
Abstract
Modulation of spinal reactive gliosis following peripheral nerve injury (PNI) is a promising strategy to restore synaptic homeostasis. Oxidized ATP (OxATP), a nonselective antagonist of purinergic P2X receptors, was found to recover a neuropathic behavior following PNI. We investigated the role of intraperitoneal (i.p.) OxATP treatment in restoring the expression of neuronal and glial markers in the mouse spinal cord after sciatic spared nerve injury (SNI). Using in vivo two-photon microscopy, we imaged Ca2+ transients in neurons and astrocytes of the dorsal horn of spinal cord at rest and upon right hind paw electrical stimulation in sham, SNI, and OxATP-treated mice. Neuropathic behavior was investigated by von Frey and thermal plantar test. Glial [glial fibrillary acidic protein (GFAP), ionized calcium-binding adaptor molecule 1 (Iba1)] and GABAergic [vesicular GABA transporter (vGAT) and glutamic acid decarboxylase 65/76 (GAD65/67)] markers and glial [glutamate transporter (GLT1) and GLAST] and neuronal amino acid [EAAC1, vesicular glutamate transporter 1 (vGLUT1)] transporters have been evaluated. In SNI mice, we found (i) increased glial response, (ii) decreased glial amino acid transporters, and (iii) increased levels of neuronal amino acid transporters, and (iv) in vivo analysis of spinal neurons and astrocytes showed a persistent increase of Ca2+ levels. OxATP administration reduced glial activation, modulated the expression of glial and neuronal glutamate/GABA transporters, restored neuronal and astrocytic Ca2+ levels, and prevented neuropathic behavior. In vitro studies validated that OxATP (i) reduced levels of reactive oxygen species (ROS), (ii) reduced astrocytic proliferation, (iii) increase vGLUT expression. All together, these data support the correlation between reactive gliosis and perturbation of the spinal synaptic homeostasis and the role played by the purinergic system in modulating spinal plasticity following PNI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is now well accepted that glial cells are active players of synaptic transmission and plasticity in the central nervous system (CNS) [1–5] through the formation of a wide neuroglial network [6–8]. Activated glial cells perturb a neuroglial network and a synaptic homeostasis through the production and release of glutamate, ATP, and cytokines, thus contributing to alteration of the glutamate/GABA balance and related uptake mechanisms [9, 10]. This suggests that reactive glial cells contribute to circuit dysfunction and induce maladaptive plasticity (i.e., anatomical and functional changes) in synaptic circuits, leading to abnormal synaptic transmission [11]. This condition is now called gliopathy [12–17] and has been implicated in the development and maintenance of persistent pain states following peripheral nerve injury (PNI) [18–20].
Evidence from experimental models indicated the activation of the purinergic system as a mechanism modulating pain perception, suggesting that purinergic neuroglial interactions are key modulators of sensory neurotransmission [21]. In particular, ATP has been implicated in neuropathic pain and inflammation [22–24] and ATP P2X receptors (P2XRs) (i) are abundantly expressed in spinal neurons and glial cells (mainly expressing P2X7 receptor) [25–27], (ii) modulate the Ca2+ increase and the intercellular propagation of astroglial Ca2+ signals [28–30], (iii) are overexpressed under conditions of reactive gliosis [31, 32], (iv) mediate production and release of cytokines [33], and (v) induce activation of matrix metalloproteinases [34] and mechanisms of cell death via caspase activation [35].
These data suggests that P2XRs are a potential target for limiting inflammatory responses, and P2XR antagonists may potentially reduce secondary damage both by directly inhibiting excitatory neuronal damage and by reducing local or systemic inflammatory responses.
To clarify the link between reactive gliosis and purinergic modulation following PNI, we used the sciatic spared nerve injury (SNI) model to induce reactive gliosis in the lumbar spinal cord. According to the methods of a recent work by our group [36], SNI animals were treated with intraperitoneal (i.p.) OxATP, a nonselective P2XR antagonist reported to attenuate proinflammatory response [37] and recover spinal glial response and neuropathic behavior following PNI.
OxATP treatment restored spinal neuroglial network homeostasis following PNI.
Moreover, two-photon in vivo microscopy was used to monitor real-time changes of intracellular Ca2+ levels in spinal neurons and astrocytes in sham and SNI mice after OxATP treatment. Ca2+ levels were significantly increased in spinal neurons and astrocytes of SNI mice and were restored after OxATP treatment. In vitro studies showed that OxATP treatment reduced astrocyte proliferation and basal levels of reactive oxygen species (ROS) in neurons and astrocytes.
Material and Methods
Animals
We used adult (25–35 g; Charles River, Italy) male C57BL6-J mice (n = 45). Animal care was in compliance with the Italian (D.L. 116/92) and EC (O.J. of E.C. L358/1 18/12/86) regulations on the care of laboratory animals.
SNI Model
Sciatic SNI was made according to the methods of Decosterd and Woolf [38]. Briefly, each mouse (n = 30) was anesthetized with chlorohydrate tiletamine (40 mg/kg) during surgery. The sciatic nerve and its three terminal branches (the sural, common peroneal, and tibial nerves) were exposed on the lateral surface of the thigh. The SNI procedure comprised axotomy and tight ligation of the tibial and common peroneal nerves leaving the sural nerve intact. For the sham-operated group (n = 15), nerves were exposed but not truncated. Muscle and skin were closed in the specific layers. Great care was taken to avoid any contact with or stretching of the intact sural nerve.
Animal Treatment
According to our previous protocol [36], after 1 day, SNI mice were treated with i.p. OxATP (6 mg/kg). OxATP was dissolved in sterile distilled water (dH2O), and 100 μl of the resulting solution (containing 0.18 mg of OxATP) was injected i.p. for each mouse. Animals were divided into three groups: (I) group OxATP (n = 15), SNI mice treated with i.p. OxATP daily from days 1 to 21; (II) vehicle (Veh) group (n = 15), SNI animals received i.p. dH2O (vehicle) for 21 days/daily; and (III) sham group (n = 15), sham-operated animals.
Spinal Cord Surgery for In Vivo Imaging
Mice belonging to each experimental group (OxATP, Veh, and sham; n = 5 for each group) were anesthetized intramuscularly with 40 mg chlorohydrate tiletamine, 15 mg xylazine, and 2.5 mg acepromazine per kg/body weight in 0.9 % NaCl solution. The anesthetized mouse was immobilized using a spine hanging device (Narishige) [39, 40]. Vertebral laminae were carefully drilled to expose the lumbar tract of spinal cord, leaving the intact dura to protect the spinal cord. Skin flaps and agar were used for building a reservoir of 37 °C normal ringer solution to prevent drying out of the spinal cord and allow the use of immersion objectives.
Pressure Injection of Ca2+-Sensitive Dye and Sulforhodamine 101
Spinal neurons and astrocytes were loaded via pressurized micropipette injection using the AM system Picospritzer 2000 [41, 42]. Oregon Green Bapta 1-AM (OGB) (50 μg) was mixed with 5 μl of pluronic acid (F-127, 20 % solution in DMSO) (Invitrogen, Italy) and then diluted in 45 μl of ACSF to the final concentration (1 mM) containing 10 μM sulforhodamine 101 (SR101) (Sigma-Aldrich, Italy) for astrocyte labeling [43]. A pressure ejection at 5–10 PSI for 60–90 s from a 2–3-μm patch pipette directly inserted into the spinal cord was used to load OGB/SR101 solution, allowing 1 h for loading.
Hind Paw Stimulation
Two subcutaneous copper needles were inserted into the right hind paw. Stimulus presentation was controlled by an analog-to-digital converter unit (AD instruments, UK) (pulse duration, 10 ms; amplitude, 1 mA; interpulse intervals, 167 ms/6 Hz and 1000 ms/1 Hz). Three 30-s resting periods (R1, R2, and R3) were allowed between two stimulations (S1 and S2) lasting 10 s [44].
In Vivo Imaging of the Mouse Spinal Cord
In vivo imaging of the living spinal cord was accomplished as recently reported by our group [45]. In brief, during imaging sessions, mice were maintained under anesthesia using 1–2 % sevoflurane at 37 °C using a heating pad. We focused in the area of superficial laminae of the dorsal horn of spinal cord at a depth of 200–300 μm from the spinal surface. Two-photon imaging of the spinal cord was performed using a custom-built two-photon laser scanning microscope. It consists of a laser, a variable attenuator filter, a scanning unit, an upright microscope, and a photomultiplier-based detection system. Laser is a tunable titanium–sapphire (Chameleon XR; coherent), whose wavelength range is from 690 to 1040 nm, power about 2.7 W, frequency 76 MHz. It was tuned at 810 nm to efficiently excite SR101 and OGB. To minimize photodamage, the excitation laser intensity was kept at a minimum for a sufficient signal-to-noise ratio (15–20 mW at the sample). The upright microscope is an Olympus BX51WI and is equipped with the water-immersion objective lens (Olympus) XLumPlanFL 20XW, 0.95 NA. The scanning unit is the modified Olympus FV300. Fluorescence detection system has two channels that allow the simultaneous detection of two fluorescence signals. In order to efficiently collect SR101 and OGB signals, our system is equipped with a 570-nm dichroic mirror and band-pass barrier filters placed in front of the PMTs (505–550 nm for OGB channel and 585–675 nm for SR101 channel).
