Moreover, an increase of inactivated phosphoser312 IRS-1 and phosphoser616 IRS-1 is observed in NFTs [185], and phosphoser636/639 IRS-1 levels have been found to be negatively correlated with episodic and working memory [186]. The levels of FoxO1 expression decline as a result of overexpression of Akt in AD [123], whereas PTEN, PP2A, and mTOR/S6K activities negatively regulate the PI3K/Akt pathway. Finally, the activation of PI3K/Akt signaling has been shown to be sustained in the brain of patients with AD, and turning off/on of this pathway modulates LTP/LTD [63]. Thus, the rather strong correlation between Akt signaling pathway with both Sox2 and proteins involved in the development of AD makes highly plausible the hypothesis that Sox2 may play an important role in AD development.
As previously mentioned, Sox2 interacts with the Wnt signaling pathway that is also supposed to be involved in the pathogenesis of AD. Indeed, loss of Wnt signaling triggers GSK3β activation, intracellular amyloid deposition, β-catenin degradation, and activation of some apoptosis pathways and ultimately leads to AD. As a reminder, GSK-3β both stimulates Aβ production and phosphorylates Tau protein [187], and it has been evidenced that PS1 mutation enhances proliferation of NPCs via an alteration of Wnt/β-catenin signaling [188]. It has also been shown that canonical Wnt/β-catenin protects hippocampal neurons from Aβ oligomers with the blockade of neuronal apoptosis and that Wnt3a increases cell survival toward Aβ-dependent neurotoxicity, inhibits GSK-3β activity and Tau phosphorylation, and prevents Aβ-induced apoptosis [189]. In the non-canonical Wnt signaling pathway, GSK3β inhibition stabilizes β-catenin, modulates mitochondrial dynamic, prevents Aβ-dependent Bcl2 augmentation, and reduces the neurotoxicity of Aβ oligomers [190]. In addition, inhibition of GSK-3β by lithium has been shown to protect rat hippocampal and cortical neurons from Aβ-induced damage through the reduction of total Aβ in brains of APPswe + PSEN1ΔE9 transgenic mice [191]. Finally, Wnt5a prevents synaptotoxicity changes induced by Aβ oligomers on PSD-95 clustering in synaptic contact [192]. Altogether, these data established that the efficacy of the Wnt/β-catenin signaling declines in AD and that Sox2, which negatively correlates with this pathway, is likely to have an impact on AD pathology.
As far as the Notch signaling pathway is concerned, it has been well established that Notch, as well as βAPP and other trans-membrane proteins, is cleaved inside the plasma membrane by γ-secretase to give rise to NICD [193]. Noteworthy, because AICD-Tip60-Fe65 complex is suppressed by NICD through physical interaction [194] and given the fact that all three partners can have an impact on NPCs fate, one could envision a putative NICD/AICD/Sox2 regulatory loop.
βAPP and Sox2
The occurrence of a functional cross-talk between Sox2 and βAPP emerged during the past years. First, Sox2 colocalizes with βAPP and Fe65 in the NSCs niche of the fetal ventricular zone, and TAG1-βAPP signaling negatively modulates neurogenesis in an AICD/Fe65-dependent manner [195]. Consistent with these findings, it has been established that Sox2 colocalizes with βAPP in both NSCs and NPCs of the adult SVZ [196]. Second, βAPP overexpression causes a rapid differentiation and alters the morphology of human ESCs, and it has been shown that the expression of Sox2 increases in βAPPWT- and βAPPSwe-overexpressing human ESCs cells when compared with controls [162]. However, a decrease of Sox2 expression has been reported in the hippocampus of βAPP-tg mice [182]. Some important issues remain to be solved, and it will be now crucial to determine, by means of genetic and pharmacological approaches, whether the observed effects are due to full-length βAPP or to some βAPP-derived metabolites. On the other hand, the described opposite effects of βAPP on Sox2 in stem cells and hippocampus mentioned above may reflect some regional or cell-specific Sox2-βAPP functional interactions in the brain. A detailed mapping of βAPP-Sox2 cross-talk in various cell lines and brain areas should answer this important question in the future.
Secretases and Sox2
Any functional cross-talk between Sox2 and secretases themselves has been documented so far, although Sox2 colocalizes with the α-secretase ADAM10 in NSCs of the SVZ [196]. However, the fact that some βAPP-derived fragments might be responsible for the observed βAPP-dependent, Sox2 regulation could illustrate an indirect involvement of these proteolytic activities. This issue deserves particular attention, and several works provide some weight to this hypothesis. First, as mentioned in a previous paragraph, transgenic mice overexpressing mutated presenilin 1 (the catalytic core of the γ-secretase complex) display a reduced number of both NSCs and NPCs in the hippocampus when compared to wild-type animals [167, 175–178]. Second, a very recent study has established that the β-secretase BACE1 can control the balance between neurogenesis and astrogenesis via the Jag1-Notch pathway and that NICD production and full-length Jag1 protein levels significantly increase in BACE1 null mice [174].
In summary, the present available data strongly support the possibility that full-length βAPP, some of the βAPP-derived metabolites and/or the proteases responsible for βAPP processing, could interact with Sox2, and we can reasonably hypothesize that this functional cross-talk could have an important role during normal adult neurogenesis and that an imbalance of this network could contribute to AD.
Conclusion
In spite of an intense multidisciplinary research, effective treatments to improve cognitive impairments in normal and pathological aging, especially Alzheimer’s disease, are urgently required. Although the boundary between normal aging and age-related neurodegenerative diseases remains to be determined, some fundamental mechanisms undergo the same pathways. Stem cell therapy, which has been considered as a treatment in neurodegenerative disorders, demonstrates some limitations like insufficient survival and stability after transplantation and graft rejection or tumor production of ESCs, iPSCs, MSCs, or NSCs following transplantation [197]. Moreover, senescence is a barrier for reprogramming. In order to identify new tracks aimed at making possible AD-targeting stem cell therapy, the modeling of iPSCs-derived neurons in AD represents an important step forward.
