
Abstract
Cigarette smoking is the major cause of preventable death and morbidity throughout the world. Many compounds are present in tobacco, but nicotine is the primary addictive one. Nicotine exerts its physiological and pharmacological roles in the brain through neuronal nicotinic acetylcholine receptors (nAChRs), which are ligand-gated ion channels consisting of five membrane-spanning subunits that can modulate the release of neurotransmitters, such as dopamine, glutamate, and GABA and mediate fast signal transmission at synapses. Considering that there are 12 nAChR subunits, it is highly likely that subunits other than α4 and β2, which have been intensively investigated, also are involved in nicotine addiction. Consistent with this hypothesis, a number of genome-wide association studies (GWAS) and subsequent candidate gene-based associated studies investigating the genetic variants associated with nicotine dependence (ND) and smoking-related phenotypes have shed light on the CHRNA5/A3/B4 gene cluster on chromosome 15, which encodes the α5, α3, and β4 nAChR subunits, respectively. These studies demonstrate two groups of risk variants in this region. The first one is marked by single nucleotide polymorphism (SNP) rs16969968 in exon 5 of CHRNA5, which changes an aspartic acid residue into asparagine at position 398 (D398N) of the α5 subunit protein sequence, and it is tightly linked SNP rs1051730 in CHRNA3. The second one is SNP rs578776 in the 3ʹ-untranslated region (UTR) of CHRNA3, which has a low correlation with rs16969968. Although the detailed molecular mechanisms underlying these associations remain to be further elucidated, recent findings have shown that α5* (where “*” indicates the presence of additional subunits) nAChRs located in the medial habenulo-interpeduncular nucleus (mHb-IPN) are involved in the control of nicotine self-administration in rodents. Disruption of α5* nAChR signaling diminishes the aversive effects of nicotine on the mHb-IPN pathway and thereby permits more nicotine consumption. To gain a better understanding of the function of the highly significant genetic variants identified in this region in controlling smoking-related behaviors, in this communication, we provide an up-to-date review of the progress of studies focusing on the CHRNA5/A3/B4 gene cluster and its role in ND.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cigarette smoking is one of the most significant public health problems in both developed and developing countries. Although new efficacious techniques for smoking cessation have helped to reduce the number of smokers significantly, there were still approximately 38 million tobacco users in the USA and 1 billion worldwide in 2012 [1]. Of these smokers, approximately 60 % are nicotine dependent [2]. The burden of smoking-related diseases and the negative economic impact on society caused by cigarette smoking is staggering. According to the World Health Organization’s report, approximately five million people each year die of smoking-related illnesses [3], making smoking the largest cause of preventable death in the world, and if the current trend continues, the worldwide death toll caused by tobacco smoking will rise to eight million annually by 2030 [4]. Moreover, smoking has various detrimental effects on physical health that often are serious, carrying significant risks of cardiovascular diseases, respiratory diseases, and lung cancer, among other ailments.
There are approximately 4000 compounds in cigarette smoke; however, nicotine is the primary component responsible for the development of nicotine dependence (ND) [5]. Nicotine exerts its pharmacological and physiological roles in the brain through neuronal nicotinic acetylcholine receptors (nAChRs), which are widely distributed in the central and peripheral nervous systems. The nAChRs are ligand-gated ion channels consisting of five membrane-spanning subunits [6] that can modulate the release of neurotransmitters such as dopamine (DA), GABA, and glutamate [7] and mediate fast signal transmission at synapses [8]. There are 12 neuronal acetylcholine receptor subunits, with nine α subunits (α2–α10) and three β subunits (β2–β4) [6, 9, 10]. These subunits arrange in numerous distinct pentameric nAChRs, resulting in receptors that differ in distribution throughout the body and in biologic functions and other pharmacologic properties [11]. Binding of nicotine to nAChRs forms the molecular basis for the reward of nicotine and, eventually, the development of ND. Thus, nAChRs represent not only plausible candidate risk factors for ND but also targets for drugs for treating ND and other psychiatric disorders.
Abundant data from twin studies demonstrate that along with environmental factors, genetic variations are responsible for ND, with an estimated heritability of about 50 % [12–16]. To identify susceptibility loci and genetic variants for ND and its related phenotypes, many studies have been conducted using various approaches such as genome-wide linkage analysis, candidate gene-based association, and genome-wide association studies (GWAS). Of the genetic variants found to be associated with ND, the variants in the CHRNA5/A3/B4 gene cluster on chromosome 15, which encodes the α5, α3, and β4 subunits [17–20], have received much attention in the past several years. Importantly, the variants in this gene cluster have been associated, not only with ND, but with lung cancer [21–23]. As a result of this genetic research, new effort has been expended to understand how variants in this region impact ND and its related phenotypes at the molecular level.
Replication of genetic association between the variants in the CHRNA5/A3/B4 gene cluster and ND increases the validity of these findings. At the same time, it stimulates interest in exploring the molecular mechanisms of variants within this gene cluster underlying ND. Of the significant variants in this gene cluster, single nucleotide polymorphism (SNP) rs16969968 appears to be the most attractive as an ND factor, as it results in an amino acid change from aspartate to asparagine at position 398 of the nicotinic receptor α5 subunit protein sequence. How the clustered nAChR subunits function in the development of ND is still unclear, although evidence from mouse models with knockout (KO) or mutations of nAChR subunits, especially the α5 subunit, suggests that disruption of α5* nAChR signaling diminishes the stimulatory effects of nicotine on the medial habenulo-interpeduncular nucleus (mHb-IPN) pathway and thereby permits consumption of greater quantities of nicotine [24]. Hence, it was thought that variants in the CHRNA5/A3/B4 gene cluster play an important role in ND through the aversive effect of nicotine on the mHb-IPN pathway, whereas there are few reports concerning the reinforcing effect of nicotine in ventral tegmental area (VTA) DA neurons [25].
To gain a better understanding of the genetic factors that contribute to ND and other smoking-related phenotypes, in this review, we first focus on the significant association between the variants detected in the CHRNA5/A3/B4 gene cluster and smoking-related phenotypes, and then present mechanisms that could explain such associations at the molecular level.
Association Between Common Variants in the CHRNA5/A3/B4 Gene Cluster and Smoking-Related Phenotypes
Nicotine Dependence
ND, as well as addiction to any other substance, is a complicated phenotype. It involves many symptoms, consisting of early-morning smoking, heavier smoking, tolerance, and ease of relapse after quitting. More importantly, the development of ND is not a sudden event; it has to go through a transition from experimental smoking with the first puff to regular smoking and finally to the establishment of ND [26]. There are a series of assessment tools for ND; the more commonly used ones are the Fagerström test for nicotine dependence (FTND) [27] and the Diagnostic and Statistical Manual for Mental Disorders (4th edition) (DSM-IV) [28]. Although both scales are commonly used to evaluate the severity of ND, there exists a limited correlation between the two tools [27], because each focuses on different aspects of ND. The FTND is a simplified measure compared with the DSM-IV, which lays particular emphasis on the number of cigarettes smoked per day (CPD) and the time from waking to the first cigarette, whereas DSM-IV mainly emphasizes the behavioral and emotional aspects of addiction. Thus, when one considers the definition of ND using the FTND, we usually choose CPD to represent it, because of its easy measurement and appropriate matching to ND.
The first report concerning the contribution of variants in the CHRNA5/A3/B4 gene cluster to ND was published by Saccone et al. in 2007 [17]. In this study, the authors examined 879 light smokers who had no symptoms of dependence, with an FTND score of 0, and 1050 heavy smokers, with an FTND score of >4.0, focusing on the transition from regular smoking to addiction. Among 3713 SNPs in more than 300 candidate genes analyzed, multiple risk SNPs were found in the CHRNA5/A3/B4 gene cluster, with the most compelling evidence for a risk allele coming from a non-synonymous SNP rs16969968 in the α5 nicotine receptor subunit gene (CHRNA5) (p = 6.4 × 10−4). Furthermore, this SNP exhibited a recessive mode of inheritance, resulting in individuals with one copy of the risk allele A having a 1.1-fold increase in the risk of developing ND once exposed to cigarette smoking, whereas there was a 2-fold increase with the AA genotype compared with subjects having no copy. Since then, numerous candidate gene-based analyses and large-scale GWAS, together with several meta-analyses [29–31] which elaborated on Vandenbergh’s literature [32] have focused on the association of polymorphisms in the CHRNA5/A3/B4 gene cluster with ND across different populations, leading to the conclusion that variants in this gene cluster contribute to the development of heavy smoking and ND [17–22, 26]. Together, these studies demonstrate two groups of risk variants in the cluster. The first one is marked by SNP rs16969968 in exon 5 of CHRNA5, which changes an aspartic acid residue into asparagine at position 398 (D398N) of the α5 subunit protein sequence, or its tightly linked SNP rs1051730 in CHRNA3. The other is SNP rs578776 in the 3′-untranslated region (UTR) of CHRNA3, which has a low linkage disequilibrium (LD) with rs16969968 (Table 1).