Breathing and heartbeat movement’s artifacts were reduced by keeping the spine blocked in the hanging apparatus and overcome by triggering image acquisition from the mouse heartbeat (PowerLab AD Instruments). Imaging frame rate is 30 Hz; frame duration is 3.3 s.
The brightness and contrast of the acquired images were adjusted. To reduce the background noise associated with photon or photomultiplier tube noise, a median filter (radius, 1 pixel) was applied to each image. ImageJ free software was used (version 1.42; NIH, USA, http://rsbweb.nih.gov/ij/).
Image Processing
Movies were imported into ImageJ, and fluorescence traces were analyzed in spinal neurons, astrocytes, and surrounding neuropil and expressed as relative change (ΔF/F) after background subtraction by exporting data to SigmaPlot 10.0 program (SPSS, Erkrath, Germany). As extensively reported [46], OGB fluorescence intensity (ΔF/F 0) directly correlated to intracellular Ca2+ levels. Neuropil region of interest (nROI) was drawn according to this main criterion—the absence of cellular body expressed as SR101 or OGB-positive elements in five consecutive planes.
Data were expressed as the mean ± standard error of the mean (SEM). Multigroup comparisons were made using an ANOVA with post hoc t tests. A p value ≤0.05 was considered statistically significant.
Behavioral Tests
Animals were habituated to the testing environment daily for at least 2 days before baseline testing. Treated SNI mice (n = 10 for each group: OxATP, Veh, and sham) were tested on day 0 (the day of the SNI); day 1 (the day of the first i.p. injection of OxATP); days 7, 14, and 21 (the day of the last i.p. injection of OxATP); and day 28, when all animals were sacrificed.
Thermal nociceptive threshold was measured using Hargreaves’ device [47]. Paw withdrawal latency in response to radiant heat (infrared) was assessed using the plantar test apparatus (Ugo Basile). The heat source was positioned under the plantar surface of the affected hind paw and activated at a setting of 4.0. The intensity of the infrared light beam was chosen to give baseline latencies of 15 s in control mice. A cutoff time of 20 s was imposed to prevent tissue damage. The injured hind limb was tested twice at each time point, with an interval of 5 min between stimulations. Mechanical allodynia was assessed using von Frey filaments (Ugo Basile). Briefly, animals were allowed to habituate for 30 min before testing. The time of response to a progressive force applied to hind paw limb (30 g in 20 s) was evaluated six times on the injured hind limb, with an interval of 5 min between stimulations. The threshold was the lowest force that evoked a consistent, brisk, withdrawal response. All testing was performed with a blind procedure.
Tissue Preparation
On day 28, mice were sacrificed. Mice were deeply anesthetized by i.p. injection of chloral hydrate (300 mg/kg body weight) and perfused transcardially with saline solution (Tris–HCl 0.1 M/EDTA 10 mM) followed by 4 % paraformaldehyde added to 0.1 % glutaraldehyde in 0.01 M phosphate buffer (PB), pH 7.4, at 4 °C. Spinal cords were removed and post fixed 2 h in the same fixative, then soaked in 30 % sucrose phosphate-buffered saline (PBS), and frozen in chilled isopentane on dry ice. Serial sections were cut at the slide microtome (25 μm thickness) and collected in cold PBS for immunohistochemistry.
Antibodies
The following antibodies have been used for immunodetection: mouse antibodies directed against glial fibrillary acidic protein (GFAP) (1:400; Sigma-Aldrich, Milan, Italy); rabbit antibodies to ionized calcium-binding adaptor molecule 1 (Iba1) (1:500; Wako Chemicals, USA); guinea pig antibodies to glutamate transporter (GLT1-1) (1:2000; Chemicon, Inc., Temecula, CA, USA); guinea pig antibodies raised against vesicular glutamate transporter 1 (vGLUT1) (1:5000; Chemicon, Inc., Temecula, CA, USA); mouse antibodies to vesicular GABA transporter (vGAT) (1:500; Synaptic Systems, Gottingen, Germany); rabbit antibodies against glutamic acid decarboxylase 65/67 (GAD65/67) (1:1000; Sigma-Aldrich, Milan, Italy); goat antibodies to neuronal glutamate transporter EAAC1 (1:2000; Chemicon, Inc., Temecula, CA, USA).
Spinal Cord Immunohistochemistry
Lumbar (L4–L6) spinal cord sections were blocked in 10 % normal serum (from animal species different from the species origin of the primary antibody used) in 0.01 M PBS/0.25 % Triton for 1 h at room temperature. Each primary antibody (GFAP, Iba1, GLAST, GLT1-1, and EAAC1) was diluted in 0.01 M PBS containing 10 % normal serum and 0.25 % Triton. Following incubation for 48 h at 4 °C, sections were washed six times (10 min each) in PBS and incubated with the appropriate biotinylated secondary antibody (1:200; Vector Labs, Inc., Burlingame, CA, USA) for 90 min at room temperature, washed in PBS, and processed using the Vectastain avidin–biotin peroxidase kit (Vector Labs, Inc., Burlingame, CA, USA) for 90 min at room temperature. Sections were then washed in 0.05 M Tris–HCl and reacted with 3,3-diaminobenzidine tetrahydrochloride (DAB; 0.5 mg/ml in Tris–HCl; Sigma) and 0.01 % hydrogen peroxide. Sections were mounted on chromealum gelatin-coated slides, dehydrated, and coverslipped. Immunofluorescence staining was performed as described previously [48]. Sections were incubated with the primary antibody (vGLUT1, vGAT, GAD65/67) for 48 h at 4 °C. Following washes with PBS, sections were incubated with the appropriate secondary antibody (Alexa Fluor 488 anti-guinea pig IgG, Alexa Fluor 546 anti-mouse IgG, Alexa Fluor 546 anti-rabbit IgG; 1:200; Invitrogen, Carlsbad, CA, USA) for 2 h. Sections were mounted and coverslipped with Vectashield (Vector Laboratories).
Confocal Microscopy
vGLUT, vGAT, and GAD65/67 expression levels were analyzed by using a laser scanning microscope Zeiss LSM 510 Meta (Oberkochen, Germany). Confocal images of dorsal horns of lumbar spinal cord were acquired and captured at a resolution of 512 × 512 pixels. The appropriate argon laser fluorescence for visualization of the vGLUT1 was used with an excitation wavelength of 488 nm and band-pass filter emission 505–530 nm. The HeNe laser fluorescence for the GAD65/67 and vGAT signal with an excitation wavelength of 546 nm and long-pass filter emission 560 nm was used. Gain, laser power, and pinhole aperture were set to a standardized level and left at that setting. A series (n = 27) of single sections were acquired at 0.54 μm apart.
Measurements and Statistical Analysis
Slides were imaged with a Zeiss Axioskop 2 light microscope equipped with high-resolution digital camera (C4742-95; Hamamatsu Photonics, Italy). Measurements of markers in the whole dorsal horn of spinal cord were accomplished using a computer-assisted image analysis system (MCID 7.0; Imaging Research, Inc., Canada). For glial markers, we preferred a morphometric approach because of the perfect visualization of single positive elements. Therefore, values of GFAP and Iba1, markers for astrocytes and microglia, respectively, were expressed as proportional areas (number of positive elements relative to the scanned area). The densitometric values of GLT1-1, GLAST, and EAAC1 were expressed as the total target measured area relative to the scanned area. For confocal images analysis, instead, we used the densitometric method by MCID (density per standardized 1 μm2 area) for quantization of vGLUT1, vGAT, and GAD65/67. Averages were obtained from five randomly selected spinal cord sections for each animal, and comparisons were made between treatments (group OxATP) versus control groups (Veh/sham). Data were exported and converted to frequency distribution histograms by using the SigmaPlot 10.0 program (SPSS Erkrath Germany). Data of all the quantitative analyses were analyzed by one-way ANOVA using all pairwise Holm–Sidak method for multiple comparisons (*p ≤ 0.01; **p ≤ 0.001). All data shown were presented as the mean ± SEM. Single images of control and treated mice were assembled, and then the same adjustments were made for brightness, contrast, and sharpness using Adobe Photoshop (Adobe Systems, San Jose, CA, USA).
Primary Cortical Neurons and Astrocytes
Cortical neurons were prepared as previously described [49] with some modifications. Briefly, cortices were dissected from neonatal (P1–P2) CD1 mice (Harlan Laboratories, Italy), washed in a dissociation medium and digested by trypsin (0.15 %) with deoxyribonuclease (DNAse, 1 mg/ml; Sigma) at 37 °C for 20 min. After mechanical dissociation, cells (1 × 106/ml) were plated onto poly-d-lysine (1 mg/ml)-coated dishes in a Neurobasal medium (NB; Invitrogen, Italy) containing B27 (Invitrogen, Italy), bFGF 10 ng/ml (Invitrogen, Italy), glutamine 1 mM (Sigma), and antibiotics (Sigma). Cultures were maintained at 37 °C in 5 % CO2 and used after 8 days in vitro (DIV).