In this context, a functional interaction between Sox2, proteins involved in AD and some signaling pathways that control normal aging and are modified in AD, is worth envisioning. Undoubtedly, Sox2 and βAPP/βAPP metabolites both interact with Wnt/β-catenin, Notch, and PI3K/Akt signaling pathways. First, during normal aging, Wnt/β-catenin signaling that promotes neurogenesis declines in neural stem cells due to the weak production of Wnt or to the increased action of negative regulators of the pathway such as Sox2. Antagonizing effect of Wnt/β-catenin on Aβ has been evidenced in AD. Second, PI3K/Akt is mostly overexpressed to address more protein synthesis and inhibits autophagy, whereas Sox2 expression, which promotes autophagy, is suppressed via the inhibition of FoxO1 by Akt. In addition, Sox2 phosphorylation is promoted by Akt signaling. However, the activity of this signaling pathway increases during aging and AD. Third, Sox2 interacts with the Notch pathway that, like βAPP, undergoes γ-secretase cleavage and with some cell cycle regulators that promote senescence.
Overall, according to the preponderant action of Sox2 in the balance between self-renewal and differentiation during neurogenesis and its involvement in some pathways correlated with normal aging and AD, it is proposed that Sox2 may play an important role in neurodegeneration and stem cell aging and that its modulation could be used in ESCs, NSCs, or iPSCs replacements that recently have been considered as a new therapeutic strategy to fight neurodegenerative diseases.
References
Gubbay J, Collignon J, Koopman P, Capel B et al (1990) A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 346:245–250. doi:10.1038/346245a0
Sinclair AH, Berta P, Palmer MS, Hawkins JR et al (1990) A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346:240–244. doi:10.1038/346240a0
Dailey L, Yuan H, Basilico C (1994) Interaction between a novel F9-specific factor and octamer-binding proteins is required for cell-type-restricted activity of the fibroblast growth factor 4 enhancer. Mol Cell Biol 14:7758–7769. doi:10.1128/MCB.14.12.7758
Pan H, Schultz RM (2011) Sox2 modulates reprogramming of gene expression in two-cell mouse embryos. Biol Reprod 85:409–416. doi:10.1095/biolreprod.111.090886
He S, Nakada D, Morrison SJ (2009) Mechanisms of stem cell self-renewal. Annu Rev Cell Dev Biol 25:377–406. doi:10.1146/annurev.cellbio.042308.113248
Ferri ALM, Cavallaro M, Braida D, Di Cristofano A et al (2004) Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 131:3805–3819. doi:10.1242/dev.01204
Suh H, Consiglio A, Ray J, Sawai T et al (2007) In vivo fate analysis reveals the multipotent and self-renewal capacities of Sox2+ neural stem cells in the adult hippocampus. Cell Stem Cell 1:515–528. doi:10.1016/j.stem.2007.09.002
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblasts cultures by defined factors. Cell 126:663–676. doi:10.1016/j. cell .2006.07.024
Takahashi K, Tanabe K, Ohnuki M, Narita M et al (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872. doi:10.1016/j. cell .2007.11.019
Sarkar A, Hochedlinger K (2013) The Sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell 12:15–30. doi:10.1016/j.stem.2012.12.007
Keramari M, Razavi J, Ingman KA, Patsch C et al (2010) Sox2 is essential for formation of trophectoderm in the preimplantation embryo. PLoS One 5:e13952. doi:10.1371/journal.pone.0013952
Avilion AA, Nicolis SK, Pevny LH, Perez L et al (2003) Multipotent cell lineages in early mouse development depend on SOX2 function. Genes Dev 17:126–140. doi:10.1101/gad.224503
Guo G, Huss M, Tong GQ, Wang C et al (2010) Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell 18:675–685. doi:10.1016/j.devcel.2010.02.012
Masui S, Nakatake Y, Toyooka Y, Shimosato D et al (2007) Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol 9:625–635. doi:10.1038/ncb1589
Thomson M, Liu SJ, Zou LN, Smith Z et al (2011) Pluripotency factors in embryonic stem cells regulate differentiation into germ layers. Cell 145:875–889. doi:10.1016/j.cell.2011.05.017
Graham V, Khudyakov J, Ellis P, Pevny L (2003) SOX2 functions to maintain neural progenitor identity. Neuron 39:749–765. doi:10.1016/S0896-6273(03)00497-5
Li HY, Say EH, Zhou XF (2007) Isolation and characterization of neural crest progenitors from adult dorsal root ganglia. Stem Cells 25:2053–2065. doi:10.1634/stemcells. 2007-0080
Lagares A, Li HY, Zhou XF, Avendano C (2007) Primary sensory neuron addition in the adult rat trigeminal ganglion: evidence for neural crest glio-neuronal precursor maturation. J Neurosci 2730:7939–7953. doi:10.1523/JNEUROSCI. 1203-07.2007
Kruger GM, Mosher JT, Bixby S, Joseph N et al (2002) Neural crest stem cells persist in the adult gut but undergo changes in self-renewal, neuronal subtype potential, and factor responsiveness. Neuron 35:657–669. doi:10.1016/S0896-6273(02)00827-9
Rask-Andersen H, Bostrom M, Gerdin B, Kinnefors A et al (2005) Regeneration of human auditory nerve. In vitro/in video demonstration of neural progenitor cells in adult human and guinea pig spiral ganglion. Hear Res 203:180–191. doi:10.1016/j.heares.2004.12.005
Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21:7153–7160
Steiner B, Klempin F, Wang L, Kott M et al (2006) Type-2 cells as link between glial and neuronal lineage in adult hippocampal neurogenesis. Glia 54:805–814. doi:10.1002/glia.20407
Ehninger D, Kempermann G (2008) Neurogenesis in the adult hippocampus. Cell Tissue Res 331:243–250. doi:10.1007/s00441-007-0478-3
Liu X, Bolteus AJ, Balkin DM, Henschel O et al (2006) GFAP-expressing cells in the postnatal subventricular zone display a unique glial phenotype intermediate between radial glia and astrocytes. Glia 54:394–410. doi:10.1002/glia.20392
Brill MS, Snapyan M, Wohlfrom H, Ninkovic J et al (2008) A dlx2- and pax6-dependent transcriptional code for periglomerular neuron specification in the adult olfactory bulb. J Neurosci 28:6439–6452. doi:10.1523/JNEUROSCI. 0700-08
Hsieh J (2012) Orchestrating transcriptional control of adult neurogenesis. Genes Dev 26:1010–1021. doi:10.1101/gad.187336.112
Ming GL, Song H (2011) Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron 70:687–702. doi:10.1016/j.neuron.2011.05.001
Lois C, Garcia-Verdugo JM, Alvarez-Buylla A (1996) Chain migration of neuronal precursors. Science 271:978–981
Remboutsika E, Elkouris M, Iulianella A, Andoniadou CL et al (2011) Flexibility of neural stem cells. Front Physiol 2:16. doi:10.3389/fphys.2011.00016
Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP et al (2011) In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 145:1142–1155. doi:10.1016/j.cell.2011.05.024
Cimadamore F, Fishwick K, Giusto E, Gnedeva K et al (2011) Human ESC-derived neural crest model reveals a key role for SOX2 in sensory neurogenesis. Cell Stem Cell 8:538–551. doi:10.1016/j.stem.2011.03.011
Sasselli V, Pachnis V, Burns AJ (2012) The enteric nervous system. Dev Biol 366:64–73. doi:10.1016/j.ydbio.2012.01.012
Heanue TA, Pachnis V (2011) Prospective identification and isolation of enteric nervous system progenitors using Sox2. Stem Cells 29:128–140. doi:10.1002/stem.557
Gershon MD (2010) Developmental determinants of the independence and complexity of the enteric nervous system. Trends Neurosci 33:446–456. doi:10.1016/j.tins.2010.06.002
Bondurand N, Natarajan D, Barlow A, Thapar N et al (2006) Maintenance of mammalian enteric nervous system progenitors by SOX10 and endothelin 3 signalling. Development 133:2075–2086. doi:10.1242/dev.02375
Metzger M, Bareiss PM, Danker T, Wagner S et al (2009) Expansion and differentiation of neural progenitors derived from the human adult enteric nervous system. Gastroenterology 137:2063–2073. doi:10.1053/j.gastro.2009.06.038
Hagedorn L, Suter U, Sommer L (1999) P0 and PMP22 mark a multipotent neural crest-derived cell type that displays community effects in response to TGF-beta family factors. Development 126:3781–3794
Perez SE, Rebelo S, Anderson DJ (1999) Early specification of sensory neuron fate revealed by expression and function of neurogenins in the chick embryo. Development 126:1715–1728
Marmigere F, Ernfors P (2007) Specification and connectivity of neuronal subtypes in the sensory lineage. Nat Rev Neurosci 8:114–127. doi:10.1038/nrn2057
Kramer I, Sigrist M, de Nooij JC, Taniuchi I et al (2006) A role for Runx transcription factor signaling in dorsal root ganglion sensory neuron diversification. Neuron 49:379–393. doi:10.1016/j.neuron.2006.01.008
Loh KM, Lim B (2011) A precarious balance: pluripotency factors as lineage specifiers. Cell Stem Cell 8:363–369. doi:10.1016/j.stem.2011.03.013
Boyer LA, Lee TI, Cole MF, Johnstone SE et al (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122:947–956. doi:10.1016/j.cell.2005.08.020
Chickarmane V, Troein C, Nuber UA, Sauro HM et al (2006) Transcriptional dynamics of the embryonic stem cell switch. PLoS Comput Biol 2:e123. doi:10.1371/journal.pcbi.0020123
Chen X, Xu H, Yuan P, Fang F et al (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133:1106–1117. doi:10.1016/j.cell.2008.04.043
Kim J, Chu J, Shen X, Wang J et al (2008) An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132:1049–1061. doi:10.1016/j.cell.2008.02.039
Kopp JL, Ormsbee BD, Desler M, Rizzino A (2008) Small increases in the level of Sox2 trigger the differentiation of mouse embryonic stem cells. Stem Cells 26:903–911. doi:10.1634/stemcells. 2007-0951
Ormsbee Golden BD, Wuebben EL, Rizzino A (2013) Sox2 expression is regulated by a negative feedback loop in embryonic stem cells that involves AKT signaling and FoxO1. PLoS One 8:e76345. doi:10.1371/journal.pone.0076345
Gao Z, Cox JL, Gilmore JM, Ormsbee BD et al (2012) Determination of protein interactome of transcription factor Sox2 in embryonic stem cells engineered for inducible expression of four reprogramming factors. J Biol Chem 287:11384–11397. doi:10.1074/jbc.M111.320143
Taranova OV, Magness ST, Fagan BM, Wu Y et al (2006) SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev 20:1187–1202. doi:10.1101/gad.1407906
Fong YW, Inouye C, Yamaguchi T, Cattoglio C et al (2011) A DNA repair complex functions as an Oct4/Sox2 coactivator in embryonic stem cells. Cell 147:120–131. doi:10.1016/j.cell.2011.08.038
