
Abstract
Generation of neurotoxic amyloid β peptides and their deposition along with neurofibrillary tangle formation represent key pathological hallmarks in Alzheimer’s disease (AD). Recent evidence suggests that inflammation may be a third important component which, once initiated in response to neurodegeneration or dysfunction, may actively contribute to disease progression and chronicity. Various neuroinflammatory mediators including complement activators and inhibitors, chemokines, cytokines, radical oxygen species and inflammatory enzyme systems are expressed and released by microglia, astrocytes and neurons in the AD brain. Degeneration of aminergic brain stem nuclei including the locus ceruleus and the nucleus basalis of Meynert may facilitate the occurrence of inflammation in their projection areas given the antiinflammatory and neuroprotective action of their key transmitters norepinephrine and acetylcholine. While inflammation has been thought to arise secondary to degeneration, recent experiments demonstrated that inflammatory mediators may stimulate amyloid precursor protein processing by various means and therefore can establish a vicious cycle. Despite the fact that some aspects of inflammation may even be protective for bystander neurons, antiinflammatory treatment strategies should therefore be considered. Non-steroidal anti-inflammatory drugs have been shown to reduce the risk and delay the onset to develop AD. While, the precise molecular mechanism underlying this effect is still unknown, a number of possible mechanisms including cyclooxygenase 2 or γ-secretase inhibition and activation of the peroxisome proliferator activated receptor γ may alone or, more likely, in concert account for the epidemiologically observed protection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Alzheimer’s disease
As the most common neurodegenerative disorders, Alzheimer’s disease (AD) currently affects 20–30 million individuals worldwide (Selkoe 2005). AD accounts for most cases of dementia that are diagnosed after the age of 60 years of life.
AD brains show two characteristic lesions: extracellular deposits of β-amyloid peptides, so called neuritic or senile plaques, and the intracellular neurofibrillary tangles (NFT) of hyperphosphorylated tau protein (Querfurth and LaFerla 2010).
Toxic β-amyloid peptides (Aβ) are generated by the sequential action of two proteases denoted as β-secretase (BACE1) and γ-secretase, which cleave the amyloid precursor protein (APP). Aβ exists with different carboxyl endings, from which Aβ1–40 and Aβ1–42 appear to be the major subtypes deposited in the brain. Aβ peptides can also be detected in normal cerebrospinal fluid and in conditioned media from various tissue culture cell lines (Shoji et al. 1992; Haass et al. 1992; Seubert et al. 1992), suggesting that it is constantly produced and constitutively secreted. The importance of Aβ formation was revealed by dominantly inherited familial forms of AD that are linked to APP mutations in or close to the β- and γ-secretase cleavage sites (Hardy and Allsop 1991).
NFT constitute intraneuronal cytoplasmic accumulations of non-membrane-bound bundles of paired helical filaments, whose main component is the hyperphosphorylated form of the Tau protein. Tau is also found in dystrophic neurites. It is found in aggregates conjugated with ubiquitin, a property that it shares with other aggregating intraneuronal proteins, such as α-synuclein. Of importance, Aβ deposits as well as NFT can also be found in other neurodegenerative diseases and even in brains of patients without any history of cognitive or other neurological deficits (Lee et al. 2001), suggesting the contribution of additional factors to fully establish the disease.
The eventual deposition of Aβ and NFT formation may not account for all clinical symptoms of AD, particularly the most early clinical symptoms arising before neuronal degeneration is evident. Inflammatory changes are observed in AD brain overall, particularly at the amyloid deposits, which are rich in activated microglia. Once stimulated, the microglia release a wide variety of pro-inflammatory mediators including cytokines, complement components, various free radicals and nitric oxide (NO), all of which potentially contribute to further neuronal dysfunction and eventually death. These create and feed a vicious cycle that could be essential in the pathological progression of AD (Griffin et al. 1998; Griffin 2000).
Inflammation and AD
Although Aβ has been considered to play a key role in AD pathogenesis (Walsh et al. 2002b; Walsh and Selkoe 2004), it remains still uncertain whether Aβ plaques and NFT are causative for AD. These doubts are being fueled by the finding that the Aβ plaque burden only poorly correlates with the progression and severity of dementia in AD. Moreover, transgenic animals that develop widespread Aβ plaque deposition in response to mutant APP overexpression show only slight cognitive deficits (Braak and Braak 1998; Davis and Laroche 2003). Furthermore, NFT may correlate better with the decline in cognitive skills, but seem to occur as a late event and in some cases possibly downstream of Aβ accumulation. However, some experimental evidence indicates that protofibrils and oligomers of Aβ1–40 and Aβ1–42, rather than concrete Aβ plaques, contribute to early dendritic and synaptic injury and thereby to neuronal dysfunction (Walsh et al. 2002a).
In addition to these direct toxic effects of Aβ1–40 and Aβ1–42 peptides, Aβ may promote neurodegeneration by parallel mechanisms including the activation of microglial cells and astrocytes. The induction of a microglia-driven inflammatory response results in the release of various inflammatory mediators including a whole array of neurotoxic cytokines (Tan et al. 1999; Heneka and O’Banion 2007). Once activated, microglia cells may also recruit astrocytes that actively enhance the inflammatory response to extracellular Aβ deposits. This neuroinflammatory component of AD is further characterized by a local cytokine-mediated acute-phase response, activation of the complement cascade and induction of inflammatory enzyme systems such as the inducible nitric oxide synthase (iNOS) and the prostanoid generating cyclooxygenase-2 (COX-2). Several lines of evidence suggest that all of these factors can contribute to neuronal dysfunction and cell death, either alone or in concert (Abbas et al. 2002; Bezzi et al. 2001a, b; Brown and Bal-Price 2003).
This review will discuss several aspects of neuroinflammation in AD focusing on the following questions:
-
1.
What stimulates the inflammatory reaction in the AD brain?
-
2.
Which cells contribute to the inflammatory component of AD and how do they interact?
-
3.
Which pro- and antiinflammatory mediators are being released in the AD brain, and what is their supposed mechanism of action?
-
4.
Are there any known pathogenetic factors in the AD brain that may facilitate the induction and persistence of neuroinflammatory mechanisms?
-
5.
Is neuroinflammation just a reaction to neurodegenerative events or does it act on neurodegenerative pathomechanisms thereby establishing a vicious and self-perpetuating cycle?
-
6.
Can antiinflammatory treatment strategies serve as a future AD therapy?
Immunostimulators in Alzheimer’s disease
While minor signs of neuroinflammation can be found in the normal aging brain, the AD brain faces a much stronger activation of inflammatory systems indicating that an increasing amount or qualitatively different immunostimulants are present. Cumulative evidence suggests that Aβ peptides play a pivotal role as inducers of neuroinflammation. However, chromogranin A and several other proteins may contribute to this induction.
Amyloid β
The concept that Aβ peptide itself can induce a local inflammatory-type response received impetus from the in vitro findings that fibrillar Aβ can bind the complement factor C1 and hence potentially activate the classical complement pathway in an antibody-independent fashion (Rogers et al. 1992). Such activated early complement factors could play an important role in the local recruitment and activation of microglial cells expressing the complement receptors CR3 and CR4 (Rozemuller et al. 1989; Eikelenboom et al. 1989). In vitro studies indicate that a certain degree of Aβ fibrillization is required for the initiation of the complement system (Snyder et al. 1994). This in vitro finding is consistent with the immunohistochemical data in AD brains showing weak or absent immunostaining for early complement components in diffuse plaques composed of non- or low-grade fibrillar Aβ peptide (Eikelenboom and Veerhuis 1996). The diffuse plaques are not associated with activated microglia and altered neurites, in contrast to the so-called classical and neuritic plaques, which are characterized by congophilic fibrillar Aβ deposits. So, the chronic inflammatory response in AD brains is seen in the plaque containing fibrillar Aβ deposits but not in the diffuse plaque with the non-congophilic low-fibrillar Aβ depositions (Rozemuller et al. 1989; Itagaki et al. 1989). For example, Aβ activates microglia by binding to the receptor for advanced glycation end products (RAGE) (Yan et al. 1998) and to other scavenger receptors (Paresce et al. 1996). Furthermore, the LPS receptor, CD14, interacts with fibrillar Aβ (Fassbender et al. 2004) and microglia kill Aβ1–42 damaged neurons by a CD14 dependent process (Bate et al. 2004). Fibrillar Aβ has been shown to increase cytokine and nitric oxide production in microglia dependent on CD14, TLR2 and TLR4 (Jana et al. 2008; Walter et al. 2007; Reed-Geaghan et al. 2009). Another signalling pathway through which Aβ can promote inflammation has been identified by Stewart et al. (2010). These authors found that Aβ triggers inflammatory signaling through heterodimer formation of Toll-like receptor 4 and 6. Assembly of this heterodimer is regulated by binding of Aβ to scavenger receptor CD36. The involvement of CD14 and/or CD36 and Toll-like receptors in Aβ induced microglia activation strongly suggests that innate immunity is linked with AD pathology. The ability of fibrillar Aβ to trigger an inflammatory response is phenocopied in murine AD models that recapitulate Aβ plaque formation (e.g. Matsuoka et al. 2001; Jimenez et al. 2008). Paradoxically, CD14 and CD36 are among the receptors that are responsible for the clearance and phagocytosis of Aβ by microglia as well (Koenigsknecht and Landreth 2004; Liu et al. 2005). It is conceivable that phagocytosis of Aβ by microglia requires inflammatory signaling, but is obviously worn out during the course of the disease, in accordance with the finding that in 8-month-old APPswe/PS1dE9 mice expression of Aβ binding receptors and degrading enzymes in microglia is reduced (Hickman et al. 2008).
Chromogranin A
Chromogranin A (CGA) represents a secretory 48–53 kDa glycoprotein which is stored and released by neurons in brain regions relevant for several neurodegenerative diseases including AD, Parkinson’s disease and amyotrophic lateral sclerosis. Of note, neuritic plaques intensely stain for CGA in AD (Rangon et al. 2003). Experiments showing that exposure of primary rat microglia to CGA resulted in rapid microglial activation characterized by profound morphological changes from an arborized to an amoeboid phenotype lead to the hypothesis that CGA may act as an important stimulator of neuroinflammation (Taupenot et al. 1996). Microglial activation was accompanied by de novo synthesis of iNOS and subsequent production of NO (Taupenot et al. 1996). Importantly, CGA was equally or more effective in stimulating iNOS derived NO release relative to microglial stimulation with bacterial lipopolysaccharide. Activation of microglial cells with CGA caused neuronal cell death, however, a direct link between NO or tumor necrosis factor α (TNFα) release and neurodegeneration has not been found in this model (Ciesielski-Treska et al. 1998a, b).
Cellular components of neuroinflammation in Alzheimer’s disease
Microglia cells represent the brain innate immune system and hence the first line of defense when challenged by bacterial, viral or fungal infection. Although these functions are of major importance and beneficial, it has become clear that microglial activation may also be evoked by endogenous proteins and can significantly contribute to neuronal damage. Along with microglia, astrocytes and even neurons are directly reacting and contributing to the chronic neuroinflammatory changes in AD.