Primary astrocytes were prepared from cortices of neonatal (P1–P2) mice (Harlan Laboratories, Italy) as previously described [50] with some modifications. After dissection, cortices were washed in Hank’s balanced salt solution supplemented with HEPES/Na pH 7.4 (10 mM), followed by dissociation in trypsin (2.5 mg/ml) with DNase (1 mg/ml) for 15 min at 37 °C. Astrocytes were maintained in 75-cm2 flasks in Basal Medium Eagle (BME, Sigma) supplemented with 10 % fetal bovine serum (FBS) and antibiotics, at 37 °C and 5 % CO2. Pure cultures (>99 %) of type 1 astrocytes were obtained by shaking flasks at 200 rpm at 37 °C to remove type 2 astrocytes, microglial cells, and oligodendrocytes. At confluence, cells were plated onto poly-d-lysine-coated dishes. Astrocytic activation was achieved by treatment with tumor necrosis factor alpha (TNF-α) (10 ng/ml). Mixed cultures of astrocytes and microglia (2 %) activated with lipopolysaccharides (LPSs, 1 μg/ml) were also used.
To evaluate the purity of primary cultures (99–99.5 %), cells were plated onto a 12-mm poly-d-lysine-coated coverslip (5000/well) and assessed by immunocytochemistry with anti-βIII tubulin (Cell Signaling), rabbit anti-GFAP (1:500, Millipore), and anti-Iba1 (1:50, Abcam) as described in [51].
Astrocyte Proliferation
Cortical astrocytes or mixed glial cells (astrocytes containing about 2 % of microglia) were plated onto 35-mm poly-d-lysine-coated dishes (5000 cells/well). Cells were changed to serum-free medium for 48 h before addition of the growth media containing TNF-α (10 ng/ml) or LPS (1 μg/ml) in the presence or absence of OxATP (100 μM). At specific time points (2, 5, 6, 7, 9, 12, and 14 days), cells were detached with trypsin (0.25 %) and the number of viable cells was counted by trypan blue exclusion.
Bromodeoxyuridine ELISA Cell Proliferation Assay
Bromodeoxyuridine (BrdU) incorporation analysis was performed by using the BrdU Cell Proliferation Assay (Chemicon) in 96 multiwell plates. Cells were plated onto poly-d-lysine-coated wells (2000 cells/well), synchronized by serum starvation for 48 h and exposed to the growth media containing TNF-α (10 ng/ml) or LPS (1 μg/ml) in the presence or absence of OxATP (100 μM). BrdU (10 μM) was added to the wells during the last 24 h of treatment, and plates were processed according to the manufacturer’s instructions. Samples were read in a microplate reader at 450 nm.
Cell Viability
Cell survival was analyzed by the methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay (Sigma). Reduction of the yellow tetrazolium salts (MTT) to the purple formazan is dependent on the activity of mitochondrial dehydrogenases by intact mitochondria and can also be taken as an index of metabolic mitochondrial activity. The assay was performed according to the manufacturer’s specifications. Briefly, cortical neurons or astrocytes (5000 cells/well) were plated in 96-well plates (EuroClone) pre-coated with poly-d-lysine. Following treatments, tetrazolium salts (0.5 mg/ml) were added directly to the culture medium for 4 h at 37 °C. After incubation, 100 μl of MTT solubilization buffer (100 μl) was added to each well for 18 h. The absorbance of samples was measured at a wavelength of 570 nm (700 nm reference wavelength) with a microplate reader (Bio-Rad). MTT conversion levels were expressed as a percentage of control.
Determination of ROS
Intracellular production of ROS in cortical neurons and astrocytes was assessed as previously described [51] by using the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Molecular Probes, Invitrogen). DCFH-DA is cleaved by cellular esterases to the nonfluorescent DCFH, which is oxidized by peroxides to the highly fluorescent compound, 2′,7′-dichlorofluorescein (DCF). DCFH-DA (10 μM) was added during the last 30 min of treatments. Cells were then washed with PBS, harvested in 0.25 % trypsin, and analyzed by FACS (FACScan, Becton-Dickinson), using the Cell Quest software (BD Bioscience). Flow cytometric measurements were taken with 10,000 cells contained in the gated regions used for calculations. Data analysis was performed with WinMDI software.
Western Blot Analysis
Total cell extracts and Western blotting (WB) were performed as previously described [19, 51, 52]. Briefly, cells were washed and scraped in ice-cold PBS and resuspended in lysis buffer (20 mM Tris pH 8.0; 137 mM NaCl; 1 % Nonidet-P40; 10 % glycerol; 1 mM dithiothreitol (DTT)) containing protease inhibitors (2 mM PMSF, 0.1 μg/ml leupeptin, 5 μg/ml aprotinin) and a phosphatase inhibitor cocktail (PhosStop, Roche). Following 20 min of incubation on ice, cellular debris were pelleted by centrifugation at 14,000g for 10 min at 4 °C. Protein concentration was determined by Bio-Rad protein assay.
Cell lysates (20–25 μg total protein) and lumbar spinal cord tissues were dissolved in loading buffer (50 mM Tris, pH 6.8; 2 % sodium dodecyl sulfate (SDS); 100 mM DTT; 10 % glycerol; 0.1 % bromophenol blue), separated on 12 % SDS-PAGE gels and transferred to nitrocellulose (Schleicher & Schuell). After blocking with 5 % nonfat milk in Tris-buffered saline with Tween (TBST) buffer (10 mM Tris, pH 7.5; 150 mM NaCl; 0.2 % Tween 20), blots were probed overnight at 4 °C with mouse vGLUT antibody (1:5000; Synaptic System) and mouse P2X7R antibody (1:500; Abcam) in TBST, followed by incubation for 1 h at RT with HRP-conjugated donkey anti-mouse IgG (1:10,000; Amersham Biotech). Detection was carried out by using the enhanced chemiluminescence system (ECL; Amersham Biosciences). Quantification of results was performed by densitometry using NIH ImageJ software. There was no significant difference in the density of actin-loading control bands between groups.
Results
OxATP-Induced Reduction of Reactive Gliosis in the Lumbar Spinal Cord Following SNI
Based on the relevance of spinal neuroglial network rearrangement following PNI, we have used the SNI model to induce reactive gliosis in the spinal cord and investigate the role of P2XRs in modulating reactive gliosis-induced perturbation of synaptic homeostasis.
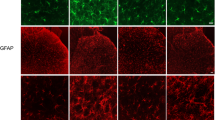
We analyzed sections of dorsal horns of lumbar spinal cord for expression levels of morphological glial markers. Analysis of spinal cord sections revealed the presence of marked gliosis 28 days after SNI, as demonstrated by the strong increase of astrocytic GFAP staining (20.56 ± 0.67) and microglial Iba1 levels (19.69 ± 0.72) (Fig. 1(a, b)) in SNI animals, as compared to the sham group (7.37 ± 0.58 and 5.40 ± 0.33 for GFAP and Iba1, respectively) (**p ≤ 0.001).

Evaluation of glial markers in the dorsal horn of spinal cord. Sections of the dorsal horns of lumbar spinal cords were prepared from sham and SNI animals treated for 21 days with OxATP (6 mg/kg) or vehicle and immunostained for GFAP (a) and Iba1 (b). Data are the mean ± SEM (n = 10 for each group); (**p ≤ 0.001, Veh vs OxATP, sham vs Veh; ANOVA and Holm–Sidak test). Scale bar = 50 μm
We then investigated the role of OxATP administration in the counteracting glial reaction following SNI. i.p. administration of OxATP for 21 days reduced GFAP and Iba1 levels to 10.90 ± 0.94 and 6.79 ± 0.62, respectively (Fig. 1(a, b)), as compared to Veh animals (**p ≤ 0.001).
These data clearly indicate that OxATP reduced both astrocytosis and microglial reaction in the lumbar spinal cord.
OxATP Reduces Astrocytic Proliferation
Proliferation of astrocytes and microglia is a key process of neuroinflammation following brain injury [53]. To assess whether the decrease of glial reaction by OxATP was due to its activity on glial proliferation, we analyzed the effect of OxATP on cortical astrocytes activated by TNF-α or mixed astroglial cells (astrocytes containing about 2 % of microglia) treated with LPS. Cells were first synchronized by serum starvation for 48 h and then switched back to the growth media containing TNF-α (10 ng/ml) or LPS (1 μg/ml) in the presence or absence of OxATP (100 μM). As shown in Fig. 2a, the cell number was dramatically increased during a 14-day time course both in astrocytes cultured in the presence of TNF-α (10 ng/ml) and, to a lesser extent, in LPS-treated glial cultures. Interestingly, we found that chronic co-treatment with OxATP (100 μM) significantly prevented astrocyte proliferation induced by TNF-α (Fig. 2a) as well as the growth of mixed glial cells treated with LPS (1 μg/ml), as compared to their corresponding treatments with TNF-α or LPS alone. The effect of OxATP on glial proliferation was further investigated in the presence of BrdU during a 10-day time course. Data in Fig. 2b show that chronic co-treatment with OxATP (100 μM) determined a strong reduction of BrdU incorporation both in TNF-α-activated astrocytes and in LPS-treated glial cultures, as compared to their corresponding treatments with TNF-α or LPS alone. These data indicate that the decrease of cell number elicited by OxATP was not the consequence of decreased survival.