The association of these SNPs with ND can be modified by different factors. For instance, Weiss et al. [19] reported that individuals who became regular smokers before the age of 16 showed a signification association between SNP rs16969968 and the severity of ND, whereas Grucza et al. [38] found that the same SNP exhibited its effects mainly on late-onset smokers, after 16 years of age. What causes such inconsistent results remains to be investigated. In addition, other environmental factors, such as parental monitoring [39], childhood adversity [40], and peer smoking [41] have been reported to influence the association between SNPs rs16969968 or rs1051730 and ND.
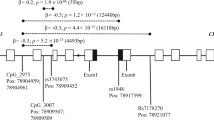
On the other hand, there are a few reports concerning the effect of common variants in CHRNB4 on ND. Three independent GWAS meta-analyses revealed the importance of the CHRNA5/A3/B4 gene cluster in influencing ND, but failed to identify any SNP in the β4 receptor subunit gene as a contributor to the genetic association signal for heavy smoking [29, 42, 43]. Thus, for the time being, we are not clear on whether common variants in CHRNB4 play any role in the development of ND, although such a role is theoretically possible because of the high LD patterns across CHRNA5, CHRNA3, and CHRNB4 (Fig. 1).

Schematic diagram of the human CHRNA5/A3/B4 cluster. Horizontal black arrows indicate the direction of transcription of each gene. Green and pink rectangles indicate exons and untranslated regions, respectively, while horizontal black lines represent introns. The genetic variants (rs1051730, rs578776, and rs16969968) significantly associated with ND are indicated by vertical arrows
Lung Cancer
Lung cancer, which can be divided into two major histopathologic types (small-cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC)), is the leading cause of cancer-related deaths throughout the world [44]. Among multiple risk factors associated with lung cancer, cigarette smoking is the most important one, as many carcinogens are present in cigarette smoke and others, such as (4-(N-nitrosomethylamino)-1-(3-pyridyl)-1-butanone) (NNK) and (Nʹ-nitrosonornicotine) (NNN), are metabolized from nicotine [45, 46]. Both of these compounds can stimulate the growth [47] or inhibit apoptosis [48] of lung cancer cells.
In parallel with the studies of ND, several SNPs within the CHRNA5/A3/B4 gene cluster seem to increase the risk of lung cancer according to several GWAS and candidate gene-based association studies [21, 22, 49, 50]. Hung et al. [22] first found that SNP rs16969968 was robustly associated with lung cancer after studying nearly 317,139 SNPs in 4614 subjects of European descent. Since then, this finding has been replicated in different ethnic populations [49, 51–53]. Furthermore, Saccone et al. [29] demonstrated the presence of a significant association between rs16969968 and lung cancer (p < 10−20) in a meta-analysis of six datasets of European-ancestry subjects (N = 13,614) [29]. However, whether the association of this SNP with lung cancer is directly or indirectly mediated by the variant’s association with ND has been the subject of extensive debate in the past several years. One group of investigators favoring a direct role of variants in the CHRNA5/A3/B4 gene cluster in lung cancer reasoned that the association was observed even in non-smokers [22] and remained significant after adjustment for smoking quantity [54, 55]. The other group, preferring an indirect role of the variant in lung cancer, argued that the studies failed to detect a significant association between the variant and lung cancer in never smokers [56]. The inaccurate measurement of uptake of carcinogens by self-reported CPD supports this view [57].
There might have some other elements, such as different ethnic backgrounds of the populations examined, sample sizes, and measurement strategies for smoking-related phenotypes, which contribute to the above-mentioned conflict. For example, the populations used in most of these studies were of European origin [21, 22], a group that has a 37–43 % frequency of the rs16969968 A allele, whereas the A nucleotide is not detected or is uncommon in African, East Asian, and Native American populations [18]. Consequently, the association between variants in the CHRNA5/A3/B4 gene cluster and lung cancer needs to be further investigated in well-designed studies, especially in other ethnic samples.
Smoking Initiation and Cessation
Cigarette smoking can be divided into three behaviors: initiation, ND, and cessation. Many variables influence the three processes, including age, education, social status, and so on. Although the variants in the CHRNA5/A3/B4 gene cluster are strongly associated with ND and smoking quantity, this region appears to play a smaller or less significant role in smoking initiation and cessation.
Thorgeirsson et al. [50] reported that the variants in CHRNA5/A3/B4 did not influence smoking initiation and experimentation. Similarly, Lips et al. [58] and Kaur-Knudsen et al. [54] also concluded that the variants in the cluster on chromosome 15 did not play a role in identifying non-smokers and smokers. At the same time, Maes et al. [59] showed that the SNPs associated with ND did not show a significant association with either smoking initiation or regular smoking in a twin study. On the other hand, Sherva et al. [60] reported an association between rs16969968 in the CHRNA5 gene and enhanced pleasurable responses to initial cigarette smoking, suggesting that phenotypes related to subjective experiences during smoking experimentation may mediate the development of ND. Meanwhile, Stephens et al. [61] conducted a meta-analysis including 56,034 subjects in 41 studies spanning nine countries, which showed a significant association of rs578776 with age of first regular tobacco use (β = 0.02; p = 0.004).
There are three main smoking cessation pharmacotherapies: varenicline, nicotine replacement therapy (NRT), and buproprion. Each has its specific pharmacologic effects. It is likely that one treatment will work for some people but not others with different genetic backgrounds. Studies of whether the variants in the CHRNA5/A3/B4 gene cluster play a role in smoking cessation has yielded inconsistent conclusions, with some studies demonstrating a significant role of SNPs in this gene cluster in quitting [58, 62–65], whereas others did not [36, 66–68]. Freathy et al. [67] showed strong evidence of an association between rs1051730 and an increased likelihood of continued smoking in pregnancy, supporting a role of genetic factors in influencing smoking cessation. Furthermore, Chen et al. [69, 70], in their two studies, demonstrated that variants in the CHRNA5 gene (rs16969968 or rs16969968–rs680244 haplotype) predicted both ND and smoking cessation. They noted that the high-risk allele of rs16969968 was associated with a lower likelihood of quitting and of cessation failure at end of treatment in the placebo group or the group without any pharmacologic treatment. However, genetic variants did not predict abstinence across active treatment conditions. Thus, Chen et al. [69, 70] suggested that pharmacological cessation treatment might mitigate the genetic risks of cessation difficulty, which might be the explanation for the inconsistent results concerning smoking cessation. Generally speaking, two types of study designs are used in smoking cessation studies. They are either prospective or retrospective, each with different sample selection. This might have different implications. The former identified the genetic risk for smoking cessation, while the latter one placed the emphasis on pharmacologic effects in persons with different genetic backgrounds. There exists a potential limitation for individual study because of differences in sample size, heterogeneity of samples, and analysis approaches, all of which should be taken into consideration in follow-up studies.
Analysis of Rare Variants in the CHRNA5/A3/B4 Gene Cluster
As mentioned above, multiple common variants in the CHRNA5/A3/B4 gene cluster have consistently been found to be significantly associated with ND and smoking-related phenotypes. Among these, a non-synonymous change (rs16969968) in CHRNA5 is the most strongly associated SNP in several GWAS [42, 71]. Additionally, a group of highly correlated SNPs, specifically rs588765, was shown to increase CHRNA5 messenger RNA (mRNA) expression, thus leading to an increased risk of ND [30, 72]. Despite these convincing results, only a small proportion of the variance (~5 %) in smoking-related behaviors can be explained by these SNPs [30]. Rare variants, generally defined as those having a minor allele frequency of <1 %, constitute another major part of genetic variants other than common ones. Thus, rare variants may well account for the inadequate explanations of the heritability of smoking-related traits, as identified by recent GWAS.
Although rare variants may play a critical role in developing or maintaining ND, the function of these variants in the CHRNA5/A3/B4 gene cluster in the risk of ND has not been intensively investigated [73]. This is, we suspect, largely because their low frequency in populations increases the difficulties in ensuring adequate statistical power. Nevertheless, Wessel et al. [74] recently investigated the contribution of rare variants in nAChR subunit genes to FTND scores in treatment-seeking smokers and observed an association of rare SNPs in CHRNA5 with the FTND score. This finding motivated the interest of Haller and her colleagues in studying rare variants in other nAChR subunit genes in relation to ND. First, the same research team undertook pooled sequencing of the coding and flanking sequences of CHRNA5, CHRNA3, CHRNB4, CHRNA6, and CHRNB3 in African-American (AA) and European-American (EA) ND smokers and in light smokers without symptoms of dependence [75]. They found that rare missense variants at conserved residues in CHRNB4 (for example, rs61737499 and rs12914008) or CHRNA3 (rs8192475 in strong LD with rs12914008) are associated with a lower risk of ND and fewer CPD in both AAs (p = 0.0025 and p = 6.6 × 10−5, respectively) and EAs (p = 0.023 and p = 0.021, respectively) [75].