Microglia
Microglia cells constitute around 10% of all cells in the nervous system. They represent the innate immune system in the brain and thus the first line of defense against invading pathogens and serve as specialized sensors for brain tissue injury (Ransohoff 2009; Hanisch and Kettenmann 2007). Under pathological situations, such as neurodegenerative disease, stroke, traumatic injury and tumor invasion, these cells become activated, migrate to and surround damaged or dead cells, and subsequently clear cellular debris from the area, similar to the phagocytic active macrophages of the peripheral immune system. During the course of neurodegeneration, not only the morphological phenotype of microglia is changing but also their overall number. To date it remains unclear whether the increase in microglia arises from local self renewal as suggested for the SOD1 transgenic mouse model of ALS and the facial axotomy model (Ajami et al. 2007) or is caused by the invasion of peripheral monocytes in response to central inflammation as shown in an AD transgenic mouse model (Simard and Rivest 2007).
Activated microglia up-regulate a variety of surface receptors, including the major histocompatibility complex and complement receptors (Liu and Hong 2003). They also undergo dramatic morphological changes from a ramified phenotype to motile activated amoeboid cells (Kreutzberg 1996). Once immunostimulated in response to neurodegenerative events, these microglia cells release a variety of proinflammatory mediators including cytokines, reactive oxygen species, complement factors, neurotoxic secretory products, free radical species and NO, all of which can contribute to neuronal dysfunction and cell death, ultimately creating a vicious cycle.
Next to the classical neuropathological features of AD, namely Aβ deposition and NFT formation, neuroinflammatory changes have been identified as the third important component of the disease. The inflammatory reactions of microglia and astrocytes are intimately associated with the pathogenesis and progress of AD. The activated microglia is associated with neuritic plaques (McGeer and McGeer 1999) and secretes a wide variety of pro-inflammatory molecules (Heneka and O’Banion 2007). Furthermore activated microglia is implicated in active phagocytosis of Aβ, thus counterbalancing the Aβ load (Frautschy et al. 1998; Bolmont et al. 2008). The latter finding, however, has been questioned by a recent study using an microglial ablation system through herpes simplex virus thymidine kinase expression under the control of the CD11b promoter and ganciclovir treatment in vivo (Grathwohl et al. 2009). In this work, ablation of microglia in an AD mouse model, that develops plaque pathology already at 2–3 month of age, did not affect plaque load. One interpretation of this finding could be that the occurrence of toxic amyloid species along with inflammatory mediators was leading to an early loss of microglial key functions including phagocytosis, which results in a paralysis of these cells. Ablation of these paralyzed microglia, of course would not result in any change of Aβ plaque load since these cells have by far earlier stopped to limit plaque build up in such a scenario.
Nevertheless, there are no doubts that the activation of microglia occurs in response to formation of amyloid plaques. Several amyloid peptides and APP itself can act as potent glial activators (Barger and Harmon 1997; Dickson et al. 1993; Schubert et al. 2000), and disruption of the APP gene and its proteolytic products delay and decrease microglial activation (DeGiorgio et al. 2002). Microglial cells have been suggested to be preferentially associated with certain amyloid plaque types indicating that plaque development and the degree of microglial reaction are interrelated (D’Andrea et al. 2004). However, it remains unclear whether Aβ plaque deposition is an absolute requirement for microglial activation, or whether this can already be evoked by soluble and toxic Aβ species. This hypothesis finds support by a recent study where focal activation of microglial cells becomes apparent at 3 month of age in APP V717I transgenic mice, which usually start to deposit Aβ in plaque-like structures much later around 10–12 months (Heneka et al. 2005a). In contrast, studies using in vivo multiphoton microscopy using 5–6 month olf B6C3-YFP transgenic mice (bearing APPswe and PS1d9x-YFP genes) suggested that microglia are recruited to Aβ plaques only after they have been formed (Meyer-Luehmann et al. 2008).
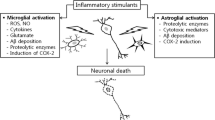
The mechanisms of microglial activation by Aβ depositions have not been fully elucidated yet, although several receptor systems are directly implicated in this process (Fig. 1). In particular, activation of microglia requires P2X7 purinoceptors and Ca2+ signaling. Exposure of cultured microglial cells to Aβ25–35 triggers Ca2+ influx, IL-1β release and P2X7-dependent membrane permeabilisation, which was absent in cells prepared from P2X7 KO mice (Sanz et al. 2009). Furthermore, intra-hippocampal injection of Aβ1–42 failed to induce microglial activation (as judged by IL-1β accumulation) in animals deficient for P2X7 receptors (Sanz et al. 2009).

Pathways of microglial activation by Aβ. Aβ is able to stimulate microglia by a variety of cell surface molecules (left), resulting in the secretion of inflammatory molecules (right). These molecules include members of the cytokine family, others are small compounds of the COX metabolism (prostaglandins) or short-lived molecules like nitric oxide
Activation of microglial cells by aggregated Aβ also involves Toll-like receptors (Udan et al. 2008; Okun et al. 2009) of the TLR2 and TLR4 type. Expression of TLR2 and TLR4 receptors were upregulated in both AD brains and in related transgenic mouse models of AD and plaque associated microglia have increased levels of mRNA coding for TLR2, -4, -5, -7 and -9 (Frank et al. 2009). A spontaneous loss-of-function mutation in the TLR4 gene significantly reduced Aβ-induced microglial activation (Walter et al. 2007). Exposure of microglial cultures to Aβ also stimulated TLR2 receptors, while inhibiting TLR9 receptors (Lotz et al. 2005). Stimulation of TLR-associated signaling systems may have dual effects in AD progression. On one hand, activation of TLRs increases microglial phagocytosis of Aβ (this involves p38 MAPK signaling and expression of G-protein-coupled formyl peptide receptor-like 2, mFPR2; the latter likely being the sensor for Aβ (Chen et al. 2006; Iribarren et al. 2005). Loss of function mutation of TLR4 or knockout of TLR2 have been found to increase plaque load in murine AD models. Increased Aβ deposition in APP/PS1 mice knockout for TLR2 was accompanied by impaired memory performance (Richard et al. 2008). Stimulation of Toll-like receptor 9 signaling with oligodeoxynucleotides has been shown to decrease plaque load and improve radial maze performance in Tg2576 mice (Scholtzova et al. 2009). It is interesting to note that activation of TLR9 triggered microglial clearance of oligomeric Aβ and attenuated neurotoxicity in neuron-microglia co-cultures exposed to oligomeric Aβ (Doi et al. 2009). At the same time, however, overstimulation of TLRs may trigger excessive release of cytokines, proteases and other cytotoxins thus promoting neural cell death (Okun et al. 2009). Aβ stimulates a nuclear factor kappa B (NFκB)-dependent pathway that is required for cytokine gene transcription (Combs et al. 2001) within activated microglia and reactive astrocytes. Not only Aβ, but also the carboxy-terminal 100 amino acids of APP (CT100), which is also present in senile plaques, can induce astrocytosis and neuronal death. CT100 exposure results in activation of the mitogen-activated protein kinase (MAPK) pathways as well as NFκB (Bach et al. 2001). Additionally, other proteins involved in APP processing have been implicated in the inflammatory response. Loss of presenilin function in presenilin conditional knockout mice leads to differential up-regulation of inflammatory markers in the cerebral cortex, such as strong microglial activation, complement component C1q, and cathepsin S (Beglopoulos et al. 2004).
Once stimulated, microglial participate in the generation and release of a wide range of inflammatory mediators including complement factors, chemokines and cytokines. The complement system represents a complex and tightly regulated attack cascade designed to destroy invaders and assist in the phagocytosis of waste, one of the key microglial tasks under physiological and pathophysiological conditions. The components of this system carry out four major functions: recognition, opsonization, inflammatory stimulation and direct killing through the membrane attack complex (MAC) (McGeer and McGeer 2002). In addition to triggering the generation of a membranolytic complex, complement proteins interact with cell surface receptors to promote a local inflammatory response that contributes to the protection and healing of the host. Microglial complement activation causes inflammation and cell damage, yet it is essential for eliminating cell debris and potentially toxic protein aggregates. The complement system consists of some 30 fluid-phase and cell-membrane associated proteins that can be activated by three different routes: the classical pathway (involving C1q, C1r, C1s, C4, C2, and C3 components) is activated primarily by the interaction of C1q with immune complexes (antibody–antigen), but activation can also be achieved after interaction of C1q with non-immune molecules such as DNA, RNA, C-reactive protein, serum amyloid P, bacterial lipopolysaccharides, some fungal and virus membranes, and most importantly, fibrillar Aβ. The initiation of the alternative pathway (involving C3, factor B, factor D, and properdin) does not require the presence of immune complexes and leads to the deposition of C3 fragments on target cells. Mannose-binding lectin (MBL), a lectin homologous to C1q, can recognize carbohydrates such as mannose and N-acetylglucosamine on pathogens and initiate the complement pathway independently of both the classical and the alternative activation pathways. Like the C1 complex in the classical pathway, MBL is associated with two serine proteases that cleave C4 and C2 components, leading to the formation of the classical C3 convertase (van Beek et al. 2003).
Microglial cells can produce complement proteins to recognize and kill pathogens locally. Studies using quantitative PCR have shown locally upregulated complement mRNA in AD brain, especially in the areas of primary pathology: entorhinal cortex, hippocampus, and midtemporal gyrus (Yasojima et al. 1999b). Numerous groups have reported the association of complement proteins of the classical pathway, particularly the MAC, with amyloid plaques and NFT in AD brains (Webster et al. 1997b). Information about the functional role comes from studies of mutant mice lacking complement proteins, which suggest that impaired phagocytosis can result in immune-mediated tissue damage and inflammation (Botto 1998; Taylor et al. 2000). However, the complement system may be Janus faced and also provide beneficial action to the brain during AD, Thus, Wyss-Coray et al. (2002) demonstrated that complement activation can protect against Aβ-induced toxicity and may reduce the accumulation or promote the clearance of senile plaques. AD mice expressing a soluble form of the complement inhibitor Crry, which blocks C3 activation, under the control of the glial fibrillary acidic protein promoter displayed higher Aβ deposition and more prominent neurodegeneration than age-matched control mice. However, more recently it was reported that transgenic mouse models of AD lacking C1q showed reduced pathology, consisting of decreased numbers of activated microglia and improved neuronal integrity, without changes in plaque. These data suggest that at stages when fibrillar plaque pathology is present, C1q exerts a detrimental effect on neuronal integrity, most likely through the activation of the classical complement cascade.
The finding that blocking C3 activation increases plaque load indicates that microglia at least to some extent has the ability to clear Aβ. Why plaque load did not alter but plaques recruited less microglia in the C1q deficient mice, is puzzling, but may point to a switch to an alternative activation state. A recent publication demonstrated that a C5a antagonist reduced plaque load and microglial activation in 3xTg mice (Fonseca et al. 2009). Since C5a is a late inflammatory marker, it may be postulated that for the clearance of Aβ some activation of microglia is necessary but that sustained strong activation leads to a neurotoxic phenotype. Targetting C5a in AD is attractive since it would leave upstream possibly beneficial complement components unaffected. It has also been shown that complement factor H expression is downregulated in AD patients via a NFkB sensitive micro RNA mediated regulation (Lukiw et al. 2008), which may result in increased complement activity on healthy host cells.