Reduction of astrocyte proliferation by OxATP. a Cell count of astrocytes or mixed glial cells treated with TNF-α (10 ng/ml) or LPS (1 μg/ml), respectively, in the presence or absence of OxATP (100 μg/ml) for the indicated times. Data are the mean ± SEM of five independent experiments. b Proliferation rate of astrocytes or mixed glial cells stimulated with TNF-α (10 ng/ml) or LPS (1 μg/ml), respectively, in the presence/absence of OxATP (100 μg/ml) for the indicated times. BrdU (10 μM) was added during the last 24 h of treatments. Data are the mean ± SEM of three independent experiments, each performed with four to six samples for each treatment
OxATP Improves Neuronal and Astrocytic Viability
The effect of OxATP on astrocytic viability was further assessed by MTT assay. We found that treatment with OxATP (100 μM) up to 72 h did not change the survival of cortical astrocytes, which instead was strongly decreased by exposure for 16 h (ON) to 200 μM H2O2, as a positive control (Fig. 3a). It was also interesting to note that OxATP (100 μM) did not affect the survival of cortical neurons, in contrast to the well-known toxic effect of glutamate (200 μM) which lead to a 50 % reduction of neuronal viability after 16-h treatments (Fig. 3a) [54]. Intriguingly, instead, flow cytometry analysis using 2′,7′-dichlorofluorescein diacetate (DCF-DA) staining revealed that treatment of cortical neurons with OxATP (100 μM) for 6 h determined a 40 % reduction of basal levels of ROS (Fig. 3b). A similar effect was also found to occur in cultured astrocytes: FACS fluorescence profiles in Fig. 3c show that OxATP (100 μM) was able to both decrease basal ROS levels and prevent ROS production induced by a 6-h exposure of astrocytes to hydrogen peroxide (H2O2, 100 μM) (Fig. 3c, d).

Effect of OxATP on neuronal and astrocytic viability. a Survival by MTT assay of cortical neurons and astrocytes following treatment with OxATP (100 μg/ml) for 72 h. Treatments with H2O2 (200 μM, dashed bar) or glutamate (200 μM, dotted bar) for 16 h were used as positive controls for astrocytes and neurons, respectively. Data, expressed as a percentage of control (CTR), are the mean ± SEM of three independent experiments, each performed with four to six samples for each treatment. b–d Measurement of ROS levels in neurons (b) and astrocytes (d) under basal conditions or following treatment with OxATP for 6 h. As a positive control, astrocytes were treated with H2O2 (100 μM) for 6 h. Cells were loaded with DCFH-DA (10 μM) during the last 30 min of treatments and analyzed by FACS. Flow cytometric measurements were taken on 10,000 cells. Data, expressed as a percentage of CTR, are the mean ± SEM of three experiments in duplicate (*p ≤ 0.05; **p ≤ 0.001, versus their respective CTR; ANOVA and Dunnett’s test). c Representative FACS fluorescence profiles of astrocytes treated with H2O2 (100 μM) in the presence or absence of OxATP (100 μM)
OxATP-Induced Modulation of Glial and Neuronal Glutamate/GABA Transporter Expression
Based on the role of astrocytes in glutamate reuptake and maintenance of synaptic homeostasis [55–57], we examined whether mechanisms of reactive gliosis following SNI involved alteration of glial transporters. IHC analyses revealed a decrease of GLAST and GLT1 expression levels in SNI animals (19.40 ± 2.19 and 37.66 ± 3.50, respectively), as compared to sham group (26.61 ± 3.02 and 78.58 ± 11.19, respectively) (*p ≤ 0.01; **p ≤ 0.001) (Fig. 4(a, b)).

Neuronal and glial amino acid transporter expression. Sections of the dorsal horns of lumbar spinal cords were prepared from sham and SNI animals treated with OxATP (6 mg/kg) or dH2O and immunostained for glial glutamate GLAST (a) and GLT1 (b) transporters or the neuronal glutamate transporter EAAC1 (c). Data are expressed as the mean ± SEM (*p ≤ 0.01; **p ≤ 0.001). Scale bar = 50 μm
These data indicate that mechanisms of reactive gliosis involve alteration of gliotransmission. Interestingly, the reduction of glial glutamate transporters (gGTs) in SNI animals was counterbalanced by an increase of the neuronal glutamate transporter EAAC1 (19.49 ± 1.30), as compared to sham group (15.07 ± 0.77) (*p ≤ 0.01; **p ≤ 0.001) (Fig. 4(c)).
In parallel with its potential in reducing astrocytes and microglial reaction, we found that i.p. administration of OxATP to SNI animals reduced the expression of GLAST and GLT1 (13.13 ± 1.70 and 28.59 ± 3.51, respectively) but also restored the expression of the neuronal transporter EAAC1 (13.46 ± 1.42) compared to Veh group (*p ≤ 0.01; **p ≤ 0.001) (Fig. 4).
To further characterize mechanisms of synaptic homeostasis linked to reactive gliosis, we also analyzed vGAT, vGLUT, and GAD65/67 expression in the spinal cord. Interestingly, no difference was found in vGLUT and GAD 65/67 levels, as revealed by the densitometric values found in sham (vGLUT, 2.88 ± 0.29; GAD 65/67, 8.77 ± 0.68) and Veh groups (vGLUT, 3.11 ± 0.28; GAD 65/67, 7.63 ± 0.84) (Fig. 5(b, c)). In contrast, analysis of spinal sections of SNI animals revealed a significant increase of vGAT (7.24 ± 0.49) compared to sham animals (5.83 ± 0.28) (*p ≤ 0.01) (Fig. 5(a)). i.p. administration of OxATP reduced vGAT (4.57 ± 0.56) and GAD65/67 (4.81 ± 0.64) expression (Fig. 5(a, c)). vGLUT expression levels, instead, were strongly increased by i.p. OxATP treatment (7.72 ± 0.65) (Fig. 5(b)) (*p < 0.01; **p ≤ 0.001).

Expression levels of vGAT, vGLUT, and GAD. Confocal microscopy images of dorsal horns of lumbar spinal cord immunostained for vGAT (a), vGLUT (b), and GAD65 (c). Sections of the dorsal horns of lumbar spinal cords were prepared from sham and SNI animals treated with OxATP (6 mg/kg) or vehicle. Data are expressed as the mean ± SEM (*p < 0.01; **p ≤ 0.001). Scale bar = 50 μm
The specific contribution of neuronal and glial components in response to purinergic inhibition was also evaluated on cortical neurons using the in vitro models of glial activation by LPS or TNF-α. WB analysis in Fig. 6a shows that vGLUT levels were dramatically reduced in cortical neurons exposed for 24 h to the conditioned medium (CM) from astrocytes activated by TNF-α (10 ng/ml) for 48 h. A similar reduction of vGLUT expression was also observed in neurons treated with CM from mixed glial cells treated with LPS (1 μg/ml) (Fig. 6a). These modifications were not observed when neurons were preincubated for 2 h with OxATP (100 μM) before addition of CM-TNF-α or CM-LPS (Fig. 6a–c). A decrease of vGLUT expression was also observed in TNF-α-activated astrocytes for 72 h (Fig. 6b) as well as in mixed glia treated with LPS for 72 h (Fig. 6b), but not after 24-h treatments (data not shown). Interestingly, under all experimental conditions, the reduction of vGLUT levels was reversed by co-treatment with OxATP (100 μM) (Fig. 6b–d). These data clearly suggest that the purinergic system, mainly through P2XRs, modulates the expression of glial and neuronal glutamate transporter vGLUT.

Expression levels of vGLUT in cortical neurons and astrocytes. a Representative Western blot (WB) of cortical neurons treated with CM from TNF-α (10 ng/ml)-activated astrocytes (CM-TNF-α) or CM from mixed glial cells treated with LPS (1 μg/ml) (CM-LPS) in the presence or absence of OxATP (100 μg/ml). b Representative WB of cortical astrocytes or mixed glial cells activated by TNF-α (10 ng/ml) or LPS (1 μg/ml), respectively, with or without OxATP (100 μg/ml). Total lysates were processed by WB using anti-vGLUT antibody. Blots were probed with β-actin to normalize for protein content. c, d Quantification of bands was achieved by NIH ImageJ software, and protein levels, normalized by the actin content, were expressed as a percentage of control (CTR) ± SEM (*p ≤ 0.05; **p ≤ 0.001, versus their respective CTR; ANOVA and Dunnett’s test)
To further investigate the specific contribution of P2XRs to glial activation, WB analysis was used to assess the levels of the glial P2X7 receptor (P2X7R) in sham, SNI, and OxATP-treated mice. In particular, as indicated in Fig. 7, P2X7R levels were found to be significantly increased in SNI animals (0.37 ± 0.01) compared to sham animals (0.19 ± 0.02) (**p ≤ 0.001). A significant decrease of P2 × 7R expression was observed after OxATP treatment (0.23 ± 0.007), as observed in sham animals.