Using HEK293 cells, Haller et al. examined whether information from this type of functional testing of rare non-synonymous variants in CHRNB4 can significantly improve the association between genotype and phenotype [76]. Consistent with the results from Liang et al. [77], the authors suggested that reduced sensitivity to activation by agonists (nicotine or ACh) results in a higher risk of ND and that, conversely, increased sensitivity reduces the risk. Moreover, an in vivo study has been conducted using models [78] where mice injected in the mHb with lentiviruses carrying the WT β4 subunit or β4 rare missense variants showed aversion to or preference for nicotine, depending on the SNP. For instance, habenular expression of the β4 gain-of-function variant rs61737499 resulted in strong aversion, whereas transduction with the β4 loss-of-function variant rs56235003 failed to induce nicotine aversion. In sum, these functional studies demonstrate the vital role of rare variants in the CHRNA5/A3/B4 gene cluster in smoking-related behaviors.
Functional Studies of the Compelling SNP rs16969968
When the association of a variant with a phenotype of interest is revealed, it represents not only an association with the tested genetic variant(s), but also an association with untested, highly correlated SNPs that could span several genes on the same chromosome. To understand the molecular mechanism of the CHRNA5/A3/B4 gene cluster associated with ND and/or lung cancer, one needs to determine which SNP might alter biological function. It appears that the most compelling SNP, rs16969968, is likely to be a biological contributor to ND, because it changes an amino acid in the α5 nicotinic receptor protein. The position of the change is in the large cytoplasmic domain adjacent to the conserved amphipathic α-helix, so it is far from the extracellular acetylcholine binding site and unlikely to influence the sensitivity of agonist binding. In such a region, the negatively charged Asp398 might promote Ca2+ permeability, whereas Asn398, replaced by an amide group instead of the negatively charged carboxyl group, might inhibit it.
Consistent with this hypothesis, recent studies have demonstrated that the D398N polymorphism affects the function of (α4β2)2α5 nAChRs [18, 79]. When the two forms of the human α5 subunit (N398 and D398) were expressed in Xenopus oocytes, using α4 and β2 subunits as a concatamer structure, (α4β2)2α5 nAChRs containing the risk allele of α5 associated with increased risk of nicotine addiction exhibited diminished agonist-evoked intracellular calcium response, reduced calcium permeability, as well as enhanced short-term desensitization compared with (α4β2)2α5 nAChRs possessing the major allele of α5 [79]. These results were qualitatively similar to those of an earlier study that involved expression in HEK293T cell of human α5 subunits with mouse α4 and β2 subunits [18]. The incorporation of α5 SNP into HEK293T cells transfected with α4β2 cDNA reduced the maximum response to a nicotinic agonist without altering its surface expression. However, these obviously different effects of rs16969968 are seen only on the (α4β2)2α5 nAChRs; whether the SNP has a similar effect on the function of (α3β4)2α5 nAChRs is unclear.
Morel et al. [25] went a step further, adopting lentiviral re-expression vectors to achieve targeted expression of mutant α5 in the VTA of the brain using a knockin mouse model. It was observed that mice with the SNP rs16969968 in the VTA yielded intermediate behavioral and electrophysiological phenotypes compared with α5 KO mice, suggesting the non-synonymous α5 variant rs16969968, frequently present in subjects of European descent, exhibits a partial loss-of-function in vivo. This leads to increased nicotine consumption in the self-administration paradigm, thus defining a critical link between this SNP, its expression in VTA DA neurons, and nicotine intake.
Besides rs16969968, there may be a second biologic mechanism in the CHRNA5/A3/B4 gene cluster associated with heavy smoking and ND, including different extents of expression of CHRNA5 mRNA in the brain [80]. Joint statistical analysis of the two loci (or haplotypes) demonstrates that the amino acid change through SNP rs16969968 and varying CHRNA5 mRNA expression tagged by rs588765 (or rs578776, rs3743078) independently contribute to ND. The risk allele of rs16969968 occurs primarily on the low mRNA expression allele of CHRNA5, whereas the non-risk allele of rs16969968 occurs on both high- and low-expression alleles tagged by rs588765 in CHRNA5. When the non-risk allele occurs against the background of low mRNA expression of CHRNA5, the risk for ND and lung cancer is significantly lower than in persons with higher mRNA expression (Fig. 2). Together, these studies reveal three levels of risk associated with CHRNA5 and at least two distinct mechanisms conferring risk for ND: altered receptor function caused by rs16969968 and variability in CHRNA5 mRNA expression.

Association of different rs16969968–rs588765 diplotypes with nicotine dependence. The bars represent odds ratios ±95 % confidence intervals using GG_CC as a reference. A the risk allele of rs16969968, C the low mRNA expression allele of CHRNA5. Adapted from the report by Wang et al. [80], with the permission of Oxford University Press, license number 3416761480752
However, there is another hypothesis, from a different perspective, to explain the vital function of SNP rs16966698. For example, Hong et al. [81] hypothesized that the smoking variance explained by the allele-modulated circuits was much higher than the smoking variance explained by the genotype alone, making brain circuit measures an intermediate marker for the convergent effects of genes. Thus, the α5 gene variant Asp398Asn is associated with a dorsal anterior cingulated-ventral striatum/extended amygdal circuit, so that the Asn “risk allele” reduced the intrinsic resting functional connectivity strength in this circuit. At the same time, the findings from this work suggest a plausible circuit-level explanation for why rs16969968 and rs578776 represent two independent smoking-related signals in the CHRNA5/A3/B4 gene cluster. The authors of this study distinguished the rs578776-related dACC-thalamus circuit, which appeared sensitive to the “state” of smoking, from the rs16969968-influenced dACC-ventral striatum circuit, predicting nicotine addiction severity.
From Association to Mechanism: Role of the α5 Subunit
Numerous genetic studies have revealed a strong association between variants in the CHRNA5/A3/B4 gene cluster and increased vulnerability to ND [17, 50], creating a need to explore the underlying mechanisms. Moreover, to determine the function of the clustered nAChR subunits, KO mice and knockdown rats have been employed primarily because of the lack of receptor agonists and antagonists with selectivity for all three subunits. So far, only α5 and β4 KO mice are available [82–84], and mice that do not express the α3 subunit usually die soon after birth as a result of multi-organ dysfunction [84]. Thus, recent studies mainly focus on the function of α5 and β4 subunits in determining the cause of the high risk of ND, with a special focus on the α5 subunit because of the functional SNP rs16969968.
The α5 nAChR subunit demonstrates a relatively discrete mRNA expression profile in the brain, with the highest densities of expression found in the mHb, which projects almost exclusively to the IPN via the fasciculus retroflexus [85, 86]. Recently, Fowler et al. [24] adopted the α5 KO mouse model (analogous to individuals with reduced α5 receptor function) to examine the underlying mechanism of ND. The α5 KO mice responded far more vigorously than wild-type (WT) mice to nicotine infusions at high doses and consumed significantly more nicotine than their WT littermates when tested under a progressive ratio schedule for reinforcement. Whereas the WT mice tried to control their nicotine intake through intravenous self-administration to achieve a consistent, desired blood concentration, KO mice did not, appearing to consume greater amounts as the dosage increased (Fig. 3). This finding leads to a hypothesis that deficient α5* nAChR signaling attenuates the negative effects of nicotine that limit its intake. Consistent with this result, the same manipulation in rats weakened the aversive effects of higher doses of nicotine but did not alter the reinforcing effects of nicotine on the brain reward system, as measured by nicotine-induced elevations and lowering of intracranial self-stimulation (ICSS) thresholds [24]. These findings are complemented by another study conducted by the same team [87], employing a conditional place preference task to represent the differential effects of nicotine dose on reward in α5 KO and WT mice [88]. Moreover, Fowler et al. showed that the mHb-IPN pathway of the KO mice was far less sensitive to nicotine-induced activation than that in WT mice by using Fos immunoreactivity as a measure of neuronal activation [24]. RNA interference-mediated knockdown of the α5 nAChR subunit in the same rat brain region also resulted in similar responses to nicotine [24]. Intriguingly, virus-mediated re-expression of the α5nAChR subunit in the MHb-IPN pathway of the KO mice abolished the increased nicotine intake seen at higher doses of nicotine [24]. Taken together, these findings indicate that the α5 receptor subunit is responsible for transmission of some aversive qualities of nicotine. In other words, nicotine-induced activation of the MHb-IPN pathway by the α5 receptor subunit results in a negative motivational signal that limits further nicotine intake. Hence, disrupted sensitivity of the MHb-IPN tract to nicotine in the α5 KO mice induces greater nicotine intake.

Increased total nicotine intake (mg/kg) in α5−/− mice compared with WT mice receiving infusions of high doses of nicotine. Data are presented as mean (±SEM) total nicotine intake at each dose. p < 0.001 indicates statistically significant differences between these groups at the same nicotine dose. Adapted from the report by Fowler et al. [24], with the permission of Nature Publishing Group, license number 3416820244064
In addition to the α5 nAChR subunit, evidence suggests that β4* nAChRs in the mHb-IPN pathway play a key role in regulating nicotine consumption. For example, Frahm et al. [89] reported that mice overexpressing the β4 subunit as a result of bacterial artificial chromosome (BAC) transgenic technology consumed far less nicotine than their WT counterparts, and this effect could be reversed by lentiviral-mediated expression of the α5 D397N variant in the mHb [89], suggesting that, similar to the α5 nAChR subunit, the β4 subunit regulates sensitivity to the aversive effects of nicotine that control the quantities of drug consumed.