In AD, unlike the aforementioned neurological disorders characterized by leukocyte infiltration, abnormal or excessive migration of inflammatory cells into the CNS has not been definitively shown to occur. Nonetheless, there is growing evidence that chemokines and chemokine receptors are upregulated in resident CNS cells in AD brain (Ransohoff 2009), and chemokines may contribute to plaque-associated inflammation and neurodegeneration. Upregulation of CXCR2 expression has been observed on some dystrophic neurites in senile plaques (Xia and Hyman 1999; Horuk et al. 1997). In addition, the expression of CCR3 and CCR5 is increased on some reactive microglia in AD, and MIP-1α is found in a subpopulation of reactive astrocytes (Xia et al. 1998). MCP-1 has also been localized to mature senile plaques and reactive microglia, but is not found in immature senile plaques. Furthermore, in vitro studies have demonstrated the ability of Aβ to stimulate the production of IL-8, MCP-1, MIP-1α and MIP-1β from human monocytes (Meda et al. 1999). For example, microglia cultured from rapid autopsies of AD and ND patients exhibit significant, dose-dependent increases in IL-8, MCP-1 and MIP-1 α after exposure to Aβ (Lue et al. 2001). Although more studies are certainly needed, it is likely that plaque-associated chemokine production plays a role in the recruitment and accumulation of microglia to A β plaques. Future studies using targeted disruption of chemokines and chemokine receptors in mouse models of AD should help clarify the role of chemokines in plaque-associated inflammation and neurodegeneration.
Microglia derived cytokines associated with AD include several interleukins (ILs), TNF-α and TGFβ amongst others. In general, cytokine production is increased in inflammatory states and they function by regulating the intensity and duration of the immune response (Tuppo and Arias 2005; Heneka and O’Banion 2007). Thus, IL-1 induces IL-6 production, stimulates iNOS activity (Rossi and Bianchini 1996), and induces the production of M-CSF (Frei et al. 1992; Aloisi et al. 1992; Thery et al. 1992). In addition, IL-1 enhances neuronal acetylcholinesterase activity, microglial activation and additional glial IL-1 production, with consequent activation, and expression of the cytokine S100β by astrocytes, thereby establishing a self propagating cycle (Griffin 2000; Mrak and Griffin 2001). IL-6 promotes astrogliosis (Selmaj et al. 1990), activates microglia (Heyser et al. 1997), and stimulates the production of acute phase proteins, like C-reactive protein and complement components (Castell et al. 1989). IL-6 knockout mice exhibit a facilitation of radial maze learning over 30 days and show a faster acquisition, suggesting a possible negative regulation of memory formation and consolidation processes by IL-6 (Braida et al. 2004). TNF-α has both pro-apoptotic and anti-apoptotic effects. This proinflammatory cytokine accounts for most of the neurotoxic activity secreted by monocytes and microglia (Combs et al. 2001). On the other hand, TNF-α has been reported to have neuroprotective properties in the AD brain.
In addition to the general role of cytokines, AD-specific interactions of certain cytokines with the APP processing pathway and Aβ may be pathophysiologically relevant. For example, IL-1 can regulate APP processing and Aβ production in vitro (Blasko et al. 1999). In turn, fibrillar Aβ has been reported to increase neurotoxic secretory products, proinflammatory cytokines and reactive oxygen species (Eikelenboom and van Gool 2004; Eikelenboom et al. 1994; McGeer and McGeer 1995). Cultured rat cortical glia exhibit elevated IL-6 mRNA after exposure to the carboxy-terminal 105 amino acids of APP (Chong 1997). IL-1, IL-6, TNF-α MIP-1α and MCP-1 increase in a dose-dependent manner after cultured microglia are incubated with Aβ (Floden and Combs 2006; Lindberg et al. 2005; Benveniste et al. 2001; Butovsky et al. 2005; Veerhuis et al. 2005; Hanisch 2002). Together, Aβ stimulated production of interleukins and other cytokines and chemokines and their feedback activation of APP production or BACE1 (Sastre et al. 2003, 2006) may establish a self perpetuating, vicious cycle. A second general category of cytokine action is manifested by inhibitory, anti-inflammatory cytokines such as IL-1 receptor antagonist (IL-1Ra), IL-4, IL-10 and TGF- β. Some of these are reportedly elevated in AD, consistent with induction of homeostatic mechanisms in neuroinflammation (Grammas and Ovase 2001; Szczepanik et al. 2001b; Rota et al. 2006). The use of anti-inflammatory cytokines such as IL-4 and TGF-β could be beneficial, because they are able to inhibit CD40 and class II MHC by restricting their expression and activity (Benveniste et al. 2001). Another potentially beneficial effect of IL-4 that counteracts AD pathology has recently been reveiled (Shimizu et al. 2008). They observed that IL-4 selectively induces the clearance of oligomeric Aβ by rat type 2 microglia (Shimizu et al. 2008), which was dependent on the expression of CD36. However, overexpression of TGFβ in transgenic mice leads to changes in the microvasculature, including age related amyloid deposition (Wyss-Coray et al. 2000), reflecting the multi-functional nature of many cytokines. In addition to the above described evidence from the analysis of human brain tissue, cell culture and transgenic animal studies, an association of AD with several polymorphisms of proinflammatory genes has been described, including IL-1 (Nicoll et al. 2000), IL-6 (Papassotiropoulos et al. 1999), TNF-α (McCusker et al. 2001; Perry et al. 2001b), and α1-antichymotrypsin, an acute phase protein (Kamboh et al. 1995). However, none of the various members of the interleukin cytokine family that are associated with AD actually map to chromosomal regions with evidence of genetic linkage (Tanzi and Bertram 2005). Thus, although inflammation and the upregulation of inflammatory mediators like the interleukins are regularly observed in AD brain, it appears less likely that variation at the genomic level of these proteins makes a large contribution to AD risk in general. Whereas overall inflammatory responses in AD may be detrimental, a few recent publications have shown that overexpression of certain proinflammatory cytokines (IL-1β, IL-6) activate microglia and reduce plaque deposition in animal models of AD (Shaftel et al. 2007; Chakrabarty et al. 2010). This is in accordance with other experimental manipulations that enhance inflammatory responses and reduce Aβ load. It is conceivable that the extent in which microglia is able to clear Aβ during the progress of AD is too limited and that boosting this ability can prevent plaque formation. However, such manipulations in themselves can create a neurotoxic environment in the brain. For instance, it has been shown that the IL-1β overexpressing mice are behaviorally impaired (Hein et al. 2010; Moore et al. 2009) and TNF-α overexpression in an animal model of AD (3xTg mice) has been shown to cause neuronal loss, but plaque load was not assessed in the latter study (Janelsins et al. 2008). Results of experiments with overexpression or knockout of cytokines or cytokine receptors have to be taken with care, moreover, since cytokine or their receptors can be found on non-inflammatory brain cells and may execute non-inflammatory functions as well. For example, knockout of the TNF death receptor in APP23 mice leads to reduced expression of BACE1, reduced Aβ production and deposition in APP23 mice (He et al. 2007). Therefore, manipulations of the inflammatory cascades should be always checked not to affect APP processing itself before any firm conclusions about an independent role of the respective inflammatory marker can be drawn. Next to complement factors, chemokines and cytokines, activated microglia can also serve as a chief source of prostanoids. Two isoforms of cyclooxygenases, the mainly constitutively expressed COX-1 and the inducible COX-2, catalyze key steps of prostanoid synthesis in mammalian cells. Downstream of both COX-1 and COX-2, several other enzymes regulate the generation of a whole spectrum of prostanoids, some of which may be neuroprotective and others neurodestructive. Thus, the composition and proportion of all prostanoids together may actually determine whether the activity of COX enzymes is beneficial or detrimental.
In vitro, LPS activated microglial cells and IL-1β-stimulated astroglial cells are capable of synthesizing COX-2 (Bauer et al. 1997; O’Banion et al. 1996; Almer et al. 2001). In contrast to peripheral monocytes, cultured rat microglia cells do not synthesize COX-2 in response to IL-1β or IL-6 (Bauer et al. 1997), suggesting that COX-2 regulation differs between CNS and peripheral cells. In rat microglial cell cultures, the major enzymatic product of COX-2 appears to be prostaglandin E2 (PGE2). Because PGE2 itself is able to induce COX-2 in microglial cells (Minghetti et al. 1997), some sort of autocrine or paracrine amplification of the COX-2 induction in microglial cells or a spreading of COX-2 expression between neurons and microglial cells seems possible. PGE2 acts on four different receptors: EP1–EP4 (Narumiya et al. 1999). EP1 and EP2 receptors have been detected in cultured microglia, while EP3 receptors are also present in activated microglia in vivo (Slawik et al. 2004). Microglial EP2 receptors inhibit phagocytosis and enhance neurotoxic activities of microglia (Shie et al. 2005a, b). PGE2 may also act on the neuronal EP2 receptor, which is involved in apoptosis, although investigations of the role of neuronal EP2 activation on neuronal cell death have yielded conflicting results and suggest a neuroprotective role of neuronal EP2 stimulation under several pathophysiological circumstances (Bilak et al. 2004; Lee et al. 2004; McCullough et al. 2004; Takadera et al. 2004). This is further exemplified in a recent report where knockout of EP2 in a double transgenic (APP/PS1) mouse led to decreased evidence of oxidative stress and descreased Aβ production, associated with lower levels of BACE (Liang et al. 2005). In conclusion, neuronal and glial secretion of PGE2 may impair phagocytotic clearance of Aβ by binding to the microglia EP2 receptor and enhancing microglial toxicity. However, the role of PGE2 in neurodegeneration may be by far more complex due to the presence of other EP receptor subtypes on microglial cells and the effects of PGE2 on other cell types. Neuronal death elicited by excitotoxins is elevated in transgenic animals with high expression of COX-2, suggesting that COX-2 expression may further interact with other pathogenic mechanisms (Kelley et al. 1999).
It should be noted that some aspects of microglia function may be beneficial, since activated microglia are able to reduce Aβ accumulation by increasing its phagocytosis, clearance and degradation (Frautschy et al. 1998; Qiu et al. 1998). Moreover, secreted Aβ1–40 and Aβ1–42 peptides are constitutively degraded by the insulin degrading enzyme (IDE), a metalloproteinase released by microglia and neural cells. Finally, microglia can also secrete several neurotrophic factors, such as the glia-derived neurotrophic factor (GDNF), which exert a well documented neuroprotective function (Liu and Hong 2003).
Finally, it has been shown that bone marrow-derived cells are able to cross the blood–brain barrier (BBB) and to differentiate into fully functional microglia afterwards (Malm et al. 2005; Simard and Rivest 2004; Hess et al. 2004). In this context it has been demonstrated that engrafted monocytes are able to infiltrate the brain and to restrict the formation of amyloid plaques (Simard et al. 2006). One has to mention that there are concerns, whether this represents an physiological event or that such a phenomenon results from the damage of the BBB during the generation of chimeric mice using either BMSC transplantation or irradiation (Mildner et al. 2007; Ajami et al. 2007).
Astrocytes
The glial involvement in the pathogenesis of AD was initially suggested by Alois Alzheimer himself. He had demonstrated that the neuritic plaques (extracellular deposits of fibrillar Aβ) together with tau NFT represent the major histopathological markers of AD. In addition, AD brains are characterized by prominent astrogliosis, mostly observed in the cells surrounding amyloid plaques with processes of activated astrocytes participating in formation of neuritic plaques (Nagele et al. 2003; Rodriguez et al. 2009).