Liu N, Lu M, Tian X, Han Z (2007) Molecular mechanisms involved in self-renewal and pluripotency of embryonic stem cells. J Cell Physiol 211:279–286. doi:10.1002/jcp.20978
Mason I (2007) Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci 8:583–596. doi:10.1038/nrn2189
Wray J, Hartmann C (2012) WNTing embryonic stem cells. Trends Cell Biol 22:159–168. doi:10.1016/j.tcb.2011.11.004
Qi X, Li TG, Hao J, Hu J et al (2004) BMP4 supports self-renewal of embryonic stem cells by inhibiting mitogen-activated protein kinase pathways. Proc Natl Acad Sci U S A 101:6027–6032. doi:10.1073/pnas.0401367101
Niwa H, Ogawa K, Shimosato D, Adachi K (2009) A parallel circuit of LIF signalling pathways maintains pluripotency of mouse ES cells. Nature 460:118–122. doi:10.1038/nature08113
Nakatake Y, Fukui N, Iwamatsu Y, Masui S et al (2006) Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol Cell Biol 26:7772–7782. doi:10.1128/MCB. 00468-06
Ravikumar B, Sarkar S, Davies JE, Futter M et al (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90:1383–1435. doi:10.1152/physrev.00030.2009
Cohen E, Dillin A (2008) The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci 9:759–767. doi:10.1038/nrn2474
Peltier J, Conway A, Keung AJ, Schaffer DV (2011) Akt increases sox2 expression in adult hippocampal neural progenitor cells, but increased sox2 does not promote proliferation. Stem Cells Dev 20:1153–1161. doi:10.1089/scd.2010.0130
Jeong CH, Cho YY, Kim MO, Kim SH et al (2010) Phosphorylation of Sox2 cooperates in reprogramming to pluripotent stem cells. Stem Cells 28:2141–2150. doi:10.1002/stem.540
Zhang X, Yalcin S, Lee DF, Yeh TY et al (2011) FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat Cell Biol 13:1092–1099. doi:10.1038/ncb2293
Paik JH, Ding Z, Narurkar R, Ramkissoon S et al (2009) FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 5:540–553. doi:10.1016/j.stem.2009.09.013
O’Neill C (2013) PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer's disease. Exp Gerontol 48:647–653. doi:10.1016/j.exger.2013.02.025
Hao J, Li TG, Qi X, Zhao DF et al (2006) WNT/beta-catenin pathway up-regulates Stat3 and converges on LIF to prevent differentiation of mouse embryonic stem cells. Dev Biol 290:81–91. doi:10.1016/j.ydbio.2005.11.011
Adachi K, Mirzadeh Z, Sakaguchi M, Yamashita T et al (2007) Beta-catenin signaling promotes proliferation of progenitor cells in the adult mouse subventricular zone. Stem Cells 25:2827–2836. doi:10.1634/stemcells. 2007-0177
Lie DC, Colamarino SA, Song HJ, Desire L et al (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437:1370–1375. doi:10.1038/nature04108
Wang YZ, Yamagami T, Gan Q, Wang Y et al (2011) Canonical Wnt signaling promotes the proliferation and neurogenesis of peripheral olfactory stem cells during postnatal development and adult regeneration. J Cell Sci 124:1553–1563. doi:10.1242/jcs.080580
Seo E, Basu-Roy U, Zavadil J, Basilico C et al (2011) Distinct functions of Sox2 control self-renewal and differentiation in the osteoblast lineage. Mol Cell Biol 31:4593–4608. doi:10.1128/MCB. 05798-11
Basu-Roy U, Seo E, Ramanathapuram L, Rapp TB et al (2012) Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene 31:2270–2282. doi:10.1038/onc.2011.405
Agathocleous M, Iordanova I, Willardsen MI, Xue XY et al (2009) A directional Wnt/beta-catenin-Sox2-proneural pathway regulates the transition from proliferation to differentiation in the Xenopus retina. Development 136:3289–3299. doi:10.1242/dev.040451
Kuwabara T, Hsieh J, Muotri A, Yeo G et al (2009) Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat Neurosci 12:1097–1105. doi:10.1038/nn.2360
Mansukhani A, Ambrosetti D, Holmes G, Cornivelli L et al (2005) Sox2 induction by FGF and FGFR2 activating mutations inhibits Wnt signaling and osteoblast differentiation. J Cell Biol 168:1065–1076. doi:10.1083/jcb.200409182
Shimojo H, Ohtsuka T, Kageyama R (2011) Dynamic expression of notch signaling genes in neural stem/progenitor cells. Front Neurosci 5:78. doi:10.3389/fnins.2011.00078
Imayoshi I, Kageyama R (2011) The role of Notch signaling in adult neurogenesis. Mol Neurobiol 44:7–12. doi:10.1007/s12035-011-8186-0
Givogri MI, de Planell M, Galbiati F, Superchi D et al (2006) Notch signaling in astrocytes and neuroblasts of the adult subventricular zone in health and after cortical injury. Dev Neurosci 28:81–91. doi:10.1159/000090755
Sun F, Mao X, Xie L, Ding M et al (2013) Notch1 signaling modulates neuronal progenitor activity in the subventricular zone in response to aging and focal ischemia. Aging Cell 12:978–987. doi:10.1111/acel.12134
Guentchev M, McKay RD (2006) Notch controls proliferation and differentiation of stem cells in a dose-dependent manner. Eur J Neurosci 23:2289–2296. doi:10.1111/j.1460-9568.2006.04766.x
Neves J, Parada C, Chamizo M, Giraldez F (2011) Jagged 1 regulates the restriction of Sox2 expression in the developing chicken inner ear: a mechanism for sensory organ specification. Development 138:735–744. doi:10.1242/dev.060657
Tomioka M, Nishimoto M, Miyagi S, Katayanagi T et al (2002) Identification of Sox-2 regulatory region which is under the control of Oct-3/4-Sox-2 complex. Nucleic Acids Res 30:3202–3213. doi:10.1093/nar/gkf435