Representative Western blot and quantitative analysis of glial P2 × 7R expression in the lumbar spinal cord in sham, SNI, and OxATP-treated mice. SNI mice showed a significant increase of P2 × 7R, compared to sham animals; OxATP treatment restored receptors’ levels (**p ≤ 0.001). Quantification of bands was achieved by NIH ImageJ software, and protein levels, normalized by the actin content, were expressed as a percentage of control (CTR) ± SEM (**p ≤ 0.001)
OxATP treatment, in conclusion, was found to modulate the expression of glial purinergic receptors.
In Vivo Ca2+ Imaging of Dorsal Horn Spinal Astrocytes
Two-photon Ca2+ imaging of astrocytes in superficial laminae (I–II) of the dorsal horn of lumbar spinal cord (Fig. 8(a)) was performed at rest and in response to right hind limb stimulation at 1 and 6 Hz in sham mice.

Sensory stimulation triggers Ca2+ increase in spinal astrocytes. a Superficial laminae of the dorsal horn of lumbar spinal cord, showing different astrocytes in rest condition (scale bar = 10 μm). nROI indicates the selected ROI for density measures of the surrounding neuropil. b, c Ca2+ levels (ΔF/F 0) at rest and during 1/6-Hz stimulation in neurons, astrocytes, and neuropil. Peripheral stimulation induces Ca2+ increase in astrocytes at 1 Hz; in contrast, neuronal and neuropil Ca2+ levels increase when stimulation occurs at 6 Hz. The difference between resting and stimulation time scales is due to different durations of the session (30 s for resting and 10 s for stimulation). Imaging frame rate is 30 Hz; frame duration is 3.3 s
Sensory stimulation at 1 Hz increased the mean Ca2+-related fluorescence value (ΔF/F 0) of astrocytes compared to rest condition (S1 vs R1 and S2 vs R2, **p ≤ 0.001) (Fig. 8(b)). Stimulation of the hind paw at 6 Hz did not evoke a significant Ca2+ increase in spinal astrocytes compared to resting (Table 1, Fig. 8(c)). i.p. treatment with OxATP in sham mice did not significantly change Ca2+ levels in spinal astrocytes (data not shown).
To test whether Ca2+ transients from astrocytes were contaminated by signals from surrounding neuropil, we assessed the stimulus-driven Ca2+ increase in absolute fluorescence (in raw gray levels) in responsive astrocytes and the surrounding neuropil (nROI), according to the methods described by Winship et al. [58].
We found that, at the stimulation frequency of astrocytic response (1 Hz), the mean increase of OGB fluorescence in astrocytes (0.53 ± 0.19) was significantly higher than that found in the neuropil ROI after stimulation in the same frames (0.12 ± 0.07) (**p ≤ 0.001) (Fig. 8(b)). This result prompted us to rule out any influence of neuropil Ca2+ dynamic on the astrocytic Ca2+ transients recorded in the dorsal horn of spinal cord.
Astrocytic Ca2+ Imaging in SNI and OxATP-Treated Mice
In SNI animals, spinal astrocytes showed a significant Ca2+ increase during 6-Hz sensory stimulation compared to the rest (S1 vs R1, **p ≤ 0.001). Then Ca2+ levels slightly decreased during the remaining imaging session did not show any clear fluctuation and remained significantly higher than the baseline (R1) at the end of the imaging session (S1 vs R3, *p ≤ 0.01) (Table 1, Fig. 9).

Average Ca2+ levels (ΔF/F 0) at rest and during 1/6-Hz stimulation in neurons and astrocytes of sham, SNI, and OxATP-treated mice. SNI induced changes of Ca2+ dynamics in neurons and astrocytes. i.p. OxATP treatment fully restored Ca2+ levels as observed in sham animals (*p ≤ 0.01; **p ≤ 0.001); (black asterisks significance for astrocytes; blue asterisks significance for neurons; red asterisks astrocytes vs neurons)
The 1-Hz stimulation of hind paw induced an increase of astrocytic Ca2+ levels only after the first stimulation, becoming significantly high at the second stimulation (S2) compared to rest (R1) (R2/S2 vs R1, **p ≤ 0.001) (Table 1, Fig. 9).
According to the role of the purinergic system in glutamate-induced Ca2+ release [59, 60] and synchronization of Ca2+ transients [61], we used OxATP treatment to evaluate changes of astrocytic and neuronal Ca2+ levels.
In SNI animals, astrocytic Ca2+ behavior and levels were restored by OxATP treatment compared to rest conditions, as observed in sham animals (Table 1, Fig. 9).
Neuronal Ca2+ Imaging in Sham, SNI, and OxATP-Treated Mice
According to our imaging protocol, we analyzed the spinal neurons in the same image planes that in sham animals showed Ca2+ responses when stimulated at 6 Hz. The mean Ca2+-related fluorescence value during rest and stimulation tasks showed that sensory stimulation increased Ca2+ concentration in neurons compared to the corresponding resting periods (S1/S2 vs R1/R3, **p ≤ 0.001) (Table 2, Fig. 8). As expected, we also observed a concomitant and significant increase of Ca2+ levels in the surrounding neuropil during stimulations (0.39 ± 0.03) compared to rest (0.16 ± 0.03; p ≤ 0.001) (Fig. 8).
A stimulus-driven Ca2+ elevation during or after 1-Hz hind limb stimulation was not observed in spinal neurons, as compared to resting values. i.p. treatment with OxATP in sham mice did not significantly change the Ca2+ levels in spinal neurons (data not shown).
In SNI animals, at both frequencies, Ca2+ levels in spinal neurons showed a significant increase compared to rest after the first stimulation (S1 vs R1, **p ≤ 0.001), progressively decreasing during the remaining imaging session. After the first stimulus (S1), Ca2+ levels were significantly higher than the baseline before stimulation (R1), but lower than those found in astrocytes in the same conditions (Table 2, Fig. 9).
i.p. OxATP treatment fully restored the neuronal Ca2+ dynamics to that observed in sham animals. We found an increase of Ca2+ levels during 6-Hz stimulations compared to rest (S1/S2 vs R1/R3, **p ≤ 0.001). Instead, a stimulus-driven Ca2+ elevation at 1 Hz was not observed in spinal neurons and we did not detect significant differences with the resting values (Table 2, Fig. 9).
OxATP Improves Behavioral Recovery Following PNI
Animals were tested for neuropathic behavior on days 0, 1, 7, 14, 21, and 28 after SNI surgery by analyzing thermal and mechanical sensitivity.
The mean baseline for normal mechanical threshold, recorded before SNI (day 0) was 27.75 ± 0.43 g. In sham animals, this value remained virtually unmodified during all 28 days (Fig. 10a). SNI mice, in contrast, showed a significant reduction in mechanical nociceptive threshold on days after surgery, presenting an early response indicative of an allodynic state (day 1, 13.68 ± 0.56 g; day 7, 13.87 ± 0.72 g; day 14, 14.46 ± 0.73 g; day 21, 14.93 ± 0.62 g; day 28, 15.25 ± 0.61 g). In the SNI mice, daily i.p. infusion of OxATP for 21 days restored mechanical sensitivity (day 7, 16.40 ± 0.47 g; day 14, 18.62 ± 0.62 g; day 21, 22.87 ± 0.41 g; day 28, 25.84 ± 0.65 g) compared to vehicle-treated animals (**p ≤ 0.001) (Fig. 10a).

Prevention of SNI-induced neuropathic behavior by i.p. OxATP treatment. Neuropathic mice were tested by von Frey (a) and plantar (b) tests for baseline sensitivity (day 0) and 1, 7, 14, 21, and 28 days after OxATP (6 mg/kg) administration. Data are the mean ± SEM (**p ≤ 0.001, Veh vs OxATP (ANOVA and Holm–Sidak test) (n = 10 for each group)
The Hargreaves test in SNI mice also showed a strong reduction of the reaction time to the thermal stimulus 1, 7, 14, 21, and 28 days after injury with a very short time response to infrared stimulation (day 1, 8.10 ± 0.14 s; day 7, 8.41 ± 0.23 s; day 14, 8.73 ± 0.28 s; day 21, 8.18 ± 0.19 s; day 28, 8.82 ± 0.23 s) compared to basal values of 14.33 ± 0.25 s, recorded in sham animals, and before surgery (Fig. 10b), indicating the onset of a hyperalgesic state. OxATP treatment for 21 days significantly restored thermal sensitivity (day 7, 11.42 ± 0.34 s; day 14, 12.96 ± 0.34 s; day 21, 13.05 ± 0.35; day 28, 13.58 ± 0.45) (**p ≤ 0.001) (Fig. 10b).