Apart from their role in the aversive effects of nicotine through the mHb-IPN pathway, the α5 and β4 nAChR subunits also have a potential action in nicotine withdrawal. Withdrawal symptoms can be divided into two classes: somatic and affective. The first ones are characterized by increased grooming, scratching, and shaking [90, 91], whereas the latter include primarily depressed mood, anxiety, difficulty concentrating, and so on [90, 92, 93]. The initiation of withdrawal can be precipitated by administration of nicotine antagonists such as mecamylamine during chronic nicotine exposure. A recent study showed that chronic nicotine-treated β4 KO mice displayed significantly milder somatic withdrawal symptoms than WT mice when the symptoms were precipitated by mecamylamine [94]. Furthermore, α5 KO mice that were dependent on nicotine (delivered through subcutaneously implanted osmotic minipumps) did not show somatic signs of nicotine withdrawal [95]. Considering that β4* and α5* nAChRs are robustly expressed in the mHb-IPN pathway and that mecamylamine was infused directly into either the mHb or the IPN of nicotine-dependent WT mice, the precipitated expression of somatic withdrawal symptoms demonstrates that these two nAChR subunits and perhaps others enriched in the mHb-IPN pathway are critical for the expression of nicotine withdrawal. On the contrary, Fowler et al. [87] concluded that the reward-inhibiting effects of precipitated nicotine withdrawal were not regulated by α5* nAChRs based on the fact that the magnitude to which mecamylamine precipitated elevations of ICSS thresholds was similar in nicotine-dependent WT and KO mice [87]. Interestingly, another study [96] showed that α5* nAChRs are more closely associated with physical signs of nicotine withdrawal than with affective symptoms, because chronic nicotine-treated α5 KO mice still appeared anxious during withdrawal.
Addiction to cigarette smoking depends not only on the attenuating aversion of high doses of nicotine and nicotine withdrawal, as described above, but also on the reinforcing effects of low doses of nicotine, the balance between the rewarding and aversive actions of the drug [90, 92]. Furthermore, although the α5 nAChR subunit is most densely expressed in the mHb-IPN pathway, its expression is also found in many other addiction-relevant brain regions; for instance, a high percentage in the VTA, which underlies the rewarding and addictive properties of drugs of abuse through the dopaminergic (DAergic) neurons [97]. Consequently, the α5* nAChRs are subjected to the same action in the VTA that explains their role in ND. However, many studies trying to identify the role of the α5 receptor subunit in the mHb-IPN pathway failed to find an effect in the VTA, especially in the dopaminergic neurons [24, 87]. There was a first report that comprehensively analyzed the role of the α5 nAChR subunit in the VTA DA system [25]. This study investigated the reinforcing effects of nicotine in drug-naive α5 KO mice by using an acute intravenous nicotine self-administration task and ex vivo and in vivo electrophysiological recording of nicotine-elicited DA cell activation. The fact that α5 KO mice, compared with WT mice, exhibited decreased sensitivity of the DAergic system and a dramatic shift to high nicotine doses in an acute nicotine injection paradigm [25] suggested a crucial role of α5* nAChRs in determining the minimum nicotine dose necessary for DA activation and thus nicotine reinforcement (Fig. 4). In addition, normal responses like those in WT mice were restored in KO mice by generalized lentiviral-mediated re-expression of the α5 subunit in all VTA cells or targeted to VTA DA cells specifically [25]. These findings have defined novel, largely unexpected roles for the α5 nAChR subunit in reinforcing the effects of nicotine, although it acts only as an accessory subunit instead of contributing to the nicotine binding site. This aspect of the research may broaden our horizons in understanding the underling mechanisms of the CHRNA5/A3/B4 gene cluster in the development of ND, although independent verification of the findings is still lacking.

Crucial role of α5* nAChRs in intravenous self-administration task (IVSA). α5−/− mice exhibited a decreased sensitivity of the DA neurons and a dramatic shift to high nicotine doses compared with WT mice. Data are presented as mean (±SEM) total nicotine intake at each dose. ***p < 0.001; **p < 0.01. Adapted from the report by Morel et al. [25], with the permission of Nature Publishing Group, license number 3416821041833
Conclusions and Future Research
Cigarette smoking continues to be a major health threat worldwide, underscoring the need to fully understand the etiology of ND. Research has implicated variants in the CHRNA5/A3/B4 gene cluster on chromosome 15 in the development of ND [17, 18, 20, 34]. There is now a compelling body of evidence linking SNPs rs16969968 (or its strongly linked SNPs) and rs578776 (or rs588765) to smoking-related phenotypes [17–20, 34]. Joint statistical analyses of the two loci mentioned above suggest the existence of two independent molecular mechanisms in ND. One is the amino acid change through SNP rs16969968, and another is differing degrees of CHRNA5 mRNA expression tagged by rs588765 (or rs578776, rs3743078) [80]. However, these findings reveal only a small portion of both common and rare variants in the CHRNA5/A3/B4 cluster. Thus, additional loci associated with smoking-related phenotypes await discovery. In particular, despite its difficulty, much attention should be paid to studies of rare variants in this gene region in order to understand in depth the genetics of ND.
There still is some controversy as to the relation between the implicated SNPs and lung cancer, although the findings from GWAS are robust [55–57]. Whether this association is direct or merely a byproduct of ND must be investigated further. Because there have been no specific pharmacological reagents for the α5, α3, or β4 nAChR subunits that are useful in elucidating such complicated relations, design of highly specific nAChRs ligands is of prime importance. Alternatively, knockin mouse model studies may directly examine the effects of variants given a constant carcinogen exposure. In other words, if, for example, SNP rs16969968 can be inserted into mice while ensuring that other conditions remain the same, the difference between the two groups of mice would be only in this SNP. Supposing that there is a difference in lung cancer between the two groups of mice, we can conclude that rs16969968 acts directly in the development of lung cancer. However, if not, we are more willing to believe that the SNP plays an indirect role.
As with the rapid development of the large-scale GWAS, extensive genomic information concerning ND is now available. This lays emphasis on the urgency of understanding the biological mechanisms of how α5, α3, and β4 nAChR subunits modulate smoking-related behaviors, which presents both opportunities and challenges. An important means to tease out the functional role of these receptor subunit genes in the neurobiology of ND is through genetic engineering technologies. Significant progress has been made in the past few years by using both in vitro and in vivo models, highlighting the importance of the α5 nAChR subunit in regulating ND. However, these functional studies so far reveal only a critical role of the α5 subunit in controlling the aversive and withdrawal effects of nicotine. How the α3 or β4 nAChR subunits function in ND has not been clarified yet, primarily because of the smaller number of functional studies of these two subunits. Even though there are a few studies suggesting a role of the α5 subunit in the rewarding effect of nicotine, most of them remain to be validated in independent studies. Thus, this part of research is in its early stages, and more relevant studies are greatly needed so as to fully understand the underlying mechanisms of ND. In addition, as discussed above, there exists a significant interaction between SNPs or haplotypes in the CHRNA5/A3/B4 gene cluster and the success of cessation measures. Those with the high-risk SNPs or haplotypes appear more biologically predisposed to having difficulty quitting without pharmacologic treatment, a problem that may be ameliorated by effective pharmacologic treatment. Thus, identification of molecular mechanisms underlying ND and responsiveness to pharmacologic treatment for ND will improve the development of novel, tailored smoking cessation therapies.