The Aβ peptide represent an activating signal for astrocytes; exposure of cultured glial cells to aggregated Aβ or to amyloid plaques isolated from human AD brains trigger reactive astrogliosis (Dewitt et al. 1998). Aβ also induces functional changes in astrocytes in vitro: Aβ1–42 and its toxic fragment Aβ25–35 induced spontaneous [Ca2+]i elevations and [Ca2+]i oscillations in astrocytes growing in mixed astroglial-neuronal cultures. The Aβ-induced [Ca2+]i oscillations lasted for many hours and were linked to neuronal death, which occurred 24 h after administration of Aβ to the cultures. Inhibition of [Ca2+]i oscillations prevented neuronal death (Abramov et al. 2003). In the same mixed culture model Aβ was also shown to induce mitochondrial depolarisation and oxidative stress in astrocytes; the release of reactive oxygen species from stressed astrocytes caused neuronal death. Recently, (Allaman et al. 2010) reported that aggregated Aβ1–42 was taken up by astrocytes and caused metabolic disturbances and production of hydrogen peroxide. Astrocytes pretreated with Aβ were toxic to neurons in co-cultures. Metabolic disturbances as well as toxicity could be prevented by a PI-3 kinase inhibitor.
The abnormalities in astroglial Ca2+ signaling were also observed in the brains of transgenic AD mice. In these experiments, employing in vivo multiphoton-confocal-microscopy, the general elevation of resting [Ca2+]i was observed throughout the astroglial syncytia. In addition, astrocytes located in the vicinity of plaques triggered spontaneous long-distance propagating Ca2+ waves, which were absent in control animals (Kuchibhotla et al. 2009).
Participation of astrocytes in plaques formation initiated the hypothesis of an Aβ-clearing role of astroglia (Nagele et al. 2003; Wyss-Coray et al. 2003) with subsequent astroglial degeneration triggered by accumulated Aβ. Indeed plating of isolated healthy astrocytes on the slices prepared from transgenic (APP) AD mice resulted in migration of astrocytes towards the plaques with subsequent accumulation and degradation of Aβ. In support of this finding, recent evidence suggests that astroglial cells are able to phagocytose Aβ peptides, a process which may depend on their apolipoprotein E (ApoE) status, suggesting that ApoE polymorphisms may influence the risk to develop AD by affecting astroglial Aβ phagocytosis. In contrast, however, endogenous astrocytes surrounding the Aβ plaques were unable to accumulate and remove Aβ (Wyss-Coray et al. 2003).
In the triple transgenic mouse model of AD [3xTg-AD; harboring the mutant genes for amyloid precursor protein (APPSwe), presenilin 1PS1M146V and tauP301L (Oddo et al. 2003)] very little, if any Aβ accumulation by reactive astrocytes was observed (Rodriguez et al. 2009). These data clearly indicate the phenotypic difference between normal astroglia and astrocytes affected by AD pathology. Another kind of phenotypic difference was observed in astrocytes from AD model expressing double mutated K670N-M671L APP; these astrocytes expressed γ-secretase becoming thus possible producers of Aβ (Hartlage-Rubsamen et al. 2003) in line with findings by others (Rossner et al. 2005; Heneka et al. 2005a). While it remains unclear to which degree astrocyte activation contributes to Aβ generation or its clearance, it seems apparent that astrocytes contribute to the inflammatory component of AD. For example, astrocytes have been shown to express iNOS and the l-arginine-supplying enzyme argininosuccinate synthetase and consequently contribute to NO- and peroxynitrite mediated neurotoxicity (Heneka et al. 2001). Although astrocytes serve as a constant and important source of neurotrophic factors under physiological conditions, in vitro and in vivo experiments suggest that chronically activated inflammatory astrocytes may not generate significant amounts of these molecules (Nagatsu and Sawada 2005).
Reactive and pathologically changed astrocytes are also responsible for failures in the functional activity of neuronal-glial-vascular units. Indeed, the vascular dysfunctions, perivascular amyloidosis and compromised blood–brain barrier are inseparable parts of AD pathology (Bell and Zlokovic 2009). How astroglial cells are participating in these changes remains, however, and open question.
The astrogliosis however is not the only astroglial reaction in the AD brains. In a recent study performed on different regions of the brains of triple-transgenic [3x-Tg-AD—(Oddo et al. 2003)] mice, both astrogliosis and astroglial atrophy were found [(Rodriguez et al. 2009); Rodriguez and Verkhratsky paper in preparation]. The decrease in complexity of astrocytes, which indicated their atrophy, begun to be observed before the formation and consolidation of neuritic plaques. In plaque infested brains the reactive astrocytes were concentrated around the Aβ plaques, whereas astroglial cells distant to the plaques had an atrophic features.
Neurons
While neurons were traditionally believed to be passive bystanders in neuroinflammation, more recent evidence suggests that neurons themselves are capable of producing inflammatory mediators. Thus, neurons can serve as source of complement, COX-2-derived prostanoids (Yermakova et al. 1999), and several cytokines including IL-1β, IL-6, and TNF-α (Botchkina et al. 1997; Breder et al. 1993; Gong et al. 1998; Murphy et al. 1999; Orzylowska et al. 1999; Suzuki et al. 1999; Tchelingerian et al. 1994; Yan et al. 1995; Hoozemans et al. 2004; Aloisi et al. 1992) and M-CSF (Du et al. 1997). Although COX-2 expression is driven by physiological synaptic activity (Yamagata et al. 1993; Yermakova and O’Banion 2000), it appears possible that neurons themselves may exacerbate local inflammatory reactions and thus contribute to their own destruction in AD. As a further factor, expression of the inflammatory induced enzyme iNOS has been described in degenerating neurons in AD brains (Vodovotz et al. 1996; Lee et al. 1999; Heneka et al. 2001), and compelling evidence exists for iNOS related long-term NO release and NO dependent peroxynitrite formation during the course of AD (Smith et al. 1997). Glial and neuronal derived NO and peroxynitrite have been demonstrated to cause neuronal dysfunction and cell death in vitro and in vivo (Boje and Arora 1992; Heneka et al. 1998). Alternatively, some of the classical pro-inflammatory mediators such as TNF-α and low concentrations of NO may actually confer neuroprotection rather than destruction in the brain and therefore constitute a defense mechanism against local inflammatory reactions.
Finally, it is reported that prostaglandin E2 is able to increase Aβ production in neuronal cells mediated by internalization of the EP4 receptor (Hoshino et al. 2009).
Pro- and antiinflammatory mediators
The neuroinflammatory response observed in AD is characterized by a whole array of pro- and antiinflammatory mediators including members of the complement cascade, chemo- and cytokines as well as inflammatory enzyme systems. Several of these factors may promote neurodegenerative mechanisms while others may rather limit ongoing inflammatory changes or even exert beneficial neurotrophic effects. Thus, not a single mediator but rather the entire spectrum of inflammatory agents will determine whether beneficial or detrimental effects prevail.
Complement
The complement system represents a complex and tightly regulated attack cascade designed to destroy invaders and assist in the phagocytosis of waste materials. The components of this system carry out four major functions: recognition, opsonization, inflammatory stimulation and direct killing through the membrane attack complex (MAC) (McGeer and McGeer 2002). In addition to triggering the generation of a membranolytic complex, complement proteins interact with cell surface receptors to promote a local inflammatory response that contributes to the protection and healing of the host. Complement activation causes inflammation and cell damage, yet it is essential for eliminating cell debris and potentially toxic protein aggregates (Shen and Meri 2003). Furthermore, the complement system may contribute to synapse remodeling through marking weak synapses for removal presumably by microglia (Stevens et al. 2007).
The complement system consists of some 30 fluid-phase and cell-membrane associated proteins that can be activated by three different routes: The classical pathway (involving C1q, C1r, C1s, C4, C2, and C3 components) is activated primarily by the interaction of C1q with immune complexes (antibody-antigen), but activation can also be achieved after interaction of C1q with non-immune molecules such as DNA, RNA, C-reactive protein, serum amyloid P, bacterial lipopolysaccharides, some fungal and virus membranes, and as already mentioned, fibrillar Aβ. The initiation of the alternative pathway (involving C3, factor B, factor D, and properdin) does not require the presence of immune complexes and leads to the deposition of C3 fragments on target cells. Mannose-binding lectin (MBL), a lectin homologous to C1q, can recognize carbohydrates such as mannose and N-acetylglucosamine on pathogens and initiate the complement pathway independently of both the classical and the alternative activation pathways. Like the C1 complex in the classical pathway, MBL is associated with two serine proteases that cleave C4 and C2 components, leading to the formation of the classical C3 convertase (van Beek et al. 2003).
Various brain cells can produce complement proteins to recognize and kill pathogens locally. Cell lines and primary cultures of human origin were used to show that glial and neuronal cells could produce most complement proteins, particularly after stimulation with inflammatory cytokines (Gasque et al. 1995). Studies using RT-PCR have shown locally upregulated complement mRNA in AD brain, especially in the areas of primary pathology: entorhinal cortex, hippocampus, and midtemporal gyrus (Yasojima et al. 1999b). Numerous groups have reported the association of complement proteins of the classical pathway, particularly the MAC, with amyloid plaques and NFT in AD brains (Webster et al. 1997a).
Studies of mutant mice lacking complement proteins suggest that impaired phagocytosis can result in immune-mediated tissue damage and inflammation (Botto 1998; Taylor et al. 2000). Wyss-Coray et al. (2002) demonstrated that complement activation can protect against Aβ-induced toxicity and may reduce the accumulation or promote the clearance of senile plaques. AD mice expressing a soluble form of the complement inhibitor Crry, which blocks C3 activation, under the control of the glial fibrillary acidic protein promoter displayed higher Aβ deposition and more prominent neurodegeneration than age-matched control mice.
However, more recently it was reported that transgenic mouse models of AD lacking C1q showed reduced pathology, consisting of decreased numbers of activated glia and improved neuronal integrity, without changes in plaque area. These data suggest that at stages when fibrillar plaque pathology is present, C1q exerts a detrimental effect on neuronal integrity, most likely through the activation of the classical complement cascade and the enhancement of inflammation (Fonseca et al. 2004). In line with this, one could hypothesize that under neurodegenerative conditions, otherwise physiological neurodevelopmental systems such as described by Stevens and colleagues (see above) may be reactivated.
Chemokines
Recent experiments have focused on understanding the role of chemokines and their receptors in AD neuroinflammation. The chemokine family consists of over 50 different molecules that confer chemotaxis, tissue extravasation, and functional modulation of leukocyte function during inflammation (Luster 1998; Owens et al. 2005). The importance of chemokine generation in AD brain is underscored by the fact that these molecules may potently regulate microglial migration and recruitment of astrocytes to the area of neuroinflammation, and thus are responsible for the extent of local inflammation. In addition, recent studies using chimeric mice grafted with green fluorescent protein expressing bone marrow cells, indicate that many of the so called “microglia” represent invading macrophages from peripheral blood (Stalder et al. 2005; Simard et al. 2006) suggesting a chemotactic stimulus from the brain. Whether this represents a technical artefact due to radiation induced alterations at the brain barrier or in fact occurs in human AD as part of the pathogenetic sequale is yet to be determined. The CXC subclass of chemokines is considered one of the two major chemokine subfamilies and its members (e.g. IL-8) are primarily chemotactic for neutrophils and endothelial cells. The conserved glutamate–leucine–arginine (ELR) motif within the receptor-binding domain of these proteins distinguishes them from non-ELR CXC chemokines such as IP-10, which primarily attract activated T cells (Strieter et al. 1995). The CC chemokine subfamily, whose members include MIP-1α, MCP-1, and RANTES, do not affect neutrophils but are chemotactic for monocytes/macrophages, T lymphocytes, basophils and eosinophils. Seven transmembrane, G-protein-coupled cell-surface receptors mediate the biological activities of chemokines and these receptors are named according to their chemokine subfamily classification. At present there are five known CXC receptors (CXCR1 to CXCR5) and nine CC receptors (CCR1 to CCR9) (Charo and Ransohoff 2006).