Miyagi S, Saito T, Mizutani K, Masuyama N et al (2004) The Sox-2 regulatory regions display their activities in two distinct types of multipotent stem cells. Mol Cell Biol 24:4207–4220. doi:10.1128/MCB. 24.10.4207-4220.2004
Sikorska M, Sandhu JK, Deb-Rinker P, Jezierski A et al (2008) Epigenetic modifications of SOX2 enhancers, SRR1 and SRR2, correlate with in vitro neural differentiation. J Neurosci Res 86:1680–1693. doi:10.1002/jnr.21635
Li H, Collado M, Villasante A, Matheu A et al (2012) p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell 11:845–852. doi:10.1016/j.stem.2012.09.014
Lodato MA, Ng CW, Wamstad JA, Cheng AW et al (2013) SOX2 co-occupies distal enhancer elements with distinct POU factors in ESCs and NPCs to specify cell state. PLoS Genet 9:e1003288. doi:10.1371/journal.pgen.1003288
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42. doi:10.1038/nrg2485
Cox JL, Mallanna SK, Luo X, Rizzino A (2010) Sox2 uses multiple domains to associate with proteins present in Sox2-protein complexes. PLoS One 5:e15486. doi:10.1371/journal.pone.0015486
Kidder BL, Palmer S (2012) HDAC1 regulates pluripotency and lineage specific transcriptional networks in embryonic and trophoblast stem cells. Nucleic Acids Res 40:2925–2939. doi:10.1093/nar/gkr1151
Baltus GA, Kowalski MP, Tutter AV, Kadam S (2009) A positive regulatory role for the mSin3A-HDAC complex in pluripotency through Nanog and Sox2. J Biol Chem 284:6998–7006. doi:10.1074/jbc.M807670200
Kuroda T, Tada M, Kubota H, Kimura H et al (2005) Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol Cell Biol 25:2475–2485. doi:10.1128/MCB. 25.6.2475-2485.2005
Mitsui K, Tokuzawa Y, Itoh H, Segawa K et al (2003) The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113:631–642. doi:10.1016/S0092-8674(03)00393-3
Ma DK, Marchetto MC, Guo JU, Ming GL et al (2010) Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat Neurosci 13:1338–1344. doi:10.1038/nn.2672
Schuettengruber B, Martinez AM, Iovino N, Cavalli G (2011) Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol 12:799–814. doi:10.1038/nrm3230
Lee TI, Jenner RG, Boyer LA, Guenther MG et al (2006) Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125:301–313. doi:10.1016/j.cell.2006.02.043
Spivakov M, Fisher AG (2007) Epigenetic signatures of stem-cell identity. Nat Rev Genet 8:263–271. doi:10.1038/nrg2046
Pollina EA, Brunet A (2011) Epigenetic regulation of aging stem cells. Oncogene 30:3105–3126. doi:10.1038/onc.2011.45
Fasano CA, Dimos JT, Ivanova NB, Lowry N et al (2007) shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell 1:87–99. doi:10.1016/j.stem.2007.04.001
He S, Iwashita T, Buchstaller J, Molofsky AV et al (2009) Bmi-1 over-expression in neural stem/progenitor cells increases proliferation and neurogenesis in culture but has little effect on these functions in vivo. Dev Biol 328:257–272. doi:10.1016/j.ydbio.2009.01.020
Molofsky AV, He S, Bydon M, Morrison SJ et al (2005) Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev 19:1432–1437. doi:10.1101/gad.1299505
Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ et al (2005) Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev 19:1438–1443. doi:10.1101/gad.1299305
Inui M, Martello G, Piccolo S (2010) MicroRNA control of signal transduction. Nat Rev Mol Cell Biol 11:252–263. doi:10.1038/nrm2868
Xu N, Papagiannakopoulos T, Pan G, Thomson JA et al (2009) MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 137:647–658. doi:10.1016/j.cell.2009.02.038
Card DA, Hebbar PB, Li L, Trotter KW et al (2008) Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol 28:6426–6438. doi:10.1128/MCB. 00359-08
Barroso-delJesus A, Romero-Lopez C, Lucena-Aguilar G, Melen GJ et al (2008) Embryonic stem cell-specific miR302-367 cluster: human gene structure and functional characterization of its core promoter. Mol Cell Biol 28:6609–6619. doi:10.1128/MCB. 00398-08
Otsubo T, Akiyama Y, Hashimoto Y, Shimada S et al (2011) MicroRNA-126 inhibits SOX2 expression and contributes to gastric carcinogenesis. PLoS One 6:e16617. doi:10.1371/journal.pone.0016617
Tay Y, Zhang J, Thomson AM, Lim B et al (2008) MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature 455:1124–1128. doi:10.1038/nature07299
Szulwach KE, Li X, Smrt RD, Li Y et al (2010) Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol 189:127–141. doi:10.1083/jcb.200908151
Altman J (1962) Are new neurons formed in the brain of adult mammals? Science 135:1127–1128
Altman J, Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124:319–335
Vadodaria KC, Gage FH (2014) Snapshot: Adult hippocampal neurogenesis. Cell 156:1114. doi:10.1016/j.cell.2014.02.029
Gage FH, Temple S (2013) Neural stem cells: generating and regenerating the brain. Neuron 80:588–601. doi:10.1016/j.neuron.2013.10.037
Bondolfi L, Ermini F, Long JM, Ingram DK et al (2004) Impact of age and caloric restriction on neurogenesis in the dentate gyrus of C57BL/6 mice. Neurobiol Aging 25:333–340. doi:10.1016/S0197-4580(03)00083-6
McDonald HY, Wojtowicz JM (2005) Dynamics of neurogenesis in the dentate gyrus of adult rats. Neurosci Lett 385:70–75. doi:10.1016/j.neulet.2005.05.022