References
Doughton S (2014) Global cigarette consumption, number of smokers climbing. Seattle Times science reporter January 7
SAMHSA (2006) Results from the 2005 National Survey on Drug Use and Health: National Findings. Substance abuse and mental health services administration: NSDUH Series H-30 DHHS Publication No. SMA 06-4194
WHO (2006) The facts about smoking and health. May 30 http://www.wpro.who.int/media_centre/fact_sheets/fs_20060530.htm
Murray CJ, Lopez AD (1997) Alternative projections of mortality and disability by cause 1990–2020: Global Burden of Disease Study. Lancet 349(9064):1498–1504. doi:10.1016/S0140-6736(96)07492-2
Stolerman IP, Jarvis MJ (1995) The scientific case that nicotine is addictive. Psychopharmacology 117(1):2–10, discussion 14–20
Le Novere N, Corringer PJ, Changeux JP (2002) The diversity of subunit composition in nAChRs: evolutionary origins, physiologic and pharmacologic consequences. J Neurobiol 53(4):447–456. doi:10.1002/neu.10153
Dani JA, Bertrand D (2007) Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol 47:699–729. doi:10.1146/annurev.pharmtox.47.120505.105214
Gotti C, Zoli M, Clementi F (2006) Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci 27(9):482–491. doi:10.1016/j.tips.2006.07.004
Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J (2001) Alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A 98(6):3501–3506. doi:10.1073/pnas.051622798
Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S (1994) Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79(4):705–715
Sargent PB (1993) The diversity of neuronal nicotinic acetylcholine receptors. Annu Rev Neurosci 16:403–443. doi:10.1146/annurev.ne.16.030193.002155
Lessov CN, Martin NG, Statham DJ, Todorov AA, Slutske WS, Bucholz KK, Heath AC, Madden PA (2004) Defining nicotine dependence for genetic research: evidence from Australian twins. Psychol Med 34(5):865–879
Lessov-Schlaggar CN, Pang Z, Swan GE, Guo Q, Wang S, Cao W, Unger JB, Johnson CA, Lee L (2006) Heritability of cigarette smoking and alcohol use in Chinese male twins: the Qingdao twin registry. Int J Epidemiol 35(5):1278–1285. doi:10.1093/ije/dyl148
Li MD (2006) The genetics of nicotine dependence. Curr Psychiatry Rep 8(2):158–164
Maes HH, Sullivan PF, Bulik CM, Neale MC, Prescott CA, Eaves LJ, Kendler KS (2004) A twin study of genetic and environmental influences on tobacco initiation, regular tobacco use and nicotine dependence. Psychol Med 34(7):1251–1261
Li MD, Cheng R, Ma JZ, Swan GE (2003) A meta-analysis of estimated genetic and environmental effects on smoking behavior in male and female adult twins. Addiction 98(1):23–31
Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, Swan GE, Goate AM, Rutter J, Bertelsen S, Fox L, Fugman D, Martin NG, Montgomery GW, Wang JC, Ballinger DG, Rice JP, Bierut LJ (2007) Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet 16(1):36–49
Bierut LJ, Stitzel JA, Wang JC, Hinrichs AL, Grucza RA, Xuei X, Saccone NL, Saccone SF, Bertelsen S, Fox L, Horton WJ, Breslau N, Budde J, Cloninger CR, Dick DM, Foroud T, Hatsukami D, Hesselbrock V, Johnson EO, Kramer J, Kuperman S, Madden PA, Mayo K, Nurnberger J Jr, Pomerleau O, Porjesz B, Reyes O, Schuckit M, Swan G, Tischfield JA, Edenberg HJ, Rice JP, Goate AM (2008) Variants in nicotinic receptors and risk for nicotine dependence. Am J Psychiatry 165(9):1163–1171
Weiss RB, Baker TB, Cannon DS, von Niederhausern A, Dunn DM, Matsunami N, Singh NA, Baird L, Coon H, McMahon WM, Piper ME, Fiore MC, Scholand MB, Connett JE, Kanner RE, Gahring LC, Rogers SW, Hoidal JR, Leppert MF (2008) A candidate gene approach identifies the CHRNA5-A3-B4 region as a risk factor for age-dependent nicotine addiction. PLoS Genet 4(7):e1000125
Stevens VL, Bierut LJ, Talbot JT, Wang JC, Sun J, Hinrichs AL, Thun MJ, Goate A, Calle EE (2008) Nicotinic receptor gene variants influence susceptibility to heavy smoking. Cancer Epidemiol Biomark Prevent Publ Am Assoc Cancer Res Cosponsored Am Soc Prevent Oncol 17(12):3517–3525. doi:10.1158/1055-9965.EPI-08-0585
Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, Wang Y, Mills G, Doheny K, Tsai YY, Chen WV, Shete S, Spitz MR, Houlston RS (2008) Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet 40(5):616–622
Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Chen C, Goodman G, Field JK, Liloglou T, Xinarianos G, Cassidy A, McLaughlin J, Liu G, Narod S, Krokan HE, Skorpen F, Elvestad MB, Hveem K, Vatten L, Linseisen J, Clavel-Chapelon F, Vineis P, Bueno-de-Mesquita HB, Lund E, Martinez C, Bingham S, Rasmuson T, Hainaut P, Riboli E, Ahrens W, Benhamou S, Lagiou P, Trichopoulos D, Holcatova I, Merletti F, Kjaerheim K, Agudo A, Macfarlane G, Talamini R, Simonato L, Lowry R, Conway DI, Znaor A, Healy C, Zelenika D, Boland A, Delepine M, Foglio M, Lechner D, Matsuda F, Blanche H, Gut I, Heath S, Lathrop M, Brennan P (2008) A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452(7187):633–637
Liu P, Vikis HG, Wang D, Lu Y, Wang Y, Schwartz AG, Pinney SM, Yang P, de Andrade M, Petersen GM, Wiest JS, Fain PR, Gazdar A, Gaba C, Rothschild H, Mandal D, Coons T, Lee J, Kupert E, Seminara D, Minna J, Bailey-Wilson JE, Wu X, Spitz MR, Eisen T, Houlston RS, Amos CI, Anderson MW, You M (2008) Familial aggregation of common sequence variants on 15q24-25.1 in lung cancer. J Natl Cancer Inst 100(18):1326–1330
Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ (2011) Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature 471(7340):597–601. doi:10.1038/nature09797
Morel C, Fattore L, Pons S, Hay YA, Marti F, Lambolez B, De Biasi M, Lathrop M, Fratta W, Maskos U, Faure P (2013) Nicotine consumption is regulated by a human polymorphism in dopamine neurons. Mol Psychiatry. doi:10.1038/mp.2013.158
Bierut LJ (2009) Nicotine dependence and genetic variation in the nicotinic receptors. Drug Alcohol Depend 104(Suppl 1):S64–S69. doi:10.1016/j.drugalcdep.2009.06.003
Heatherton TF, Kozlowski LT, Frecker RC, Fagerstrom KO (1991) The Fagerstrom test for nicotine dependence: a revision of the Fagerstrom Tolerance Questionnaire. Br J Addict 86(9):1119–1127
Vazquez FL, Torres A, Otero P, Diaz O (2011) Prevalence, comorbidity, and correlates of DSM-IV axis I mental disorders among female university students. J Nerv Ment Dis 199(6):379–383. doi:10.1097/NMD.0b013e31821cd29c
Tobacco and Genetics Consortium. (2010) Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet 42(5):441–447. doi: 10.1038/ng.571
Saccone NL, Culverhouse RC, Schwantes-An TH, Cannon DS, Chen X, Cichon S, Giegling I, Han S, Han Y, Keskitalo-Vuokko K, Kong X, Landi MT, Ma JZ, Short SE, Stephens SH, Stevens VL, Sun L, Wang Y, Wenzlaff AS, Aggen SH, Breslau N, Broderick P, Chatterjee N, Chen J, Heath AC, Heliovaara M, Hoft NR, Hunter DJ, Jensen MK, Martin NG, Montgomery GW, Niu T, Payne TJ, Peltonen L, Pergadia ML, Rice JP, Sherva R, Spitz MR, Sun J, Wang JC, Weiss RB, Wheeler W, Witt SH, Yang BZ, Caporaso NE, Ehringer MA, Eisen T, Gapstur SM, Gelernter J, Houlston R, Kaprio J, Kendler KS, Kraft P, Leppert MF, Li MD, Madden PA, Nothen MM, Pillai S, Rietschel M, Rujescu D, Schwartz A, Amos CI, Bierut LJ (2010) Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS Genet 6(8). doi:10.1371/journal.pgen.1001053
Ware JJ, van den Bree MB, Munafo MR (2011) Association of the CHRNA5-A3-B4 gene cluster with heaviness of smoking: a meta-analysis. Nicotine Tobacco Res Off J Soc Res Nicotine Tobacco 13(12):1167–1175. doi:10.1093/ntr/ntr118
Vandenbergh DJ, Schlomer GL (2014) Finding genomic function for genetic associations in nicotine addiction research: the ENCODE project's role in future pharmacogenomic analysis. Pharmacol Biochem Behav 123:34–44. doi:10.1016/j.pbb.2014.01.009