While it has been reported that chemokines exert physiological action in healthy brain (Hesselgesser and Horuk 1999), the majority of studies have focused on the expression pattern of chemokines and their respective receptors in neurological diseases such as multiple sclerosis, traumatic brain injury and stroke. All of these disorders share disruption of the blood brain barrier as an important pathogenetic event subsequently allowing peripheral leukocytes to infiltrate the lesion site (Glabinski and Ransohoff 1999). In contrast, no convincing evidence exists for blood brain barrier disruption or significant leukocyte infiltration in the AD brain. However, several chemokines and chemokine receptors have been found to be upregulated in the AD brain (Xia and Hyman 1999), and chemokines may play an important role for recruiting microglia and astroglia to sites of Aβ deposition. Thus, Aβ stimulated human monocytes generate IL-8, MCP-1, MIP-1α and MIP-1β in vitro (Smits et al. 2002), and microglia cultured from rapid autopsies of AD and non-demented patients revealed an increased expression of IL-8, MCP-1 and MIP-1α after experimental exposure to Aβ (Lue et al. 2001). Neuropathological studies have found MCP-1 (Ishizuka et al. 1997) and increased expression of CCR3 and CCR5 in reactive microglia (Xia et al. 1998). Supporting the hypothesis that astrocytes actively contribute to the inflammatory disease component, MIP-1β has been detected in reactive astrocytes nearby Aβ plaques (Xia et al. 1998).
More recently a direct modulation of neurotoxicity by chemokines has been proposed, thus modulation of the CX3CR1/CX3CL1 system has been shown to influence neuronal survival in rodent models of neurodegeneration. While in the SOD1 mouse model for ALS, the MPTP neurotoxin model of Parkinson`s disease and in a model of generalized inflammation CX3CR1 knockout mice showed an increased neuronal loss (Cardona et al. 2006), the same transgenic knockout prevented neuron loss in an triple transgenic mouse model of AD (Fuhrmann et al. 2010).
Inflammatory cytokines
The cytokine class of inflammatory mediators is secreted by microglia and astrocytes surrounding β-amyloid neuritic plaques. Cytokines associated with AD include several interleukins (ILs), TNF-α and TGF-β along with several others. Their production is increased in inflammatory states and they function by regulating the intensity and duration of the immune response.
In astrocytes, IL-1β induces IL-6 production, stimulates iNOS activity (Rossi and Bianchini 1996), and induces the production of M-CSF (Frei et al. 1992; Aloisi et al. 1992; Thery et al. 1992). In addition, IL-1β enhances neuronal acetylcholinesterase activity, microglial activation and additional IL-1α production, with consequent astrocyte activation, and expression of the cytokine S100β by astrocytes, thereby establishing a self propagating cycle (Griffin 2000; Mrak and Griffin 2001). IL-6 promotes astrogliosis (Selmaj et al. 1990), activates microglia (Heyser et al. 1997), and stimulates the production of acute phase proteins (Castell et al. 1989). IL-6 knockout mice exhibit a facilitation of radial maze learning over 30 days and show a faster acquisition, suggesting a possible involvement of IL-6 in memory processes (Braida et al. 2004). TNF-α has both pro-apoptotic and anti-apoptotic effects. This proinflammatory cytokine accounts for most of the neurotoxic activity secreted by monocytes and microglia (Combs et al. 2001). On the other hand, TNF-α has been reported to have neuroprotective properties in the AD brain.
In addition to the general role of cytokines, AD-specific interactions of certain cytokines and chemokines with Aβ may be pathophysiologically relevant. For example, TNFα can regulate APP processing and Aβ production in vitro (Blasko et al. 1999). In turn, fibrillar Aβ has been reported to increase neurotoxic secretory products, proinflammatory cytokines and reactive oxygen species as evidenced by a various groups and publications. Cultured rat cortical glia exhibit elevated IL-6 mRNA after exposure to the carboxy-terminal 105 amino acids of APP (Chong 1997). IL-1, IL-6, TNF-α MIP-1α and MCP-1 increase in a dose-dependent manner after cultured microglia are incubated with Aβ (Floden and Combs 2006; Lindberg et al. 2005; Benveniste et al. 2001; Butovsky et al. 2005; Veerhuis et al. 2005; Hanisch 2002; Lue et al. 2001; Lee et al. 2002). Production of IL-6 and M-CSF by human neurons is reportedly stimulated by glycation endproduct-modified tau and Aβ. Additionally, Aβ is able to stimulate a NFκB-dependent pathway that is required for cytokine production (Combs et al. 2001). The production of interleukins and other cytokines and chemokines may also lead to microglial activation, astrogliosis, and further secretion of proinflammatory molecules and amyloid, thus perpetuating the cascade (Ho et al. 2005).
A second general category of cytokine action is manifested by inhibitory, anti-inflammatory cytokines such as IL-1 receptor antagonist (IL-1Ra), IL-4, IL-10 and TGF-β. Some of these are reportedly elevated in AD, consistent with induction of homeostatic mechanisms in neuroinflammation (Grammas and Ovase 2001; Rota et al. 2006; Szczepanik et al. 2001a). The presence of anti-inflammatory cytokines such as IL-4 and TGF-β could be beneficial, because they are able to inhibit CD40 and class II MHC by restricting their expression and activity (Benveniste et al. 2001). However, overexpression of TGFβ in transgenic mice leads to changes in the microvasculature, including age-related amyloid deposition (Wyss-Coray et al. 2000), reflecting the multi-functional nature of many cytokines.
Even so AD mouse models are widely used to study in vivo consequences APP overproduction, only a limited number of studies have characterized neuroinflammatory changes in these animals. All but one study used APP695 transgenic mice (Tg2576), and results were controversial. Mehlhorn et al. (2000) analyzed APP695 transgenic animals from 2 to 14 months of age but failed to detect mRNA levels of several cytokines including IL-1-α/β, IL-6, IL-10, IL-12 and IFN-γ using ribonuclease protection assay. In the same study, IL-1-β-positive astrocytes were detected in close proximity to Aβ deposition, whereas immunohistochemistry for TNF-α, IL-1-α, IL-6, and MCP-1 was negative. In contrast, Sly and colleagues detected TNF-α mRNA as early as 6 month of age (Sly et al. 2001), and Abbas and colleagues detected IFN-γ and IL-12 mRNA and protein levels by in situ hybridization and immunohistochemistry in 9-month-old APP 695 transgenic mice (Abbas et al. 2002). Moreover, IL-1-β, TNF-α and IL-10 were found by immunohistochemistry in 12–13-month old animals (Benzing et al. 1999; Apelt and Schliebs 2001). In our own study, IL-1β as well as IL-6 were detectable in CD11b positive microglia of APPV717I transgenic mice as early as at 3 month of age. The variability of findings in the same transgenic mouse line is likely caused by different techniques employed and indicates the difficulty of assessing inflammatory changes in these animal models. Differences between various APP mouse models, instead, may arise from time dependent changes of Aβ secretion. In contrast to the above described animal models, nearly all cytokines that have been studied, including IL-1α/β, IL-6, TNF-α, IL-8, and TGF-β, seem to be upregulated in AD compared to healthy age-matched individuals.
In addition to these primarily immunohistological evaluations, an association of AD with several polymorphisms of proinflammatory genes has been described, including IL-1 (Nicoll et al. 2000), IL-6 (Papassotiropoulos et al. 1999), TNF-α (McCusker et al. 2001; Perry et al. 2001a), and α1-antichymotrypsin, an acute phase protein (Kamboh et al. 1995). However, none of the various members of the interleukin cytokine family that are associated with AD actually map to chromosomal regions with evidence of genetic linkage (Tanzi and Bertram 2005). Thus, although inflammation and the upregulation of inflammatory mediators like the interleukins are regularly observed in AD brain, it appears less likely that variation at the genomic level of these proteins makes a large contribution to AD risk in general. An update on the genetic information can be found under http://www.alzgene.org
Cyclooxygenase and prostanoids
The two isoforms of cyclooxygenases, the mainly constitutively expressed COX-1 and the inducible COX-2, catalyze key steps of prostanoid synthesis in mammalian cells (O’Banion 1999). Downstream of both COX-1 and COX-2, several other enzymes regulate the generation of a whole spectrum of prostanoids, some of which may be neuroprotective and others neurotoxic. Thus, the composition and proportion of all prostanoids together may actually determine whether the activity of COX enzymes is beneficial or detrimental.
In vitro, LPS activated microglial cells and IL-1β-stimulated astroglial cells are capable of synthesizing COX-2 (O’Banion et al. 1996; Bauer et al. 1997). In contrast to peripheral monocytes, cultured rat microglia cells do not synthesize COX-2 in response to IL-1 or IL-6 (Bauer et al. 1997), suggesting that COX-2 regulation differs between CNS and peripheral cells. In rat microglial cell cultures, the major enzymatic product of COX-2 appears to be prostaglandin E2 (PGE2). Because PGE2 itself is able to induce COX-2 in microglial cells (Minghetti et al. 1997), an autocrine or paracrine amplification of COX-2 in microglial cells and perhaps other cell types is possible. PGE2 acts on four different receptors: EP1–EP4 (Narumiya et al. 1999). EP1 and EP2 receptors have been detected in cultured microglia, while EP3 receptors are also present in activated microglia in vivo (Slawik et al. 2004). Microglial EP2 receptors inhibit phagocytosis and enhance neurotoxic activities of microglia (Shie et al. 2005a; Shie et al. 2005b). In cultured rat and human astrocytes, EP2 and EP4 receptors are present and may potentiate glial cytokine production (Fiebich et al. 2001). PGE2 may also act on the neuronal EP2 receptor, which is involved in apoptosis, although investigations of the role of neuronal EP2 activation on neuronal cell death have yielded conflicting results and suggest a neuroprotective role of neuronal EP2 stimulation under several pathophysiological circumstances (Bilak et al. 2004; Lee et al. 2004; McCullough et al. 2004; Takadera et al. 2004). Indeed, deletion of EP2 in a double transgenic AD mouse model led to decreased oxidative stress and amyloid burden (Liang et al. 2005). In conclusion, neuronal and glial secretion of PGE2 may impair phagocytotic clearance of Aβ by binding to the microglia EP2 receptor and enhancing microglial toxicity. However, the role of PGE2 in neurodegeneration may be far more complex due to the presence of other EP receptor subtypes on microglial cells and the effects of PGE2 on other cell types. Neuronal death elicited by excitotoxins is elevated in transgenic animals with high expression of COX-2, suggesting that COX-2 expression may further interact with other pathogenetic mechanisms (Kelley et al. 1999).
COX-2 is upregulated in many inflammatory disorders (Dubois et al. 1998). However, data about COX-2 expression in AD brain are conflicting. Several investigators have detected increased levels of COX-2 mRNA and protein staining in AD tissue (Ho et al. 2001; Kitamura et al. 1999; Oka and Takashima 1997; Yasojima et al. 1999a) while others have found decreased COX-2 expression, particularly in late stage AD (O’Banion et al. 1997; Yermakova and O’Banion 2001; Hoozemans et al. 2004). In addition to differences in tissue sources and methodologies employed, a well controlled post mortem study indicated a higher variability of COX-2 mRNA in the brains of AD patients compared to age matched controls (Lukiw and Bazan 1997). Furthermore, COX-2 expression is not restricted to microglial or astroglial cells in AD brain; indeed it is predominantly observed in neurons of AD brains and age matched controls (O’Banion et al. 1997).