These data suggest that antagonism of the purinergic system prevents neuropathic behavior.
Discussion
Decades of studies regarding the mechanisms of neuropathic pain following PNI have demonstrated that not only altered neuronal plasticity but also aberrant functions of glial cells are involved.
Neuroglial plastic changes following PNI have been showed to induce both neuronal/astrocytic activation and alteration of neuroglial interactions, determining maladaptive synaptic plasticity in the spinal somatosensory system that seems to be directly responsible for neuronal hyperexcitability and enhanced/aberrant synaptic transmission that sustain neuropathic pain [62, 63].
As we previously demonstrated [9, 10, 12, 19, 20], we here confirm that SNI was associated with (1) the onset and persistence of reactive gliosis in the lumbar spinal cord (Fig. 1) and (2) changes in the expression of glial and neuronal neurotransmitter transporters as demonstrated by the reduction of gGTs (GLT1 and GLAST) and the increase of both neuronal glutamate transporters EAAC1 (Fig. 4) and vGAT (Fig. 5).
We speculate that the reduction of gGTs might be correlated to glial activation (phenotypic changes), and the increase of EAAC1 might represent the neuronal attempt to reduce extracellular glutamate levels and prevent further excitatory glutamate transmission and excitotoxicity. Similarly, it is conceivable that the increase of vGAT might be an adaptive response of the CNS to counteract hyperexcitability of glutamate transmission, enhancing the GABAergic tone [9, 10, 13]. Behavioral data, indeed, confirmed the onset of neuropathic pain syndrome, as shown by reduced mechanical and thermal paw withdrawal thresholds (Fig. 10).
To directly investigate the spinal plastic changes following PNI and evaluate their modulation by the purinergic system, we used two-photon microscopy to assess in vivo Ca2+ levels and functionally visualize living astrocytes and neurons in the spinal cord both under resting and sensory stimulation conditions.
Furthermore, we measured Ca2+ modifications following PNI and the role played by the purinergic system.
Recently, an increasing number of in vivo studies have demonstrated that astrocytic Ca2+ dynamics play a key role for synaptic plasticity and modulate synaptic transmission by triggering neurotransmitter release [58, 64, 65], suggesting that astrocytes actively regulate neuroglial network. Moreover, hind limb-selective sensory-evoked Ca2+ signals in cortical astrocytes were found to operate on a timescale similar to neurons and ex vivo studies showed that astrocytic Ca2+ signaling is critical to the bidirectional communication between neurons and astrocytes [5, 6]. Our data suggest that peripheral sensory stimulation is associated with Ca2+ increase in spinal dorsal horn neurons and astrocytes of sham animals (Fig. 8). PNI-induced reactive gliosis alters Ca2+ dynamics in spinal neurons and astrocytes (Figs. 8 and 9) with a persistent increase of Ca2+ levels, when stimulated at both frequencies.
It has been reported that purinergic-mediated glial functions make a critical contribution to pathologically enhanced pain processing in the dorsal horns [21, 36, 66], suggesting that glial purinergic receptors might be promising targets for treating neuropathic pain [67]. Among purinergic receptors, the P2XR subtype was reported to modulate synaptic plasticity [68], intracellular Ca2+ concentrations [69], neurotransmitter release, and glial response [70] and, nonetheless, to increase GFAP expression with elongation of astrocytic processes, cellular proliferation, microglial activation, and release of proinflammatory cytokines (e.g., TNF-α, IL-1β) [71–73].
To better understand the role of the purinergic system in modulating/restoring glial reaction and its involvement in the maintenance of synaptic homeostasis, we used the systemic administration of OxATP (a nonselective P2XR antagonist) to inhibit P2X7R signaling, limit inflammatory response, and confer neuroprotection [71, 74].
The aim of the current work was to further understand the morpho-functional molecular correlates of pain recovery. In line with the results of a recent work by our group [36] that validated the experimental design (the fixed dose of 6 mg/kg of OxATP and the i.p. administration) and the efficacy of this dialdehydic compound in the prevention of the neuropathic behavior.
In particular, we here confirm that i.p. administration of OxATP for 21 days restores GFAP and Iba1 levels (Fig. 1), showing that this treatment is associated to the (i) reduction of gGT expression (Fig. 4), (ii) increased vGLUT expression (Fig. 5), (iii) restored EAAC1 and vGAT levels (Figs. 4 and 5), and (iv) recovery of Ca2+ dynamics in neurons and astrocytes (Figs. 8 and 9), suggesting that the purinergic system plays a key role in regulating neuroglial reaction as well as homeostasis of spinal Ca2+ levels.
These morphological and molecular changes were fitly paralleled by the behavioral recovery of mice, as mechanical and thermal paw withdrawal thresholds were fully restored by OxATP treatment (Fig. 10). Accordingly, a recent work first demonstrated that persistent pain and LTP of spinal nociceptive pathways was correlated to hyperalgesia [75], suggesting that modulation of spinal plasticity represents a putative therapeutic strategy to chronic pain.
In line with other works, phenotypic changes of reactive glial cells include upregulation of P2XRs, in particular the P2X7 receptor (Fig. 7) [28–30], and selective blockade of P2XRs has been shown to attenuate microglial activation and inflammation [24, 37, 76]. P2XR inhibition by OxATP reduced glial proliferation induced by TNF-α and LPS (Fig. 2) without affecting cell survival (Fig. 3), downregulated P2X7 (Fig. 7), while lowered neuronal and astrocytic ROS production, both under basal conditions and following oxidative stress (Fig. 3). This effect might be linked to the role of OxATP in modulating Ca2+ influx through P2XRs [77].
Together, these data suggest an anti-inflammatory and anti-gliotic action of OxATP, resulting from the blockade of P2XR expression and activity.
In line with its potential anti-gliosis effect [36], we here report that OxATP treatment for 21 days induced a significant reduction of gGT levels in dorsal horns of spinal cord (Fig. 4), most likely due to the decreased glutamate release.
Therefore, we suggest that the inhibition of P2XRs may allow the recovery of astrocytic gGT function, modulating the glutamatergic and GABAergic transmission and increasing the transporter levels, as previously postulated [78].
Actually, OxATP-mediated blockade of neuronal presynaptic P2XRs [79] has been previously shown to reduce glutamate and GABA release [80], vesicular fusion, and exocytosis [81]. Accordingly, in the spinal cord of OxATP-treated mice, we found a significant increase of vGLUT expression, as demonstrated also in vitro. Restoring vGLUT levels involved both astrocytic and neuronal compartments, while vGLUT expression was dramatically reduced by TNF-α/LPS and CM from activated glia. Thus OxATP-mediated reduction of glutamate release may account for the adaptive downregulation of gGTs (GLAST and GLT1) and restored glutamatergic and GABAergic function, as indicated by the reduction of expression levels of vGAT and GAD65/67 (Fig. 5).
Taken together, our results indicate that OxATP, through P2XRs, modulates glial and neuronal glutamate/GABA transporters following PNI, inducing long-lasting plastic changes of spinal neuroglial circuitry. It is now well accepted that modifications of neuroglial network strongly contribute to mental disorders [82–84] and neurodegenerative diseases [11, 85–88]. Therefore, targeting P2XRs may be helpful to contrast neuroinflammatory reaction and restore neuroglial homeostasis.
In conclusion, our findings confirm the importance of phenotypic glial changes following PNI, based on (i) reduction of gGTs with a concurrent impaired glutamatergic function and perturbation of synaptic circuitry homeostasis and (ii) further decrease of glutamate uptake, thus affecting glial function of synaptic protection against excitotoxicity.
Moreover, we show that sensory stimulation is associated with Ca2+ increase in both spinal neurons and astrocytes, though Ca2+ dynamics are altered in condition of reactive gliosis. Restoring Ca2+ levels antagonizing P2XRs supports that Ca2+ behavior is firmly modulated by the purinergic system in conditions of reactive gliosis and maladaptive plasticity.
Therefore, the purinergic system seems to play a key role in regulating neuroglial reaction and spinal Ca2+ dynamics. Moreover, our data strongly support the relevance of OxATP in modulating glial protective function and its role in preserving the synaptic circuitry through modulation of glutamatergic and GABAergic components.
In conclusion, these data extend current understanding of reactive gliosis mechanisms in synaptic plasticity following PNI, shedding light on the emerging role of the purinergic system and glial components (astrocytes and microglia) in modulating homeostasis of synaptic circuits both under physiological and pathological conditions.