Saccone NL, Saccone SF, Hinrichs AL, Stitzel JA, Duan W, Pergadia ML, Agrawal A, Breslau N, Grucza RA, Hatsukami D, Johnson EO, Madden PA, Swan GE, Wang JC, Goate AM, Rice JP, Bierut LJ (2009) Multiple distinct risk loci for nicotine dependence identified by dense coverage of the complete family of nicotinic receptor subunit (CHRN) genes. Am J Med Genet B Neuropsychiatr Genet 150B(4):453–466. doi:10.1002/ajmg.b.30828
Saccone NL, Wang JC, Breslau N, Johnson EO, Hatsukami D, Saccone SF, Grucza RA, Sun L, Duan W, Budde J, Culverhouse RC, Fox L, Hinrichs AL, Steinbach JH, Wu M, Rice JP, Goate AM, Bierut LJ (2009) The CHRNA5-CHRNA3-CHRNB4 nicotinic receptor subunit gene cluster affects risk for nicotine dependence in African-Americans and in European-Americans. Cancer Res 69(17):6848–6856. doi:10.1158/0008-5472.CAN-09-0786
Winterer G, Mittelstrass K, Giegling I, Lamina C, Fehr C, Brenner H, Breitling LP, Nitz B, Raum E, Muller H, Gallinat J, Gal A, Heim K, Prokisch H, Meitinger T, Hartmann AM, Moller HJ, Gieger C, Wichmann HE, Illig T, Dahmen N, Rujescu D (2010) Risk gene variants for nicotine dependence in the CHRNA5-CHRNA3-CHRNB4 cluster are associated with cognitive performance. Am J Med Genet B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet 153B(8):1448–1458. doi:10.1002/ajmg.b.31126
Sarginson JE, Killen JD, Lazzeroni LC, Fortmann SP, Ryan HS, Schatzberg AF, Murphy GM Jr (2011) Markers in the 15q24 nicotinic receptor subunit gene cluster (CHRNA5-A3-B4) predict severity of nicotine addiction and response to smoking cessation therapy. Am J Med Genet B Neuropsychiatric Genet Off Publ Int Soc Psychiatric Genet 156B(3):275–284. doi:10.1002/ajmg.b.31155
Siedlinski M, Cho MH, Bakke P, Gulsvik A, Lomas DA, Anderson W, Kong X, Rennard SI, Beaty TH, Hokanson JE, Crapo JD, Silverman EK, Investigators CO, Investigators E (2011) Genome-wide association study of smoking behaviours in patients with COPD. Thorax 66(10):894–902. doi:10.1136/thoraxjnl-2011-200154
Grucza RA, Johnson EO, Krueger RF, Breslau N, Saccone NL, Chen LS, Derringer J, Agrawal A, Lynskey M, Bierut LJ (2010) Incorporating age at onset of smoking into genetic models for nicotine dependence: evidence for interaction with multiple genes. Addict Biol 15(3):346–357. doi:10.1111/j.1369-1600.2010.00220.x
Chen LS, Johnson EO, Breslau N, Hatsukami D, Saccone NL, Grucza RA, Wang JC, Hinrichs AL, Fox L, Goate AM, Rice JP, Bierut LJ (2009) Interplay of genetic risk factors and parent monitoring in risk for nicotine dependence. Addiction 104(10):1731–1740
Xie P, Kranzler HR, Zhang H, Oslin D, Anton RF, Farrer LA, Gelernter J (2012) Childhood adversity increases risk for nicotine dependence and interacts with alpha5 nicotinic acetylcholine receptor genotype specifically in males. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol 37(3):669–676. doi:10.1038/npp.2011.240
Johnson EO, Chen LS, Breslau N, Hatsukami D, Robbins T, Saccone NL, Grucza RA, Bierut LJ (2010) Peer smoking and the nicotinic receptor genes: an examination of genetic and environmental risks for nicotine dependence. Addiction 105(11):2014–2022. doi:10.1111/j.1360-0443.2010.03074.x
Thorgeirsson TE, Gudbjartsson DF, Surakka I, Vink JM, Amin N, Geller F, Sulem P, Rafnar T, Esko T, Walter S, Gieger C, Rawal R, Mangino M, Prokopenko I, Magi R, Keskitalo K, Gudjonsdottir IH, Gretarsdottir S, Stefansson H, Thompson JR, Aulchenko YS, Nelis M, Aben KK, den Heijer M, Dirksen A, Ashraf H, Soranzo N, Valdes AM, Steves C, Uitterlinden AG, Hofman A, Tonjes A, Kovacs P, Hottenga JJ, Willemsen G, Vogelzangs N, Doring A, Dahmen N, Nitz B, Pergadia ML, Saez B, De Diego V, Lezcano V, Garcia-Prats MD, Ripatti S, Perola M, Kettunen J, Hartikainen AL, Pouta A, Laitinen J, Isohanni M, Huei-Yi S, Allen M, Krestyaninova M, Hall AS, Jones GT, van Rij AM, Mueller T, Dieplinger B, Haltmayer M, Jonsson S, Matthiasson SE, Oskarsson H, Tyrfingsson T, Kiemeney LA, Mayordomo JI, Lindholt JS, Pedersen JH, Franklin WA, Wolf H, Montgomery GW, Heath AC, Martin NG, Madden PA, Giegling I, Rujescu D, Jarvelin MR, Salomaa V, Stumvoll M, Spector TD, Wichmann HE, Metspalu A, Samani NJ, Penninx BW, Oostra BA, Boomsma DI, Tiemeier H, van Duijn CM, Kaprio J, Gulcher JR, Consortium E, McCarthy MI, Peltonen L, Thorsteinsdottir U, Stefansson K (2010) Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet 42(5):448–453. doi:10.1038/ng.573
Liu JZ, Tozzi F, Waterworth DM, Pillai SG, Muglia P, Middleton L, Berrettini W, Knouff CW, Yuan X, Waeber G, Vollenweider P, Preisig M, Wareham NJ, Zhao JH, Loos RJ, Barroso I, Khaw KT, Grundy S, Barter P, Mahley R, Kesaniemi A, McPherson R, Vincent JB, Strauss J, Kennedy JL, Farmer A, McGuffin P, Day R, Matthews K, Bakke P, Gulsvik A, Lucae S, Ising M, Brueckl T, Horstmann S, Wichmann HE, Rawal R, Dahmen N, Lamina C, Polasek O, Zgaga L, Huffman J, Campbell S, Kooner J, Chambers JC, Burnett MS, Devaney JM, Pichard AD, Kent KM, Satler L, Lindsay JM, Waksman R, Epstein S, Wilson JF, Wild SH, Campbell H, Vitart V, Reilly MP, Li M, Qu L, Wilensky R, Matthai W, Hakonarson HH, Rader DJ, Franke A, Wittig M, Schafer A, Uda M, Terracciano A, Xiao X, Busonero F, Scheet P, Schlessinger D, St Clair D, Rujescu D, Abecasis GR, Grabe HJ, Teumer A, Volzke H, Petersmann A, John U, Rudan I, Hayward C, Wright AF, Kolcic I, Wright BJ, Thompson JR, Balmforth AJ, Hall AS, Samani NJ, Anderson CA, Ahmad T, Mathew CG, Parkes M, Satsangi J, Caulfield M, Munroe PB, Farrall M, Dominiczak A, Worthington J, Thomson W, Eyre S, Barton A, Wellcome Trust Case Control C, Mooser V, Francks C, Marchini J (2010) Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet 42(5):436–440. doi: 10.1038/ng.572
Albuquerque EX, Pereira EF, Alkondon M, Rogers SW (2009) Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89(1):73–120. doi:10.1152/physrev.00015.2008
Hecht SS (1999) Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst 91(14):1194–1210
Shields PG (2002) Molecular epidemiology of smoking and lung cancer. Oncogene 21(45):6870–6876. doi:10.1038/sj.onc.1205832
Schuller HM (1989) Cell type specific, receptor-mediated modulation of growth kinetics in human lung cancer cell lines by nicotine and tobacco-related nitrosamines. Biochem Pharmacol 38(20):3439–3442
Maneckjee R, Minna JD (1990) Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci U S A 87(9):3294–3298
Amos CI, Gorlov IP, Dong Q, Wu X, Zhang H, Lu EY, Scheet P, Greisinger AJ, Mills GB, Spitz MR (2010) Nicotinic acetylcholine receptor region on chromosome 15q25 and lung cancer risk among African Americans: a case–control study. J Natl Cancer Inst 102(15):1199–1205. doi:10.1093/jnci/djq232
Thorgeirsson TE, Geller F, Sulem P, Rafnar T, Wiste A, Magnusson KP, Manolescu A, Thorleifsson G, Stefansson H, Ingason A, Stacey SN, Bergthorsson JT, Thorlacius S, Gudmundsson J, Jonsson T, Jakobsdottir M, Saemundsdottir J, Olafsdottir O, Gudmundsson LJ, Bjornsdottir G, Kristjansson K, Skuladottir H, Isaksson HJ, Gudbjartsson T, Jones GT, Mueller T, Gottsater A, Flex A, Aben KK, de Vegt F, Mulders PF, Isla D, Vidal MJ, Asin L, Saez B, Murillo L, Blondal T, Kolbeinsson H, Stefansson JG, Hansdottir I, Runarsdottir V, Pola R, Lindblad B, van Rij AM, Dieplinger B, Haltmayer M, Mayordomo JI, Kiemeney LA, Matthiasson SE, Oskarsson H, Tyrfingsson T, Gudbjartsson DF, Gulcher JR, Jonsson S, Thorsteinsdottir U, Kong A, Stefansson K (2008) A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 452(7187):638–642