Interestingly, COX-1 is prominently expressed by microglia in rodent and human brain (Yermakova et al. 1999; Hoozemans et al. 2001) and appears to be modestly elevated in AD brain (Yermakova et al. 1999). Whether microglial COX-1 contributes to neuroinflammation in AD has not been established; however, COX-1 activity has been linked to PGE2 production in several experimental models of acute brain injury (Candelario-Jalil et al. 2003; Moore et al. 2005).
NO synthase, nitric oxide and free radicals
NO is a gaseous free radical, which is generated through the conversion of l-arginine to l-citrulline by enzymes of the nitric oxide synthase family (Bredt and Snyder 1990). Ca2+/calmodulin-dependent constitutive isoforms are present in neuronal and endothelial cells, and produce NO in a highly regulated manner. iNOS is rapidly expressed in macrophages, microglia and astrocytes upon stimulation with lipopolysaccharide (LPS) or several cytokines (Galea et al. 1992; Corradin et al. 1993; Simmons and Murphy 1992; Stuehr and Marletta 1985). This isoform produces large amounts of NO in a Ca2+-independent manner for prolonged periods of time. NO generated by iNOS is cytotoxic for invading microorganisms and tumor cells (Moncada et al. 1992). However, induction of iNOS may also have deleterious consequences for the host since vasodilatation, organ dysfunction and septic shock are partly mediated by an overproduction of NO (Thiemermann 1994). The consequences of iNOS induction in glial cells, however, seem to depend on a variety of factors including the type of cell cultures used. Both deleterious effects on neurons and unaffected neuronal viability after iNOS induction in mixed glial-neuronal cultures have been reported (Chao et al. 1996; Dawson et al. 1994; Demerle-Pallardy et al. 1993; Skaper et al. 1995). Importantly, iNOS expression and NO generation have been described in several brain pathologies including demyelinating diseases (Willenborg et al. 1999a, b), cerebral ischemia (del Zoppo et al. 2000), AIDS dementia (Hori et al. 1999), amyotrophic lateral sclerosis (Almer et al. 1999), and AD (Vodovotz et al. 1996; Wallace et al. 1997; Weldon et al. 1998; Lee et al. 1999; Heneka et al. 2001).
In addition to glial iNOS, neuronal iNOS may impact neurological disorders. For example, Vodovotz et al. (1996) reported that NFT-bearing neurons in affected brain regions of patients suffering from AD express iNOS. Further support for a role for iNOS in the inflammatory pathomechanisms involved in AD, is provided by reports of increased nitrotyrosine staining in AD brains, indicating sustained exposure and oxidative damage by peroxynitrite, an intermediate NO reaction product (Smith et al. 1997). In addition to NFTs and SPs, eosinophilic rod-like inclusions (Hirano bodies) are observed in AD brains and iNOS-immunoreactivity has been detected in association with Hirano bodies in the pyramidal layer of the CA1 region of the hippocampus and to a lesser extent in the stratum lacunosum (Lee et al. 1999). In that study, control brains showed only occasional iNOS-positive staining associated with rare Hirano bodies, while other studies, as well as our own experiments, failed to detect iNOS in control brains (MTH, unpublished observations).
To further characterize the pathway involved in neuronal iNOS expression in AD, we investigated expression of the enzyme argininosuccinate synthetase (AS) and its possible colocalization with iNOS in AD brain (Heneka et al. 2001). AS is the rate limiting enzyme in the metabolic pathway that recycles the iNOS substrate l-arginine from its catalytic byproduct l-citrulline. Several brain areas of AD patients showed a marked increase in AS expression in neurons and GFAP-positive astrocytes. Occasionally, AS expression was also detected in CD 68-positive activated microglia cells. Expression of AS was colocalized with iNOS immunoreactivity in neurons and glia. These results suggest that neurons and glial cells in AD not only express iNOS, but also AS. Because an adequate supply of L-arginine is indispensable for long-term NO generation by iNOS, coinduction of AS could enable cells to sustain NO generation, which could subsequently damage the iNOS expressing neurons as well as surrounding tissue.
Inflammation-permissive factors in AD
Occurrence and deposition of aggregated, misfolded or phosphorylated proteins may play the pivotal role for the induction and ongoing stimulation of inflammation in the AD brain. However, since some of these proteins may well occur in the normal aging brain without evoking such a dramatic immune response, it may be hypothesized that several other changes in AD are facilitating inflammation. Loss of aminergic brains stem nuclei, such as the locus ceruleus and the nucleus basalis of Meynert may result in an impaired control of neuroinflammation in the AD brain.
Locus ceruleus cell death
The locus ceruleus (LC) is located in the pontine tegmentum and serves as the main subcortical site for the synthesis of noradrenaline (NA) (Freedman et al. 1975). Ascending noradrenergic axons of the dorsal portion of the LC preferentially project to the hippocampus, the frontal and entorhinal cortex and to a minor extent to various other brain regions. Neuronal cell death of aminergic brain stem nuclei such as the LC and the dorsal raphe nucleus is a well defined, very early feature of AD that was first described by Forno (1966) and later confirmed by several groups (Mann et al. 1980, 1982; Wilcock et al. 1988). In AD, the central and dorsal portion of the LC show the most extensive loss of cells (Marcyniuk et al. 1986). LC loss and the subsequent degeneration of ascending noradrenergic axons lead to decreased NA levels in the LC projection areas (Iversen et al. 1983) whereas adrenergic receptors are upregulated in response to noradrenergic deafferentiation (Kalaria et al. 1989).
Besides its role as a classical neurotransmitter, NA acts as a potent suppressor of inflammatory gene transcription within the brain (Feinstein et al. 2002b; Marien et al. 2004). LC loss and subsequently decreasing NA levels may therefore be permissive for inflammatory mechanisms, which are otherwise controlled by physiologically released NA. Specifically, NA has been shown to suppress the generation and secretion of several inflammatory molecules including microglial synthesis of TNF-α and astrocytic expression of class II antigens. Studies from ourselves and others show that NA can also inhibit LPS- and cytokine-dependent iNOS expression in astrocytes and microglial cells, mediated by the activation of β-adrenergic receptors, and increases in cAMP (Gavrilyuk et al. 2002).
Since the initial neuropathological description, a number of studies have also demonstrated significant correlation of LC cell death or decreased cortical NA levels with severity and duration of dementia in AD (German et al. 1992; Yates et al. 1983). Interestingly, LC loss correlates better with the clinical course of the disease and the severity of dementia than loss of the nucleus basalis of Meynert and perturbation of the cholinergic system (Zarow et al. 2003). It has been argued that LC degeneration may occur as a consequence of primary degenerative changes in the cortical projection areas. However, even aged APP transgenic mice that do show an intense Aβ plaque load do not reveal any significant reduction of LC cell numbers or cortical NA levels, suggesting that LC cell death occurs independently of Aβ deposition and not in response to neurodegenerative events in its projection areas. This assumption is further supported by a recent finding that shows significant LC degeneration even in patients suffering from mild cognitive impairment, a clinical phase widely regarded as a prestage of AD (Grudzien et al. 2007).
Experimentally induced noradrenergic depletion can be achieved by systemic treatment of animals with the selective noradrenergic neurotoxin N-(2-chloroethyl)-N-ethyl-2 bromobenzylamine (DSP4) (Fritschy and Grzanna 1989). DSP4 causes widespread degeneration of LC axon terminals, decreased activity, and loss of LC neurons (Fritschy and Grzanna 1989; Olpe et al. 1983). Moreover DSP4 also impairs electrophysiological functions of remaining LC neurons (Magnuson et al. 1993) and NA depletion by DSP4 has been demonstrated to markedly increase neurodegeneration induced by N-metyhl-4 phenyl 1,2,3,6-tetrahydropyridine (Mavridis et al. 1991) and cerebral ischemia (Nishino et al. 1991).
Using a similar model we showed that DSP4 treatment of rats caused degeneration of noradrenergic projections and cell death of LC neurons, but did not affect substantia nigra neuron survival. Local application of Aβ into the rat cortex resulted in a greater and prolonged IL-1β expression in microglial cells in noradrenergic depleted animals as compared to controls with an intact NA system. Likewise expression levels of IL-6, iNOS and COX-2 were significantly increased in NA depleted cortices. Interestingly, iNOS expression was completely restricted to microglial cells in controls, whereas NA depleted animals showed widespread iNOS expression in pyramidal neurons (Heneka et al. 2002; Heneka et al. 2003), a phenomenon previously reported in AD brains (Heneka and Feinstein 2001). Noradrenergic depletion in APP transgenic mice led to a similar picture, with increased glial activation and amyloid deposition, as well as evidence of increased neurodegeneration (Heneka et al. 2006). Additional in vitro and in vivo experiments assessing microglial migration and phagocytosis found that NA, while suppressing inflammatory gene transcription, increased microglial migration and phagocytic capacity at the same time (Heneka et al. 2010). Of note, microglial migration and Aβ phagocytosis, which was impaired in response to induced NA deficiency, could be re-established by treatment of animals with the NA precursor l-threo-DOPS. This may indicate that NA serves as an important factor that switches activated microglial cells from a cytokine releasing phenotype to a more mobile and phagocyting one. Thus, LC loss and noradrenergic depletion of cortical and hippocampal areas may drive neuropathological and inflammatory changes into a vicious, sustained cycle in AD. In keeping with this, balancing the NA deficit in AD may actively interact with disease pathogenesis and may open a new therapeutic avenue.
Basal forebrain cell death
Similar to cell death observed in the locus ceruleus, the basal forebrain nucleus (Ncl. basalis of Meynert) degenerates in AD. The neuronal loss observed here is thought to be the major factor for the subsequent decrease of acetylcholine (ACh) in the cortical projection regions of its neurons. The hypothesis that the loss of ACh is permissive for neuroinflammatory events in cortical projection areas was suggested by finding that efferent stimulation of the vagus nerve decreases the release of TNF-α and various other proinflammatory mediators by macrophages of the gastrointestinal tract (Borovikova et al. 2000; Wang et al. 2003; Pavlov and Tracey 2005). This effect has been attributed to the presence of the α7 subunit of the ACh receptor. Interestingly, glial cells of the CNS such as astrocytes and microglia cells express the α7 subunit (Graham et al. 2003) and expression of the α7 subunit is increased in astrocytes derived from AD patients compared to age-matched controls (Teaktong et al. 2003).
Diabetes mellitus
Diabetes mellitus (DM) is characterized by either an impaired production of insulin due to primary islet cell death or by insulin resistance of normally insulin responsive cells. The latter form is often observed at later stages of life and termed non-insulin dependent DM (NIDDM), because most of these patients can achieve control over the blood glucose level without subcutaneous insulin administration. NIDDM often becomes apparent at a similar time as AD and is an established risk factor for the development of AD (Ristow 2004). While the connecting and underlying mechanisms are yet unclear, one may hypothesize that impaired immunological defences of NIDDM patients and frequent peripheral infections contribute to the course of AD. Specifically, frequent infections result in higher levels of circulating cytokines and bacterial cell wall components such as lipopolysaccharides. Animal experiments suggest that the peripheral administration of lipopolysaccharides can contribute and enhance existing brain inflammation especially within the hippocampus (Semmler et al. 2005; Weberpals et al. 2009), and ultimately lead to an increased rate of Aβ plaque deposition (Sheng et al. 2003). In addition, recent work by Fishel et al. (2005) demonstrated that mild hyperinsulinemia in humans provoked an increase in cytokines and prostanoids in the CSF, suggesting stimulation of inflammatory brain circuits in NIDDM.