References
Pascual O, Ben AS, Rostaing P, Triller A, Bessis A (2012) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A 109:E197–E205
Perea G, Araque A (2007) Astrocytes potentiate transmitter release at single hippocampal synapses. Science 317:1083–1086
Angulo MC, Kozlov AS, Charpak S, Audinat E (2004) Glutamate released from glial cells synchronizes neuronal activity in the hippocampus. J Neurosci 24:6920–6927
Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G (2004) Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron 43:729–743
Haydon PG (2001) GLIA: listening and talking to the synapse. Nat Rev Neurosci 2:185–193
Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N (2010) Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat Rev Neurosci 11:87–99
Verkhratsky A (2010) Physiology of neuronal-glial networking. Neurochem Int 57:332–343
Allen NJ, Barres BA (2009) Neuroscience: glia—more than just brain glue. Nature 457:675–677
Cirillo G, Colangelo AM, Bianco MR, Cavaliere C, Zaccaro L, Sarmientos P, Alberghina L, Papa M (2012) BB14, a nerve growth factor (NGF)-like peptide shown to be effective in reducing reactive astrogliosis and restoring synaptic homeostasis in a rat model of peripheral nerve injury. Biotechnol Adv 30:223–232
Cirillo G, Bianco MR, Colangelo AM, Cavaliere C, De Luca D, Zaccaro L, Alberghina L, Papa M (2011) Reactive astrocytosis-induced perturbation of synaptic homeostasis is restored by nerve growth factor. Neurobiol Dis 41:630–639
Papa M, De Luca C, Petta F, Alberghina L, Cirillo G (2014) Astrocyte-neuron interplay in maladaptive plasticity. Neurosci Biobehav Rev 42C:35–54
Colangelo AM, Cirillo G, Lavitrano ML, Alberghina L, Papa M (2012) Targeting reactive astrogliosis by novel biotechnological strategies. Biotechnol Adv 30:261–271
Gwak YS, Hulsebosch CE (2011) GABA and central neuropathic pain following spinal cord injury. Neuropharmacology 60:799–808
Gwak YS, Kang J, Unabia GC, Hulsebosch CE (2012) Spatial and temporal activation of spinal glial cells: role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp Neurol 234:362–372
Hulsebosch CE (2008) Gliopathy ensures persistent inflammation and chronic pain after spinal cord injury. Exp Neurol 214:6–9
Lobsiger CS, Cleveland DW (2007) Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci 10:1355–1360
Pekny M, Nilsson M (2005) Astrocyte activation and reactive gliosis. Glia 50:427–434
Milligan ED, Watkins LR (2009) Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci 10:23–36
Colangelo AM, Bianco MR, Vitagliano L, Cavaliere C, Cirillo G, De Gioia L, Diana D, Colombo D, Redaelli C, Zaccaro L, Morelli G, Papa M, Sarmientos P, Alberghina L, Martegani E (2008) A new nerve growth factor-mimetic peptide active on neuropathic pain in rats. J Neurosci 28:2698–2709
Cirillo G, Cavaliere C, Bianco MR, De Simone A, Colangelo AM, Sellitti S, Alberghina L, Papa M (2010) Intrathecal NGF administration reduces reactive astrocytosis and changes neurotrophin receptors expression pattern in a rat model of neuropathic pain. Cell Mol Neurobiol 30:51–62
Inoue K, Tsuda M (2012) Purinergic systems, neuropathic pain and the role of microglia. Exp Neurol 234:293–301
Gourine AV, Dale N, Llaudet E, Poputnikov DM, Spyer KM, Gourine VN (2007) Release of ATP in the central nervous system during systemic inflammation: real-time measurement in the hypothalamus of conscious rabbits. J Physiol 585:305–316
Khakh BS, North RA (2006) P2X receptors as cell-surface ATP sensors in health and disease. Nature 442:527–532
Peng W, Cotrina ML, Han X, Yu H, Bekar L, Blum L, Takano T, Tian GF, Goldman SA, Nedergaard M (2009) Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc Natl Acad Sci U S A 106:12489–12493
Illes P, Verkhratsky A, Burnstock G, Franke H (2012) P2X receptors and their roles in astroglia in the central and peripheral nervous system. Neuroscientist 18:422–438
Koles L, Leichsenring A, Rubini P, Illes P (2011) P2 receptor signaling in neurons and glial cells of the central nervous system. Adv Pharmacol 61:441–493
Wang X, Arcuino G, Takano T, Lin J, Peng WG, Wan P, Li P, Xu Q, Liu QS, Goldman SA, Nedergaard M (2004) P2X7 receptor inhibition improves recovery after spinal cord injury. Nat Med 10:821–827
Zhou D, Chen ML, Zhang YQ, Zhao ZQ (2010) Involvement of spinal microglial P2X7 receptor in generation of tolerance to morphine analgesia in rats. J Neurosci 30:8042–8047
Hamilton N, Vayro S, Kirchhoff F, Verkhratsky A, Robbins J, Gorecki DC, Butt AM (2008) Mechanisms of ATP- and glutamate-mediated calcium signaling in white matter astrocytes. Glia 56:734–749
Narcisse L, Scemes E, Zhao Y, Lee SC, Brosnan CF (2005) The cytokine IL-1beta transiently enhances P2X7 receptor expression and function in human astrocytes. Glia 49:245–258
Rodriguez-Zayas AE, Torrado AI, Rosas OR, Santiago JM, Figueroa JD, Miranda JD (2012) Blockade of P2 nucleotide receptors after spinal cord injury reduced the gliotic response and spared tissue. J Mol Neurosci 46:167–176
Rodriguez-Zayas AE, Torrado AI, Miranda JD (2010) P2Y2 receptor expression is altered in rats after spinal cord injury. Int J Dev Neurosci 28:413–421
Shiratori M, Tozaki-Saitoh H, Yoshitake M, Tsuda M, Inoue K (2010) P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J Neurochem 114:810–819
Delarasse C, Auger R, Gonnord P, Fontaine B, Kanellopoulos JM (2011) The purinergic receptor P2X7 triggers alpha-secretase-dependent processing of the amyloid precursor protein. J Biol Chem 286:2596–2606
Hanley PJ, Kronlage M, Kirschning C, del RA, Di VF, Leipziger J, Chessell IP, Sargin S, Filippov MA, Lindemann O, Mohr S, Konigs V, Schillers H, Bahler M, Schwab A (2012) Transient P2X7 receptor activation triggers macrophage death independent of Toll-like receptors 2 and 4, caspase-1, and pannexin-1 proteins. J Biol Chem 287:10650–10663
Bianco MR, Cirillo G, Petrosino V, Marcello L, Soleti A, Merizzi G, Cavaliere C, Papa M (2012) Neuropathic pain and reactive gliosis are reversed by dialdehydic compound in neuropathic pain rat models. Neurosci Lett 530:85–90
Beigi RD, Kertesy SB, Aquilina G, Dubyak GR (2003) Oxidized ATP (oATP) attenuates proinflammatory signaling via P2 receptor-independent mechanisms. Br J Pharmacol 140:507–519
Decosterd I, Woolf CJ (2000) Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87:149–158
Johannssen HC, Helmchen F (2010) In vivo Ca2+ imaging of dorsal horn neuronal populations in mouse spinal cord. J Physiol 588:3397–3402
Davalos D, Lee JK, Smith WB, Brinkman B, Ellisman MH, Zheng B, Akassoglou K (2008) Stable in vivo imaging of densely populated glia, axons and blood vessels in the mouse spinal cord using two-photon microscopy. J Neurosci Methods 169:1–7
Stosiek C, Garaschuk O, Holthoff K, Konnerth A (2003) In vivo two-photon calcium imaging of neuronal networks. Proc Natl Acad Sci U S A 100:7319–7324
Golshani P, Portera-Cailliau C (2008) In vivo 2-photon calcium imaging in layer 2/3 of mice. J Vis Exp
Nimmerjahn A, Kirchhoff F, Kerr JN, Helmchen F (2004) Sulforhodamine 101 as a specific marker of astroglia in the neocortex in vivo. Nat Methods 1:31–37
Sanganahalli BG, Herman P, Blumenfeld H, Hyder F (2009) Oxidative neuroenergetics in event-related paradigms. J Neurosci 29:1707–1718
Cirillo G, De Luca D, Papa M (2012) Calcium imaging of living astrocytes in the mouse spinal cord following sensory stimulation. Neural Plast 2012:425818
Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ (2009) Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323:1211–1215
Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32:77–88
Papa M, Boscia F, Canitano A, Castaldo P, Sellitti S, Annunziato L, Taglialatela M (2003) Expression pattern of the ether-a-gogo-related (ERG) K+ channel-encoding genes ERG1, ERG2, and ERG3 in the adult rat central nervous system. J Comp Neurol 466:119–135
Gullo F, Maffezzoli A, Dossi E, Wanke E (2009) Short-latency cross- and autocorrelation identify clusters of interacting cortical neurons recorded from multi-electrode array. J Neurosci Methods 181:186–198
Consonni A, Morara S, Codazzi F, Grohovaz F, Zacchetti D (2011) Inhibition of lipopolysaccharide-induced microglia activation by calcitonin gene related peptide and adrenomedullin. Mol Cell Neurosci 48:151–160