Jaworowska E, Trubicka J, Lener MR, Masojc B, Zlowocka-Perlowska E, McKay JD, Renard H, Oszutowska D, Wokolorczyk D, Lubinski J, Grodzki T, Serwatowski P, Nej-Wolosiak K, Toloczko-Grabarek A, Sikorski A, Slojewski M, Jakubowska A, Cybulski C, Lubinski J, Scott RJ (2011) Smoking related cancers and loci at chromosomes 15q25, 5p15, 6p22.1 and 6p21.33 in the Polish population. PLoS One 6(9):e25057. doi:10.1371/journal.pone.0025057
Shiraishi K, Kohno T, Kunitoh H, Watanabe S, Goto K, Nishiwaki Y, Shimada Y, Hirose H, Saito I, Kuchiba A, Yamamoto S, Yokota J (2009) Contribution of nicotine acetylcholine receptor polymorphisms to lung cancer risk in a smoking-independent manner in the Japanese. Carcinogenesis 30(1):65–70. doi:10.1093/carcin/bgn257
Timofeeva MN, McKay JD, Smith GD, Johansson M, Byrnes GB, Chabrier A, Relton C, Ueland PM, Vollset SE, Midttun O, Nygard O, Slimani N, Romieu I, Clavel-Chapelon F, Boutron-Ruault MC, Fagherazzi G, Kaaks R, Teucher B, Boeing H, Weikert C, Bueno-de-Mesquita HB, van Gils C, Peeters PH, Agudo A, Barricarte A, Huerta JM, Rodriguez L, Sanchez MJ, Larranaga N, Khaw KT, Wareham N, Allen NE, Travis RC, Gallo V, Norat T, Krogh V, Masala G, Panico S, Sacerdote C, Tumino R, Trichopoulou A, Lagiou P, Trichopoulos D, Rasmuson T, Hallmans G, Riboli E, Vineis P, Brennan P (2011) Genetic polymorphisms in 15q25 and 19q13 loci, cotinine levels, and risk of lung cancer in EPIC. Cancer Epidemiol Biomark Prevent Publ Am Assoc Cancer Res Cosponsored Am Soc Prevent Oncol 20(10):2250–2261. doi:10.1158/1055-9965.EPI-11-0496
Kaur-Knudsen D, Bojesen SE, Tybjaerg-Hansen A, Nordestgaard BG (2011) Nicotinic acetylcholine receptor polymorphism, smoking behavior, and tobacco-related cancer and lung and cardiovascular diseases: a cohort study. J Clin Oncol Off J Am Soc Clin Oncol 29(21):2875–2882. doi:10.1200/JCO.2010.32.9870
Wassenaar CA, Dong Q, Wei Q, Amos CI, Spitz MR, Tyndale RF (2011) Relationship between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 variation and smoking behaviors and lung cancer risk. J Natl Cancer Inst 103(17):1342–1346. doi:10.1093/jnci/djr237
Girard N, Lou E, Azzoli CG, Reddy R, Robson M, Harlan M, Orlow I, Yatabe Y, Nafa K, Ladanyi M, Viale A, Kris MG, Riely G, Miller V, Klein RJ, Matsuo K, Pao W (2010) Analysis of genetic variants in never-smokers with lung cancer facilitated by an Internet-based blood collection protocol: a preliminary report. Clin Cancer Res Off J Am Assoc Cancer Res 16(2):755–763. doi:10.1158/1078-0432.CCR-09-2437
Munafo MR, Timofeeva MN, Morris RW, Prieto-Merino D, Sattar N, Brennan P, Johnstone EC, Relton C, Johnson PC, Walther D, Whincup PH, Casas JP, Uhl GR, Vineis P, Padmanabhan S, Jefferis BJ, Amuzu A, Riboli E, Upton MN, Aveyard P, Ebrahim S, Hingorani AD, Watt G, Palmer TM, Timpson NJ, Group ES, Davey Smith G (2012) Association between genetic variants on chromosome 15q25 locus and objective measures of tobacco exposure. J Natl Cancer Inst 104(10):740–748. doi:10.1093/jnci/djs191
Lips EH, Gaborieau V, McKay JD, Chabrier A, Hung RJ, Boffetta P, Hashibe M, Zaridze D, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Field JK, Liloglou T, Xinarianos G, McLaughlin J, Liu G, Skorpen F, Elvestad MB, Hveem K, Vatten L, Study E, Benhamou S, Lagiou P, Holcatova I, Merletti F, Kjaerheim K, Agudo A, Castellsague X, Macfarlane TV, Barzan L, Canova C, Lowry R, Conway DI, Znaor A, Healy C, Curado MP, Koifman S, Eluf-Neto J, Matos E, Menezes A, Fernandez L, Metspalu A, Heath S, Lathrop M, Brennan P (2010) Association between a 15q25 gene variant, smoking quantity and tobacco-related cancers among 17 000 individuals. Int J Epidemiol 39(2):563–577. doi:10.1093/ije/dyp288
Maes HH, Neale MC, Chen X, Chen J, Prescott CA, Kendler KS (2011) A twin association study of nicotine dependence with markers in the CHRNA3 and CHRNA5 genes. Behav Genet 41(5):680–690. doi:10.1007/s10519-011-9476-z
Sherva R, Wilhelmsen K, Pomerleau CS, Chasse SA, Rice JP, Snedecor SM, Bierut LJ, Neuman RJ, Pomerleau OF (2008) Association of a single nucleotide polymorphism in neuronal acetylcholine receptor subunit alpha 5 (CHRNA5) with smoking status and with 'pleasurable buzz' during early experimentation with smoking. Addiction 103(9):1544–1552
Stephens SH, Hartz SM, Hoft NR, Saccone NL, Corley RC, Hewitt JK, Hopfer CJ, Breslau N, Coon H, Chen X, Ducci F, Dueker N, Franceschini N, Frank J, Han Y, Hansel NN, Jiang C, Korhonen T, Lind PA, Liu J, Lyytikainen LP, Michel M, Shaffer JR, Short SE, Sun J, Teumer A, Thompson JR, Vogelzangs N, Vink JM, Wenzlaff A, Wheeler W, Yang BZ, Aggen SH, Balmforth AJ, Baumeister SE, Beaty TH, Benjamin DJ, Bergen AW, Broms U, Cesarini D, Chatterjee N, Chen J, Cheng YC, Cichon S, Couper D, Cucca F, Dick D, Foroud T, Furberg H, Giegling I, Gillespie NA, Gu F, Hall AS, Hallfors J, Han S, Hartmann AM, Heikkila K, Hickie IB, Hottenga JJ, Jousilahti P, Kaakinen M, Kahonen M, Koellinger PD, Kittner S, Konte B, Landi MT, Laatikainen T, Leppert M, Levy SM, Mathias RA, McNeil DW, Medland SE, Montgomery GW, Murray T, Nauck M, North KE, Pare PD, Pergadia M, Ruczinski I, Salomaa V, Viikari J, Willemsen G, Barnes KC, Boerwinkle E, Boomsma DI, Caporaso N, Edenberg HJ, Francks C, Gelernter J, Grabe HJ, Hops H, Jarvelin MR, Johannesson M, Kendler KS, Lehtimaki T, Magnusson PK, Marazita ML, Marchini J, Mitchell BD, Nothen MM, Penninx BW, Raitakari O, Rietschel M, Rujescu D, Samani NJ, Schwartz AG, Shete S, Spitz M, Swan GE, Volzke H, Veijola J, Wei Q, Amos C, Cannon DS, Grucza R, Hatsukami D, Heath A, Johnson EO, Kaprio J, Madden P, Martin NG, Stevens VL, Weiss RB, Kraft P, Bierut LJ, Ehringer MA (2013) Distinct loci in the CHRNA5/CHRNA3/CHRNB4 gene cluster are associated with onset of regular smoking. Genet Epidemiol 37(8):846–859. doi:10.1002/gepi.21760
De Ruyck K, Nackaerts K, Beels L, Werbrouck J, De Volder A, Meysman M, Salhi B, Van Meerbeeck J, Thierens H (2010) Genetic variation in three candidate genes and nicotine dependence, withdrawal and smoking cessation in hospitalized patients. Pharmacogenomics 11(8):1053–1063. doi:10.2217/pgs.10.75
Breetvelt EJ, Numans ME, Aukes MF, Hoeben W, Strengman E, Luykx JJ, Bakker SC, Kahn RS, Ophoff RA, Boks MP (2012) The association of the alpha-5 subunit of the nicotinic acetylcholine receptor gene and the brain-derived neurotrophic factor gene with different aspects of smoking behavior. Psychiatr Genet 22(2):96–98. doi:10.1097/YPG.0b013e32834c0c75
Breitling LP, Dahmen N, Mittelstrass K, Illig T, Rujescu D, Raum E, Winterer G, Brenner H (2009) Smoking cessation and variations in nicotinic acetylcholine receptor subunits alpha-5, alpha-3, and beta-4 genes. Biol Psychiatry 65(8):691–695. doi:10.1016/j.biopsych.2008.10.004
Breitling LP, Twardella D, Hoffmann MM, Witt SH, Treutlein J, Brenner H (2010) Prospective association of dopamine-related polymorphisms with smoking cessation in general care. Pharmacogenomics 11(4):527–536. doi:10.2217/pgs.10.1
Baker TB, Weiss RB, Bolt D, von Niederhausern A, Fiore MC, Dunn DM, Piper ME, Matsunami N, Smith SS, Coon H, McMahon WM, Scholand MB, Singh N, Hoidal JR, Kim SY, Leppert MF, Cannon DS (2009) Human neuronal acetylcholine receptor A5-A3-B4 haplotypes are associated with multiple nicotine dependence phenotypes. Nicotine Tob Res 11(7):785–796. doi:10.1093/ntr/ntp064
Freathy RM, Ring SM, Shields B, Galobardes B, Knight B, Weedon MN, Smith GD, Frayling TM, Hattersley AT (2009) A common genetic variant in the 15q24 nicotinic acetylcholine receptor gene cluster (CHRNA5-CHRNA3-CHRNB4) is associated with a reduced ability of women to quit smoking in pregnancy. Hum Mol Genet 18(15):2922–2927. doi:10.1093/hmg/ddp216
Munafo MR, Johnstone EC, Walther D, Uhl GR, Murphy MF, Aveyard P (2011) CHRNA3 rs1051730 genotype and short-term smoking cessation. Nicot Tobacco Res Off J Soc Res Nicot Tobacco 13(10):982–988. doi:10.1093/ntr/ntr106