Leuner B, Kozorovitskiy Y, Gross CG, Gould E (2007) Diminished adult neurogenesis in the marmoset brain precedes old age. Proc Natl Acad Sci U S A 104:17169–17173. doi:10.1073/pnas.0708228104
Hattiangady B, Shetty AK (2008) Aging does not alter the number or phenotype of putative stem/progenitor cells in the neurogenic region of the hippocampus. Neurobiol Aging 29:129–147. doi:10.1016/j.neurobiolaging.2006.09.015
Sharpless NE, DePinho RA (2007) How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol 8:703–713. doi:10.1038/nrm2241
Ferron SR, Marques-Torrejon MA, Mira H, Flores I et al (2009) Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J Neurosci 29:14394–14407. doi:10.1523/JNEUROSCI. 3836-09.2009
Encinas JM, Michurina TV, Peunova N, Park JH et al (2011) Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 8:566–579. doi:10.1016/j.stem.2011.03.010
Artegiani B, Calegari F (2012) Age-related cognitive decline: can neural stem cells help us? Aging 4:176–186
Jang YY, Sharkis SJ (2007) A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 110:3056–3063. doi:10.1182/blood-2007-05-087759
Le Belle JE, Orozco NM, Paucar AA, Saxe JP et al (2011) Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8:59–71. doi:10.1016/j.stem.2010.11.028
Walton NM, Shin R, Tajinda K, Heusner CL et al (2012) Adult neurogenesis transiently generates oxidative stress. PLoS One 7:e35264. doi:10.1371/journal.pone.0035264
McCord AM, Jamal M, Shankavaram UT, Lang FF et al (2009) Physiologic oxygen concentration enhances the stem-like properties of CD133+ human glioblastoma cells in vitro. Mol Cancer Res 7:489–497. doi:10.1158/1541-7786.MCR-08-0360
Shyh-Chang N, Daley GQ, Cantley LC (2013) Stem cell metabolism in tissue development and aging. Development 140:2535–2547. doi:10.1242/dev.091777
Kloet DE, Burgering BM (2011) The PKB/FOXO switch in aging and cancer. Biochim Biophys Acta 1813:1926–1937. doi:10.1016/j.bbamcr.2011.04.003
Tyson JJ, Csikasz-Nagy A, Novak B (2002) The dynamics of cell cycle regulation. BioEssays 24:1095–1109. doi:10.1002/bies.10191
Ben-Porath I, Weinberg RA (2005) The signals and pathways activating cellular senescence. Int J Biochem Cell Biol 37:961–976. doi:10.1016/j.biocel.2004.10.013
Campisi J, d'Adda di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8:729–740. doi:10.1038/nrm2233
Reinhardt HC, Schumacher B (2012) The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet 28:128–136. doi:10.1016/j.tig.2011.12.002
Meletis K, Wirta V, Hede SM, Nister M et al (2006) p53 suppresses the self-renewal of adult neural stem cells. Development 133:363–369. doi:10.1242/dev.02208
Marques-Torrejon MA, Porlan E, Banito A, Gomez-Ibarlucea E et al (2013) Cyclin-dependent kinase inhibitor p21 controls adult neural stem cell expansion by regulating Sox2 gene expression. Cell Stem Cell 12:88–100. doi:10.1016/j.stem.2012.12.001
Li DM, Sun H (1998) PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci U S A 95:15406–15411. doi:10.1073/pnas.95.26.15406
Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI et al (2004) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114:1299–1307. doi:10.1172/JCI22475
Molofsky AV, Slutsky SG, Joseph NM, He S et al (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443:448–452. doi:10.1038/nature05091
Abdouh M, Chatoo W, El Hajjar J, David J et al (2012) Bmi1 is down-regulated in the aging brain and displays antioxidant and protective activities in neurons. Plos One 7:e31870. doi:10.1371/journal.pone.0031870
Li H, Collado M, Villasante A, Strati K et al (2009) The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460:1136–1139. doi:10.1038/nature08290
Bass AJ, Watanabe H, Mermel CH, Yu S et al (2009) SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 41:1238–1242. doi:10.1038/ng.465
Hussenet T, Dali S, Exinger J, Monga B et al (2010) SOX2 is an oncogene activated by recurrent 3q26.3 amplifications in human lung squamous cell carcinomas. PLoS One 5:e8960. doi:10.1371/journal.pone.0008960
Ring KL, Tong LM, Balestra ME, Javier R et al (2012) Direct reprogramming of mouse and human fibroblasts into multipotent neural stem cells with a single factor. Cell Stem Cell 11:100–109. doi:10.1016/j.stem.2012.05.018
Kim I, Rodriguez-Enriquez S, Lemasters JJ (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462:245–253. doi:10.1016/j.abb.2007.03.034
Cho YY, Kim DJ, Lee HS, Jeong CH et al (2013) Autophagy and cellular senescence mediated by Sox2 suppress malignancy of cancer cells. PLoS One 8:e57172. doi:10.1371/journal.pone.0057172
Miranda CJ, Braun L, Jiang Y, Hester ME et al (2012) Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell 11:542–552. doi:10.1111/j.1474-9726.2012.00816.x
Florian MC, Nattamai KJ, Dorr K, Marka G et al (2013) A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature 503:392–396. doi:10.1038/nature12631
Okamoto M, Inoue K, Iwamura H, Terashima K et al (2011) Reduction in paracrine Wnt3 factors during aging causes impaired adult neurogenesis. FASEB J 25:3570–3582. doi:10.1096/fj.11-184697
St Laurent G 3rd, Hammell N, McCaffrey TA (2010) A LINE-1 component to human aging: do LINE elements exact a longevity cost for evolutionary advantage? Mech Ageing Dev 131:299–305. doi:10.1016/j.mad.2010.03.008
Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW et al (2009) L1 retrotransposition in human neural progenitor cells. Nature 460:1127–1131. doi:10.1038/nature08248
Gasior SL, Wakeman TP, Xu B, Deininger PL (2006) The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol 357:1383–1393. doi:10.1016/j.jmb.2006.01.089
Maxwell PH, Burhans WC, Curcio MJ (2011) Retrotransposition is associated with genome instability during chronological aging. Proc Natl Acad Sci U S A 108:20376–20381. doi:10.1073/pnas.1100271108
Wallace NA, Belancio VP, Deininger PL (2008) L1 mobile element expression causes multiple types of toxicity. Gene 419:75–81. doi:10.1016/j.gene.2008.04.013
De Strooper B (2010) Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev 90:465–494. doi:10.1152/physrev.00023.2009
Wang H, Li R, Shen Y (2013) β-secretase: its biology as a therapeutic target in diseases. Trends Pharmacol Sci 34:215–225. doi:10.1016/j.tips.2013.01.008
Wolfe MS (2009) γ-secretase in biology and medicine. Semin Cell Dev Biol 20:219–224. doi:10.1016/j.semcdb.2008.12.011
Vincent B, Checler F (2012) α-secretase in Alzheimer’s disease and beyond: mechanistic, regulation and function in the shedding of membrane proteins. Curr Alzheimer Res 9:140–156. doi:10.2174/156720512799361646
Jin K, Peel AL, Mao XO, Xie L et al (2004) Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A 101:343–347. doi:10.1073/pnas.2634794100
Deng W, Aimone JB, Gage FH (2010) New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11:339–350. doi:10.1038/nrn2822
Wang Q, Matsumoto Y, Shindo T, Miyake K et al (2006) Neural stem cells transplantation in cortex in a mouse model of Alzheimer's disease. J Med Investig 53:61–69. doi:10.2152/jmi.53.61
Wu S, Sasaki A, Yoshimoto R, Kawahara Y et al (2008) Neural stem cells improve learning and memory in rats with Alzheimer's disease. Pathobiology 75:186–194. doi:10.1159/000124979
Moghadam FH, Alaie H, Karbalaie K, Tanhaei S et al (2009) Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in Alzheimerian rats. Differentiation 78:59–68. doi:10.1016/j.diff.2009.06.005
Park D, Lee HJ, Joo SS, Bae DK et al (2012) Human neural stem cells over-expressing choline acetyltransferase restore cognition in rat model of cognitive dysfunction. Exp Neurol 234:521–526. doi:10.1016/j.expneurol.2011.12.040
Livesey FJ (2014) Human stem cell models of dementia. Hum Mol Genet 23:R35–R39. doi:10.1093/hmg/ddu302
Donovan MH, Yazdani U, Norris RD, Games D et al (2006) Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer's disease. J Comp Neurol 495:70–83197. doi:10.1002/cne.20840
Rodriguez JJ, Jones VC, Tabuchi M, Allan SM et al (2008) Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One 3:e2935198. doi:10.1371/journal.pone.0002935
Hamilton A, Holscher C (2012) The effect of ageing on neurogenesis and oxidative stress in the APP(swe)/PS1(deltaE9) mouse model of Alzheimer's disease. Brain Res 1449:83–93. doi:10.1016/j.brainres.2012.02.015
Freude KK, Penjwini M, Davis JL, LaFerla FM et al (2011) Soluble amyloid precursor protein induces rapid neural differentiation of human embryonic stem cells. J Biol Chem 286:24264–24274. doi:10.1074/jbc.M111.227421
Young-Pearse TL, Suth S, Luth ES, Sawa A et al (2010) Biochemical and functional interaction of disrupted-in-schizophrenia 1 and amyloid precursor protein regulates neuronal migration during mammalian cortical development. J Neurosci 30:10431–10440. doi:10.1523/JNEUROSCI. 1445-10.2010
Shariati SA, Lau P, Hassan BA, Muller U et al (2013) APLP2 regulates neuronal stem cell differentiation during cortical development. J Cell Sci 126:1268–1277. doi:10.1242/jcs.122440
Porayette P, Gallego MJ, Kaltcheva MM, Bowen RL et al (2009) Differential processing of amyloid-β precursor protein directs human embryonic stem cell proliferation and differentiation into neuronal precursor cells. J Biol Chem 284:23806–23817. doi:10.1074/jbc.M109.026328
Lopez-Toledano MA, Shelanski ML (2004) Neurogenic effects of β-amyloid peptide in the development of neural stem cells. J Neurosci 24:5439–5444. doi:10.1523/JNEUROSCI. 0974-04.2004
Zhang C, McNeil E, Dressler L, Siman R (2007) Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer's disease. Exp Neurol 204:77–87. doi:10.1016/j.expneurol.2006.09.018
He P, Shen Y (2009) Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer's disease. J Neurosci 29:6545–6557. doi:10.1523/JNEUROSCI. 0421-09.2009
Caille I, Allinquant B, Dupont E, Bouillot C et al (2004) Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 131:2173–2181. doi:10.1242/dev.01103
Baratchi S, Evans J, Tate WP, Abraham WC et al (2012) Secreted amyloid precursor proteins promote proliferation and glial differentiation of adult hippocampal neural progenitor cells. Hippocampus 22:1517–1527. doi:10.1002/hipo.20988
Gakhar-Koppole N, Hundeshagen P, Mandl C, Weyer SW et al (2008) Activity requires soluble amyloid precursor protein alpha to promote neurite outgrowth in neural stem cell-derived neurons via activation of the MAPK pathway. Eur J Neurosci 28:871–882. doi:10.1111/j.1460-9568.2008.06398.x