Cytokine driven feedback mechanisms
Apart from self-propagation and direct cytopathic effect on neurons, cytokines may more directly contribute to AD related neurodegeneration. Thus, studies performed in transgenic animals suggest that cerebral amyloid deposition is increased under inflammatory conditions (Games et al. 1995; Guo et al. 2002). Moreover, these animals do not develop amyloid plaques unless inflammation is induced suggesting that inflammatory molecules either raise the susceptibility for Aβ deposition and aggregation or directly influence the APP processing pathway.
Several lines of evidence suggest that cytokines may promote Aβ formation, aggregation and deposition at multiple levels. For example, IL-1α has been implicated in the transformation of diffuse β-amyloid aggregates into β-amyloid plaques. Furthermore, when the amyloid plaque associated proteins α1-antichymotrypsin (ACT) or apolipoprotein E were added to preparations of synthetic Aβ peptide in vitro, increased polymerization of Aβ into amyloid filaments was observed (Ma et al. 1994). In addition, cytokines are able to transcriptionally upregulate BACE1 mRNA, protein and enzymatic activity (Sastre et al. 2003). BACE1 and presenilin-1 are key enzymes in neuronal Aβ formation since in their absence, Aβ synthesis is either abolished or considerably reduced (Walter et al. 2001). These results are in line with data concerning increased expression and activity of BACE1 in NT2 neurons exposed to oxidative stress (Tamagno et al. 2002), in experimental traumatic brain injury (Blasko et al. 2004), and in reactive astrocytes in chronic models of gliosis (Hartlage-Rubsamen et al. 2003). Prolonged cytokine treatment can also influence βAPP maturation and secretion in various cell types (Blasko et al. 2000; Blasko et al. 1999). Finally, cytokines have been shown to increase APP expression. For example, TGF-β treatment of human astrocytes markedly elevated APP mRNA levels, and also increased the half-life of APP message by at least fivefold (Amara et al. 1999). In addition, IL-1α and IL-1β increased APP synthesis by up to sixfold in primary human astrocytes and by 15-fold in human astrocytoma cells without changing the steady-state levels of APP mRNA (Rogers et al. 1999).
Cytokines may also be involved in NFT formation since chronic IL-1β release from implanted capsules led to phosphorylation of neurofilaments and increased phospho-tau immunoreactivity in the rat hippocampus (Sheng et al. 2000). Similar studies have been reported in cortical neuron cultures (Li et al. 2003). The possibility that IL-1 represents a link between Aβ formation with consequent microglial activation and tau phosphorylation was recently supported by work in the triple transgenic mouse model of AD. In these animals, tau phosphorylation appears to proceed amyloid deposition (Oddo et al. 2003, 2006). However, phosphorylated tau-epitopes were observed in young animals that were chronically treated with intraperitoneal LPS prior to the time that plaques appear (Kitazawa et al. 2005). These changes in tau were associated with increased levels of IL-1β and appeared to result from activation of cdk5 kinase.
Functional and structural consequences of neuroinflammation in AD
Over the past decade, numerous lines of accumulating evidence have strongly implicated that neuroinflammation contributes to AD pathogenesis. Irregardless at which time point of the disease it occurs, it seems clear that once initiated, inflammatory pathomechanisms can affect the AD brain. Functional and structural consequences may be differentiated to understand the various levels at which inflammation may contribute to AD.
Inflammation triggered functional impairment
LTP represents a key element of memory formation and consolidation. Since several cytokines including TNF-α, IL-1β and IFN-γ are able to suppress hippocampal LTP, the presence of these inflammatory molecules alone may be sufficient to induce neuronal dysfunction without structurally affecting neurons (Tancredi et al. 1992; Tancredi et al. 2000; Murray and Lynch 1998). Similar to cytokines, immunostimulated and iNOS derived NO also impairs LTP (Wang et al. 2004). Since iNOS expression is a long-lasting event, surrounding neurons face a sustained production of high NO concentrations. Functional impairment by increased cytokine and nitric oxide production may be of special relevance for the early stages of AD, when patients present with mild cognitive decline, often at a time when brain MRI scans fail to detect any atrophy of cortical or limbic structures.
Structural damage
In addition to neuronal dysfunction, several inflammation-evoked molecules may directly exert cytotoxic effects on neurons as already described. Despite descriptions of paradoxical protective effects for some of these mediators such as C5a (Osaka et al. 1999; Pasinetti et al. 1996; Tocco et al. 1997) or TNF-α (Feuerstein et al. 1994; Barger et al. 1995), the majority of experiments suggest that cytokines contribute to neuronal cell death. Further support for a more cytopathic role of these molecules comes from transgenic animal experiments showing that mice expressing these inflammatory proteins under brain-specific promoters invariably exhibit profound pathologic changes (Probert et al. 1995; Stalder et al. 1998; Wyss-Coray et al. 1997).
In addition to cytokine and complement generation, microglia and astrocytes may contribute by other means to neurodegeneration: While resting glial cells serve as an important source of various trophic factors such as GDNF, BDNF and others, chronic inflammatory activation may decrease the generation and release of these factors. It has therefore been suggested that inflammatory activation also leads to a significant trophic factor withdrawal which further contributes to neurodegeneration (Nagatsu and Sawada 2005).
Excitotoxic mechanisms significantly contribute to the neuronal loss in AD (Barger et al. 1993; Hensley et al. 1994; Smithswintosky et al. 1994). It is important to note, that the cytotoxic effects of iNOS derived NO and several proinflammatory molecules are not simply additive but potentiate NMDA or kainate induced excitotoxicity (Hewett et al. 1994; Morimoto et al. 2002).
Anti-inflammatory treatment strategies
Neuroinflammatory changes may even occur at early stages in the AD brain and significantly contribute to the pathogenesis of the disease. This raises the question whether therapeutic strategies can be developed which successfully target the ongoing inflammation.
NSAIDs as preventive treatment for AD
Epidemiological studies have suggested a beneficial effect of non-steroidal anti-inflammatory drugs (NSAIDs) in AD (Rogers et al. 1993; McGeer et al. 1996; Anthony et al. 2000; Breitner et al. 1995; Breitner 1996; Szekely et al. 2004; Stewart et al. 1997; Beard et al. 1998; In t’Veld et al. 2001). In particular, long-term NSAID therapy seem to delay the onset and progression of the disease, reduced symptomatic severity, and significantly slowed the rate of cognitive impairment (Rich et al. 1995), and at least one early treatment study with indomethacin suggested modest benefit in a small number of AD patients (Rogers et al. 1993). The epidemiological literature suggests an association between treatment duration and response for NSAIDs in preventing AD, with at least 2 years of exposure necessary to obtain full benefit (Breitner and Zandi 2001). Thus, the benefit may be greater the longer NSAIDs are taken (Etminan et al. 2003). Studies from aged controls and post mortem AD patients, both on reported NSAID medication, show that long-term NSAID therapy reduces the degree of plaque associated inflammation (Mackenzie and Munoz 1998; Mackenzie 2001; Alafuzoff et al. 2000). Nevertheless, these beneficial effects are limited to certain NSAIDS since naproxen and celecoxib were not able to modify AD (ADAPT Research Group et al. 2007).
Some of these beneficial effects have been further investigated in animal studies using APP overexpressing mice that display amyloid as well as inflammatory components of the disease. Several studies have demonstrated that the NSAID ibuprofen acts to reduce astrocyte and microglial activation and cytokine production in APP transgenic mice (Lim et al. 2000; Yan et al. 2003; Heneka et al. 2005b). Six-month treatment with NSAIDs in Tg2576 significantly delayed AD symptoms, including a decrease of 40–50% in amyloid deposition (Lim et al. 2000) and improved performance in behavioral tasks (Lim et al. 2001). This effect was also observed in a short-term administration of a subset of NSAIDs to young Tg2576 APP mice, which lowered the soluble levels of Aβ1–42 (Weggen et al. 2001; Eriksen et al. 2003). Moreover, treatment in the same mice with Meclofen, S-flurbiprofen or indomethacin reduced Aβ1–42 levels (Eriksen et al. 2003). Additionally, it has been shown that long-term treatment with ibuprofen and indomethacin significantly decreased Aβ1–40 and Aβ1–42 levels in both cortex and hippocampus of APP transgenic (Tg2576) mice (Yan et al. 2003; Sung et al. 2004).
To date, the underlying mechanisms by which NSAIDs prevent AD are unclear, however evidence for several mechanisms has been put forward and there is the distinct possibility that actions at multiple rather than a single level of AD relevant pathology account for the observed beneficial effects of NSAIDs. Potential mechanisms include:
-
(1)
Protection against Aβ aggregation: It has been shown that certain NSAIDs may alter the β-sheet conformation of Aβ affecting the aggregation of Aβ peptides in vitro (Agdeppa et al. 2003; Thomas et al. 2001). Another report indicates that NSAIDs could induce the expression of amyloid binding proteins such as transthyretin, subsequently decreasing Aβ aggregation (Ray et al. 1998).
-
(2)
Effect on amyloid precursor protein (APP processing: The protective effect of NSAIDs has been associated with decreased secretion of Aβ peptides and soluble APP, although there is still debate about the molecular mechanism involved (Weggen et al. 2001; Sastre et al. 2003). In particular, a subset of NSAIDs appears to directly affect the generation of Aβ1–42, which is the most amyloidogenic form of Aβ. This subset of NSAIDs was shown to shift the cleavage products of APP to shorter, less fibrillogenic forms, indicating that NSAIDs could have an allosteric inhibitory effect on γ-secretase by altering PS1 conformation (Lleo et al. 2004).
-
(3)
Inhibition of an alternate pathway: Ibuprofen has been shown to reduce pro-amyloidogenic α1-antichymotrypsin, an effect likely mediated by decreasing IL-1β (Morihara et al. 2005).
-
(4)
Inhibition of cyclooxygenases: The canonical targets of NSAIDs are COX-1 and -2. Prostaglandin E2 levels are increased fivefold in the CSF of probable AD patients (Montine et al. 1999) and COX-2 products are associated with neurodegeneration as discussed above.
-
(5)
Several NSAIDs target the peroxisome proliferator activated receptor γ (PPARγ) (Lehmann et al. 1997), a nuclear hormone receptor studied for more than a decade by endocrinologists for its ability to increase insulin sensitivity. As described in more detail below, some of the beneficial effects ascribed to NSAID medication may be mediated by PPARγ activation.
-
(6)
Prevention of ectopic neuronal cell cycle events, which indicate early neuronal vulnerability along with reduction of microglial activation. Importantly, this effect was independent of any modulation of the APP processing or steady state levels of Aβ1–40 or Aβ1–42 (Varvel et al. 2009).
Despite these multiple mechanisms, recent clinical trials with selective COX-2 inhibitors and the mixed COX-1/COX-2 inhibitor naproxen have been uniformly disappointing (Reines et al. 2004; Thal et al. 2005). This suggests that pathological changes may be too far advanced by the time of clinical diagnosis. Alternatively, off-target effects of NSAIDs may be critical and not maximally achieved with the specific drugs used in recent trials. Along these lines, neither COX-2 inhibitors nor naproxen inhibit Aβ1–42 generation in vitro or in vivo (Eriksen et al. 2003).