Bianco MR, Berbenni M, Amara F, Viggiani S, Fragni M, Galimberti V, Colombo D, Cirillo G, Papa M, Alberghina L, Colangelo AM (2011) Cross-talk between cell cycle induction and mitochondrial dysfunction during oxidative stress and nerve growth factor withdrawal in differentiated PC12 cells. J Neurosci Res 89:1302–1315
Cavaliere C, Cirillo G, Bianco MR, Rossi F, De Novellis V, Maione S, Papa M (2007) Gliosis alters expression and uptake of spinal glial amino acid transporters in a mouse neuropathic pain model. Neuron Glia Biol 3:141–153
Robel S, Berninger B, Götz M (2011) The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci 12:88–104
Bachis A, Colangelo AM, Vicini S, Doe PP, De Bernardi MA, Brooker G, Mocchetti I (2001) Interleukin-10 prevents glutamate-mediated cerebellar granule cell death by blocking caspase-3-like activity. J Neurosci 21(9):3104–3112
Henneberger C, Rusakov DA (2010) Synaptic plasticity and Ca2+ signalling in astrocytes. Neuron Glia Biol 1–6
Hamilton NB, Attwell D (2010) Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci 11:227–238
Ni Y, Parpura V (2009) Dual regulation of Ca2+-dependent glutamate release from astrocytes: vesicular glutamate transporters and cytosolic glutamate levels. Glia 57:1296–1305
Winship IR, Plaa N, Murphy TH (2007) Rapid astrocyte calcium signals correlate with neuronal activity and onset of the hemodynamic response in vivo. J Neurosci 27:6268–6272
Jiang M, Chen G (2009) Ca2+ regulation of dynamin-independent endocytosis in cortical astrocytes. J Neurosci 29:8063–8074
Bardoni R, Ghirri A, Zonta M, Betelli C, Vitale G, Ruggieri V, Sandrini M, Carmignoto G (2010) Glutamate-mediated astrocyte-to-neuron signalling in the rat dorsal horn. J Physiol 588:831–846
Koizumi S (2010) Synchronization of Ca2+ oscillations: involvement of ATP release in astrocytes. FEBS J 277:286–292
Gwak YS, Hulsebosch CE (2009) Remote astrocytic and microglial activation modulates neuronal hyperexcitability and below-level neuropathic pain after spinal injury in rat. Neuroscience 161:895–903
Wang W, Wang W, Mei X, Huang J, Wei Y, Wang Y, Wu S, Li Y (2009) Crosstalk between spinal astrocytes and neurons in nerve injury-induced neuropathic pain. PLoS One 4:e6973
Fellin T, Carmignoto G (2004) Neurone-to-astrocyte signalling in the brain represents a distinct multifunctional unit. J Physiol 559:3–15
Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86:1009–10031
Trang T, Beggs S, Salter MW (2012) ATP receptors gate microglia signaling in neuropathic pain. Exp Neurol 234:354–361
Tsuda M, Tozaki-Saitoh H, Inoue K (2012) Purinergic system, microglia and neuropathic pain. Curr Opin Pharmacol 12:74–79
Pankratov Y, Lalo U, Krishtal OA, Verkhratsky A (2009) P2X receptors and synaptic plasticity. Neuroscience 158:137–148
Nakatsuka T, Tsuzuki K, Ling JX, Sonobe H, Gu JG (2003) Distinct roles of P2X receptors in modulating glutamate release at different primary sensory synapses in rat spinal cord. J Neurophysiol 89:3243–3252
He WJ, Cui J, Du L, Zhao YD, Burnstock G, Zhou HD, Ruan HZ (2012) Spinal P2X(7) receptor mediates microglia activation-induced neuropathic pain in the sciatic nerve injury rat model. Behav Brain Res 226:163–170
Choi HB, Ryu JK, Kim SU, McLarnon JG (2007) Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J Neurosci 27:4957–4968
Neary JT, Kang Y, Shi YF, Tran MD, Wanner IB (2006) P2 receptor signalling, proliferation of astrocytes, and expression of molecules involved in cell-cell interactions. Novartis Found Symp 276:131–143
Neary JT, Kang Y (2005) Signaling from P2 nucleotide receptors to protein kinase cascades induced by CNS injury: implications for reactive gliosis and neurodegeneration. Mol Neurobiol 31:95–103
Fulgenzi A, Dell’Antonio G, Foglieni C, Dal Cin E, Ticozzi P, Franzone JS, Ferrero ME (2005) Inhibition of chemokine expression in rat inflamed paws by systemic use of the antihyperalgesic oxidized ATP. BMC Immunol 6:18
Bonin RP, De Koninck Y (2014) A spinal analog of memory reconsolidation enables reversal of hyperalgesia. Nat Neurosci 17(8):1043–1045
Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CB, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN (2005) Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain 114:386–396
Kim SY, Moon JH, Lee HG, Kim SU, Lee YB (2007) ATP released from beta-amyloid-stimulated microglia induces reactive oxygen species production in an autocrine fashion. Exp Mol Med 39:820–827
Liu YP, Yang CS, Chen MC, Sun SH, Tzeng SF (2010) Ca(2+)-dependent reduction of glutamate aspartate transporter GLAST expression in astrocytes by P2X(7) receptor-mediated phosphoinositide 3-kinase signaling. J Neurochem 113:213–227
Deuchars SA, Atkinson L, Brooke RE, Musa H, Milligan CJ, Batten TF, Buckley NJ, Parson SH, Deuchars J (2001) Neuronal P2X7 receptors are targeted to presynaptic terminals in the central and peripheral nervous systems. J Neurosci 21:7143–7152
Sperlagh B, Kofalvi A, Deuchars J, Atkinson L, Milligan CJ, Buckley NJ, Vizi ES (2002) Involvement of P2X7 receptors in the regulation of neurotransmitter release in the rat hippocampus. J Neurochem 81:1196–1211
Anderson CM, Nedergaard M (2006) Emerging challenges of assigning P2X7 receptor function and immunoreactivity in neurons. Trends Neurosci 29:257–262
Cavaliere C, Cirillo G, Bianco MR, Adriani W, De Simone A, Leo D, Perrone-Capano C, Papa M (2012) Methylphenidate administration determines enduring changes in neuroglial network in rats. Eur Neuropsychopharmacol 22:53–63
Leo D, Adriani W, Cavaliere C, Cirillo G, Marco EM, Romano E, Di Porzio U, Papa M, Perrone-Capano C, Laviola G (2009) Methylphenidate to adolescent rats drives enduring changes of accumbal Htr7 expression: implications for impulsive behavior and neuronal morphology. Genes Brain Behav 8:356–368
Musholt K, Cirillo G, Cavaliere C, Bianco MR, Bock J, Helmeke C, Braun K, Papa M (2009) Neonatal separation stress reduces glial fibrillary acidic protein- and S100beta-immunoreactive astrocytes in the rat medial precentral cortex. Dev Neurobiol 69:203–211
Cirillo G, Maggio N, Bianco MR, Vollono C, Sellitti S, Papa M (2010) Discriminative behavioral assessment unveils remarkable reactive astrocytosis and early molecular correlates in basal ganglia of 3-nitropropionic acid subchronic treated rats. Neurochem Int 56:152–160
Giovannoni R, Maggio N, Bianco MR, Cavaliere C, Cirillo G, Lavitrano M, Papa M (2007) Reactive astrocytosis and glial glutamate transporter clustering are early changes in a spinocerebellar ataxia type 1 transgenic mouse model. Neuron Glia Biol 3:335–351
Heneka MT, O’Banion MK, Terwel D, Kummer MP (2010) Neuroinflammatory processes in Alzheimer’s disease. J Neural Transm 117:919–947
Zagami CJ, Beart PM, Wallis N, Nagley P, O’Shea RD (2009) Oxidative and excitotoxic insults exert differential effects on spinal motoneurons and astrocytic glutamate transporters: implications for the role of astrogliosis in amyotrophic lateral sclerosis. Glia 57:119–135
Acknowledgments
This work was supported by grants from the Italian Minister of University and Research (PRIN2007) to M.P. and A.M.C.; Sys-BioNet–Italian Roadmap ESFRI Infrastructures to L.A., A.M.C., and M.P.; FIRB-ITALBIONET and NEDD to L.A.; and Blueprint Pharma s.r.l. and PRIMM s.r.l. Research work in the authors’ laboratory is funded by grants from the Regione Campania (L.R. N.5 Bando 2003 and Prog. Spec art 12 E.F. 2000 to M.P.), CNR (Neurobiotecnologie 2003 to M.P.), and Associazione Levi-Montalcini (fellowships to G.C.).
Conflict of Interest
The authors declare that this article has no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Giovanni Cirillo and Anna Maria Colangelo equally contributed to this work.
Rights and permissions
About this article

Cite this article
Cirillo, G., Colangelo, A.M., Berbenni, M. et al. Purinergic Modulation of Spinal Neuroglial Maladaptive Plasticity Following Peripheral Nerve Injury. Mol Neurobiol 52, 1440–1457 (2015). https://doi.org/10.1007/s12035-014-8943-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-014-8943-y