Chen LS, Baker TB, Piper ME, Breslau N, Cannon DS, Doheny KF, Gogarten SM, Johnson EO, Saccone NL, Wang JC, Weiss RB, Goate AM, Bierut LJ (2012) Interplay of genetic risk factors (CHRNA5-CHRNA3-CHRNB4) and cessation treatments in smoking cessation success. Am J Psychiatry 169(7):735–742. doi:10.1176/appi.ajp.2012.11101545
Chen LS, Bach RG, Lenzini PA, Spertus JA, Bierut LJ, Cresci S (2014) CHRNA5 variant predicts smoking cessation in patients with acute myocardial infarction. Nicotine Tobacco Res Off J Soc Res Nicotine Tobacco. doi:10.1093/ntr/ntu059
Bierut LJ (2011) Genetic vulnerability and susceptibility to substance dependence. Neuron 69(4):618–627. doi:10.1016/j.neuron.2011.02.015
Wang JC, Grucza R, Cruchaga C, Hinrichs AL, Bertelsen S, Budde JP, Fox L, Goldstein E, Reyes O, Saccone N, Saccone S, Xuei X, Bucholz K, Kuperman S, Nurnberger J Jr, Rice JP, Schuckit M, Tischfield J, Hesselbrock V, Porjesz B, Edenberg HJ, Bierut LJ, Goate AM (2009) Genetic variation in the CHRNA5 gene affects mRNA levels and is associated with risk for alcohol dependence. Mol Psychiatry 14(5):501–510. doi:10.1038/mp.2008.42
Doyle GA, Chou AD, Saung WT, Lai AT, Lohoff FW, Berrettini WH (2014) Identification of CHRNA5 rare variants in African-American heavy smokers. Psychiatr Genet 24(3):102–109. doi:10.1097/YPG.0000000000000029
Wessel J, McDonald SM, Hinds DA, Stokowski RP, Javitz HS, Kennemer M, Krasnow R, Dirks W, Hardin J, Pitts SJ, Michel M, Jack L, Ballinger DG, McClure JB, Swan GE, Bergen AW (2010) Resequencing of nicotinic acetylcholine receptor genes and association of common and rare variants with the Fagerstrom test for nicotine dependence. Neuropsychopharmacology 35(12):2392–2402. doi:10.1038/npp.2010.120
Haller G, Druley T, Vallania FL, Mitra RD, Li P, Akk G, Steinbach JH, Breslau N, Johnson E, Hatsukami D, Stitzel J, Bierut LJ, Goate AM (2012) Rare missense variants in CHRNB4 are associated with reduced risk of nicotine dependence. Hum Mol Genet 21(3):647–655. doi:10.1093/hmg/ddr498
Haller G, Li P, Esch C, Hsu S, Goate AM, Steinbach JH (2014) Functional characterization improves associations between rare non-synonymous variants in CHRNB4 and smoking behavior. PLoS One 9(5):e96753. doi:10.1371/journal.pone.0096753
Liang Y, Salas R, Marubio L, Bercovich D, De Biasi M, Beaudet AL, Dani JA (2005) Functional polymorphisms in the human beta4 subunit of nicotinic acetylcholine receptors. Neurogenetics 6(1):37–44. doi:10.1007/s10048-004-0199-7
Slimak MA, Ables JL, Frahm S, Antolin-Fontes B, Santos-Torres J, Moretti M, Gotti C, Ibanez-Tallon I (2014) Habenular expression of rare missense variants of the beta4 nicotinic receptor subunit alters nicotine consumption. Front Hum Neurosci 8:12. doi:10.3389/fnhum.2014.00012
Kuryatov A, Berrettini W, Lindstrom J (2011) Acetylcholine receptor (AChR) alpha5 subunit variant associated with risk for nicotine dependence and lung cancer reduces (alpha4beta2)(2)alpha5 AChR function. Mol Pharmacol 79(1):119–125. doi:10.1124/mol.110.066357
Wang JC, Cruchaga C, Saccone NL, Bertelsen S, Liu P, Budde JP, Duan W, Fox L, Grucza RA, Kern J, Mayo K, Reyes O, Rice J, Saccone SF, Spiegel N, Steinbach JH, Stitzel JA, Anderson MW, You M, Stevens VL, Bierut LJ, Goate AM, Collaborators C, Collaborators G (2009) Risk for nicotine dependence and lung cancer is conferred by mRNA expression levels and amino acid change in CHRNA5. Hum Mol Genet 18(16):3125–3135. doi:10.1093/hmg/ddp231
Hong LE, Hodgkinson CA, Yang Y, Sampath H, Ross TJ, Buchholz B, Salmeron BJ, Srivastava V, Thaker GK, Goldman D, Stein EA (2010) A genetically modulated, intrinsic cingulate circuit supports human nicotine addiction. Proc Natl Acad Sci U S A 107(30):13509–13514. doi:10.1073/pnas.1004745107
Wang N, Orr-Urtreger A, Chapman J, Rabinowitz R, Nachman R, Korczyn AD (2002) Autonomic function in mice lacking alpha5 neuronal nicotinic acetylcholine receptor subunit. J Physiol 542(Pt 2):347–354
Wang N, Orr-Urtreger A, Chapman J, Rabinowitz R, Korczyn AD (2003) Deficiency of nicotinic acetylcholine receptor beta 4 subunit causes autonomic cardiac and intestinal dysfunction. Mol Pharmacol 63(3):574–580
Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou CN, Patrick J, Role L, De Biasi M, Beaudet AL (1999) Megacystis, mydriasis, and ion channel defect in mice lacking the alpha3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A 96(10):5746–5751
De Biasi M, Salas R (2008) Influence of neuronal nicotinic receptors over nicotine addiction and withdrawal. Exp Biol Med 233(8):917–929. doi:10.3181/0712-MR-355
Sheffield EB, Quick MW, Lester RA (2000) Nicotinic acetylcholine receptor subunit mRNA expression and channel function in medial habenula neurons. Neuropharmacology 39(13):2591–2603
Fowler CD, Tuesta L, Kenny PJ (2013) Role of alpha5* nicotinic acetylcholine receptors in the effects of acute and chronic nicotine treatment on brain reward function in mice. Psychopharmacology. doi:10.1007/s00213-013-3235-1
Jackson KJ, Marks MJ, Vann RE, Chen X, Gamage TF, Warner JA, Damaj MI (2010) Role of alpha5 nicotinic acetylcholine receptors in pharmacological and behavioral effects of nicotine in mice. Pharmacol Exp Therap 334(1):137–146. doi:10.1124/jpet.110.165738
Frahm S, Slimak MA, Ferrarese L, Santos-Torres J, Antolin-Fontes B, Auer S, Filkin S, Pons S, Fontaine JF, Tsetlin V, Maskos U, Ibanez-Tallon I (2011) Aversion to nicotine is regulated by the balanced activity of beta4 and alpha5 nicotinic receptor subunits in the medial habenula. Neuron 70(3):522–535. doi:10.1016/j.neuron.2011.04.013
Kenny PJ, Markou A (2001) Neurobiology of the nicotine withdrawal syndrome. Pharmacol Biochem Behav 70(4):531–549
Damaj MI, Kao W, Martin BR (2003) Characterization of spontaneous and precipitated nicotine withdrawal in the mouse. J Pharmacol Exp Therapeut 307(2):526–534. doi:10.1124/jpet.103.054908
Doherty K, Kinnunen T, Militello FS, Garvey AJ (1995) Urges to smoke during the first month of abstinence: relationship to relapse and predictors. Psychopharmacology 119(2):171–178
Parrott AC (1993) Cigarette smoking: effects upon self-rated stress and arousal over the day. Addict Behav 18(4):389–395
Salas R, Pieri F, De Biasi M (2004) Decreased signs of nicotine withdrawal in mice null for the beta4 nicotinic acetylcholine receptor subunit. J Neurosci Off Soc Neurosci 24(45):10035–10039
Salas R, Sturm R, Boulter J, De Biasi M (2009) Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci Off J Soc Neurosci 29(10):3014–3018. doi:10.1523/JNEUROSCI. 4934-08.2009
Jackson KJ, Martin BR, Changeux JP, Damaj MI (2008) Differential role of nicotinic acetylcholine receptor subunits in physical and affective nicotine withdrawal signs. J Pharmacol Exp Therapeut 325(1):302–312. doi:10.1124/jpet.107.132977
Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP (2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci Off J Soc Neurosci 21(5):1452–1463
Acknowledgments
We thank Dr. David L. Bronson for excellent editing of this manuscript. The preparation of this review was supported in part by the Research Center for Air Pollution and Health of Zhejiang University, Ministry of Science and Technology of China (2012AA020405), National Natural Science Foundation of China grant 81273223, Young Scientists Fund of National Science Foundation of China (81301140), and NIH grant DA012844.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Wen, L., Jiang, K., Yuan, W. et al. Contribution of Variants in CHRNA5/A3/B4 Gene Cluster on Chromosome 15 to Tobacco Smoking: From Genetic Association to Mechanism. Mol Neurobiol 53, 472–484 (2016). https://doi.org/10.1007/s12035-014-8997-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8997-x