Ghosal K, Stathopoulos A, Pimplikar SW (2010) APP intracellular domain impairs adult neurogenesis in transgenic mice by inducing neuroinflammation. PLoS One 5:e11866. doi:10.1371/journal.pone.0011866
Apostolova LG, Green AE, Babakchanian S, Hwang KS et al (2012) Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment (MCI), and Alzheimer Disease. Alzheimer Dis Assoc Disord 26:17–27. doi:10.1097/WAD.0b013e3182163b62
Hu X, He W, Luo X, Tsubota KE et al (2013) BACE1 regulates hippocampal astrogenesis via the Jagged1-Notch pathway. Cell Rep 4:40–49. doi:10.1016/j.celrep.2013.06.005
Wang R, Dineley KT, Sweatt JD, Zheng H (2004) Presenilin 1 familial Alzheimer's disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience 126:305–312. doi:10.1016/j.neuroscience.2004.03.048
Wen PH, Hof PR, Chen X, Gluck K et al (2004) The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol 188:224–237. doi:10.1016/j.expneurol.2004.04.002
Choi SH, Veeraraghavalu K, Lazarov O, Marler S et al (2008) Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron 59:568–580. doi:10.1016/j.neuron.2008.07.033
Veeraraghavalu K, Sisodia SS (2013) Mutant presenilin 1 expression in excitatory neurons impairs enrichment-mediated phenotypes of adult hippocampal progenitor cells. Proc Natl Acad Sci U S A 110:9148–9153. doi:10.1073/pnas.1302106110
Ma Z, Li Q, Zhang Z, Zheng Y (2013) A disintegrin and metalloprotease 10 in neuronal maturation and gliogenesis during cortex development. Neural Regen Res 8:24–30. doi:10.3969/j.issn. 1673-5374.2013.01.003
Katakowski M, Chen J, Zhang ZG, Santra M et al (2007) Stroke-induced subventricular zone proliferation is promoted by tumor necrosis factor-α-converting enzyme protease activity. J Cereb Blood Flow Metab 27:669–678. doi:10.1038/sj.jcbfm.9600390
Chen X, Chen L, Zhang R, Yi Y et al (2013) ADAM17 regulates self-renewal and differentiation of U87 glioblastoma stem cells. Neurosci Lett 537:44–49. doi:10.1016/j.neulet.2013.01.021
Crews L, Adame A, Patrick C, Delaney A et al (2010) Increased BMP6 levels in the brains of Alzheimer's disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J Neurosci 30:12252–12262. doi:10.1523/JNEUROSCI. 1305-10.2010
Griffin RJ, Moloney A, Kelliher M, Johnston JA et al (2005) Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer's disease pathology. J Neurochem 93:105–117. doi:10.1111/j.1471-4159.2004.02949.x
Aoyagi N, Uemura K, Kuzuya A, Kihara T et al (2010) PI3K inhibition causes the accumulation of ubiquitinated presenilin 1 without affecting the proteasome activity. Biochem Biophys Res Commun 391:1240–1245. doi:10.1016/j.bbrc.2009.12.051
Moloney AM, Griffin RJ, Timmons S, O'Connor R et al (2010) Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 31:224–243. doi:10.1016/j.neurobiolaging.2008.04.002
Talbot K, Wang HY, Kazi H, Han LY et al (2012) Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122:1316–1338. doi:10.1172/JCI59903
Phiel CJ, Wilson CA, Lee VM, Klein PS (2003) GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 423:435–439. doi:10.1038/nature01640
Chevallier NL, Soriano S, Kang DE, Masliah E et al (2005) Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol 167:151–159. doi:10.1016/S0002-9440(10)62962-8
Alvarez AR, Godoy JA, Mullendorff K, Olivares GH et al (2004) Wnt-3a overcomes beta-amyloid toxicity in rat hippocampal neurons. Exp Cell Res 297:186–196. doi:10.1016/j.yexcr.2004.02.028
Silva-Alvarez C, Arrazola MS, Godoy JA, Ordenes D et al (2013) Canonical Wnt signaling protects hippocampal neurons from Abeta oligomers: role of non-canonical Wnt-5a/Ca(2+) in mitochondrial dynamics. Front Cell Neurosci 7:97. doi:10.3389/fncel.2013.00097
Inestrosa NC, Toledo EM (2008) The role of Wnt signaling in neuronal dysfunction in Alzheimer's Disease. Mol Neurodegener 3:9. doi:10.1186/1750-1326-3-9
Cerpa W, Farias GG, Godoy JA, Fuenzalida M et al (2010) Wnt-5a occludes Abeta oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol Neurodegener 5:3. doi:10.1186/1750-1326-5-3
Kopan R, Goate A (2000) A common enzyme connects notch signaling and Alzheimer's disease. Genes Dev 14:2799–2806. doi:10.1101/gad.836900
Kim SY, Kim MY, Mo JS, Park HS (2007) Notch1 intracellular domain suppresses APP intracellular domain-Tip60-Fe65 complex mediated signaling through physical interaction. Biochim Biophys Acta 1773:736–746. doi:10.1016/j.bbamcr.2007.02.001
Ma QH, Futagawa T, Yang WL, Jiang XD et al (2008) A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol 10:283–294. doi:10.1038/ncb1690
Demars M, Hu YS, Gadadhar A, Lazarov O (2010) Impaired neurogenesis is an early event in the etiology of familial Alzheimer's disease in transgenic mice. J Neurosci Res 88:2103–2117. doi:10.1002/jnr.22387
Kim SU, Lee HJ, Kim YB (2013) Neural stem cell-based treatment for neurodegenerative diseases. Neuropathology 33:491–504. doi:10.1111/neup.12020
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sarlak, G., Vincent, B. The Roles of the Stem Cell-Controlling Sox2 Transcription Factor: from Neuroectoderm Development to Alzheimer’s Disease?. Mol Neurobiol 53, 1679–1698 (2016). https://doi.org/10.1007/s12035-015-9123-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9123-4