PPARγ as target for NSAIDs
Ibuprofen, indomethacin and naproxen are among the five most prescribed NSAIDs, which have potentially decreased the risk for AD (In t’Veld et al. 2001). Interestingly, they are all agonists of the PPARγ (Lehmann et al. 1997). PPARs represent ligand-activated transcription factors that belong to a nuclear receptor superfamily and two isoforms, i.e. PPARγ1 (Kliewer et al. 1994) and PPARγ2 (Tontonoz et al. 1994) are formed from the same gene by alternative mRNA splicing. PPARγ2 is specifically expressed in adipose tissue and differs from PPARγ1 by the presence of 30 additional N-terminal amino acids that confer a tissue-specific transactivation function. PPARγ1 is the predominant, if not the only, isoform in all other tissues, including skeletal muscle and liver (Li et al. 2000).
PPARγ forms heterodimers with retinoid X receptors (RXR) (Tugwood et al. 1992) and upon ligand activation, the PPAR/RXR heterodimer recruits coactivators and binds to sequence-specific PPAR response elements (PPRE) present in the promoter region of several target genes. Alternatively, PPARγ can inhibit specific gene expression without direct binding to the gene promoter, since transrepression of several genes, i.e. iNOS and COX-2, is achieved in part by antagonizing the activities of transcription factors STAT1, NF-κB and AP-1 (Li et al. 2000; Daynes and Jones 2002; Kelly et al. 2004; Heneka et al. 2003).
PPARγ is involved in several cellular functions, including control of glucose homeostasis, regulation of systemic insulin sensitivity, cell differentiation and cholesterol metabolism (Vamecq and Latruffe 1999; Patsouris et al. 2004). The PPARγ gene knockout animal is embryonic lethal, due to essential roles in adipose, kidney and placental development (Barak et al. 1999). A role in the regulation of immune and inflammatory responses was suggested by the findings that PPARγ is expressed in macrophages and that receptor activation results in the inhibition of various inflammatory events, such as the production of IL-1β, TNF-α, IL-6 and iNOS (Ricote et al. 1998). PPARγ also appears to be involved in proliferation and production of IL-2 by T-lymphocytes and IFN-γ expression in murine CD4 and CD8 cells (Cunard et al. 2004).
In the brain, several anti-inflammatory effects of NSAIDs may be in part mediated through the activation of PPARγ (Landreth and Heneka 2001), since it has been shown that PPARγ agonists protect neurons from cytokine-mediated death (Heneka et al. 1999). Combs et al. (2000) reported that PPARγ agonists, including ibuprofen, inhibited Aβ-mediated microglial activation and neurotoxicity using in vitro models. Similar anti-inflammatory effects of PPARγ agonists and ibuprofen were also observed following infusion of immunostimulants into rodent brain (Heneka et al. 2000). In line with these findings, recent studies have documented the salutary effects of PPARγ-agonists in animal models of multiple sclerosis (Feinstein et al. 2002a; Niino et al. 2001; Diab et al. 2004; Natarajan and Bright 2002) and Parkinson’s disease (Dehmer et al. 2004; Breidert et al. 2002) and Amyotrophic lateral sclerosis (Schutz et al. 2005; Kiaei et al. 2005). The potent anti-inflammatory effects of PPARγ-agonists suggest that they may have beneficial effects in treating other CNS diseases with an inflammatory component.
PPARγ activation is achieved by binding to a specific receptor binding pocket by various endogenous and synthetic ligands (Lehmann et al. 1997; Yki-Jarvinen 2004). Thus, PPARγ is stimulated best by 9-HODE (hydroxyoctadeca-9Z,11E-dienoic acid, 13-HODE and 15-deoxy-Δ12,14-prostaglandin J2 (15dPGJ2, although there is now considerable controversy as to whether 15d-PGJ2 is actually a biologically relevant PPARγ activator or the majority of its action is mediated by inhibition of IκB kinase (IKKα and subsequently NFκB activation (Rossi et al. 2000). However, it has been reported recently that 15d-PGJ2 is produced in vivo, and in large quantities by macrophages in vitro. Synthetic PPARγ ligands are used for their potent antidiabetic effects. In the United States, two ligands of the thiazolidinedione (TZD class, rosiglitazone and pioglitazone, have been approved for the treatment of NIDDM. Both substances bind PPARγ with high affinity and enhance insulin-mediated glucose uptake by increasing insulin sensitivity. In addition to receptor mediated effects some of these substances exert anti-inflammatory effects independently of PPARγ activation (Chawla et al. 2001; Feinstein et al. 2005).
Two in vivo investigations on the effects of PPARγ activation in APP transgenic mice have been reported. An acute 7 day oral treatment of 10-month-old APPV717I mice with the PPARγ agonist pioglitazone or the NSAID ibuprofen resulted in a reduced number of activated microglia and reactive astrocytes in the hippocampus and frontal cortex (Heneka et al. 2005b). Drug treatment reduced expression of the proinflammatory enzymes COX-2 and iNOS and the levels of BACE1. The same mice presented decreased Aβ42 levels, while a non-statistically significant reduction of about 20–25% in Aβ40 levels was found. Furthermore, intracellular Aβ staining was reduced in mice treated with ibuprofen or pioglitazone, indicating that PPARγ activation is involved in the regulation of Aβ generation (Sastre et al. 2006). A different study indicated that treatment of 11-month-old Tg2576 mice overexpressing human APP with the NSAID ibuprofen and PPARγ agonist pioglitazone for 16 weeks, only modestly reduced SDS-soluble Aβ levels and did not affect amyloid plaque burden (Yan et al. 2003). Since only 20% of pioglitazone crosses the blood brain barrier, and the study by Heneka et al. utilized twice the concentration used by Yan and colleagues, the observed difference may be explained by the drug concentrations applied.
All PPARs have been shown to be present in the CNS and to exhibit both unique and overlapping patterns of expression in various areas and at different developmental stages. The levels of expression of PPARγ in post-mortem brain sections from AD patients have been examined (Sastre et al. 2006) and immunohistochemical assessment of frontal cortex revealed that PPARγ is expressed in astrocytes and neurons. It has previously been suggested that AD brains contain increased levels of PPARγ in the cytosolic fraction compared to healthy controls (Kitamura et al. 1999). These results are in contrast with our own analysis showing a 40% reduction of PPARγ protein levels in AD patients compared to controls. In addition, PPARγ protein levels and its binding to a PPRE in the BACE1 promoter were decreased in AD brains (Sastre et al. 2006). Combined, these findings point to a direct role of PPARγ in the regulation of BACE1 transcription and activity in AD, ultimately facilitating Aβ generation.
Conclusions and future directions
Increasing evidence suggests that inflammation significantly contributes to the pathogenesis of AD. The generation and secretion of proinflammatory mediators may interact at multiple levels with neurodegenerative mechanisms. Thus, several proinflammatory cytokines cannot only induce neuropathic mechanisms and thereby contribute to neuronal death, but are also able to influence classical neurodegenerative pathways such as APP processing. The concomitant release of anti-inflammatory mediators may partly antagonize this action ultimately contributing to the chronicity of the disease. Several AD specific mechanisms such as locus ceruleus and nucleus basalis Meynert cell death may facilitate the occurrence of neuroinflammation. Future studies need to determine the underlying mechanisms and means by which the course of the disease can be influenced. Additional information on how inflammatory mediators and excitotoxic factors potentiate their detrimental effects is required. Additionally, more information is needed regarding the extent to which inflammatory mediators functionally impair cognition and memory.
Clinically, novel approaches to visualize early neuroinflammation in the human brain are needed to improve the monitoring and control of therapeutic strategies that target inflammatory and other pathological mechanisms. Similarly, more insight in the role of genetic factors that transmit a disposition for the disease should improve the detection of people at risk to develop AD.
References
Abbas N, Bednar I, Mix E, Marie S, Paterson D, Ljungberg A, Morris C, Winblad B, Nordberg A, Zhu J (2002) Up-regulation of the inflammatory cytokines IFN-gamma and IL-12 and down-regulation of IL-4 in cerebral cortex regions of APP(SWE) transgenic mice. J Neuroimmunol 126:50–57
Abramov AY, Canevari L, Duchen MR (2003) Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 23:5088–5095
ADAPT Research Group, Lyketsos CG, Breitner JC, Green RC, Martin BK, Meinert C, Piantadosi S, Sabbagh M (2007) Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 68:1800–1808
Agdeppa ED, Kepe V, Petri A, Satyamurthy N, Liu J, Huang SC, Small GW, Cole GM, Barrio JR (2003) In vitro detection of (S)-naproxen and ibuprofen binding to plaques in the Alzheimer’s brain using the positron emission tomography molecular imaging probe 2-(1-[6-[(2-[(18)F]fluoroethyl)(methyl)amino]-2-naphthyl]ethylidene)malono nitrile. Neuroscience 117:723–730
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543
Alafuzoff I, Overmyer M, Helisalmi S, Soininen H (2000) Lower counts of astroglia and activated microglia in patients with Alzheimer’s disease with regular use of non-steroidal anti-inflammatory drugs. J Alzheimers Dis 2:37–46
Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ (2010) Amyloid-{beta} aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 30:3326–3338
Almer G, Vukosavic S, Romero N, Przedborski S (1999) Inducible nitric oxide synthase up-regulation in a transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem 72:2415–2425
Almer G, Guegan C, Teismann P, Naini A, Rosoklija G, Hays AP, Chen C, Przedborski S (2001) Increased expression of the pro-inflammatory enzyme cyclooxygenase-2 in amyotrophic lateral sclerosis. Ann Neurol 49:176–185
Aloisi F, Care A, Borsellino G, Gallo P, Rosa S, Bassani A, Cabibbo A, Testa U, Levi G, Peschle C (1992) Production of hemolymphopoietic cytokines (Il-6, Il-8, colony-stimulating factors) by normal human astrocytes in response to Il-1-beta and tumor-necrosis-factor-alpha. J Immunol 149:2358–2366
Amara FM, Junaid A, Clough RR, Liang BH (1999) TGF-beta(1), regulation of Alzheimer amyloid precursor protein mRNA expression in a normal human astrocyte cell line: mRNA stabilization. Mol Brain Res 71:42–49
Anthony JC, Breitner JC, Zandi PP, Meyer MR, Jurasova I, Norton MC, Stone SV (2000) Reduced prevalence of AD in users of NSAIDs and H2 receptor antagonists: the Cache County study. Neurology 54:2066–2071
Apelt J, Schliebs R (2001) Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res 894:21–30
Bach JH, Chae HS, Rah JC, Lee MW, Park CH, Choi SH, Choi JK, Lee SH, Kim YS, Kim KY, Lee WB, Suh YH, Kim SS (2001) C-terminal fragment of amyloid precursor protein induces astrocytosis. J Neurochem 78:109–120
Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM (1999) PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell 4:585–595
Barger SW, Harmon AD (1997) Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388:878–881
Barger SW, Smithswintosky VL, Rydel RE, Mattson MP (1993) Beta-amyloid precursor protein mismetabolism and loss of calcium homeostasis in Alzheimers-disease. Alzheimers Dis: Amyloid Precusor Proteins, Signal Transduct, Neuronal Transplant 695:158–164
Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP (1995) Tumor necrosis factors alpha and beta protect neurons against amyloid beta-peptide toxicity: evidence for involvement of a kappa B-binding factor and attenuation of peroxide and Ca2+ accumulation. Proc Natl Acad Sci USA 92:9328–9332
Bate C, Veerhuis R, Eikelenboom P, Williarns A (2004) Microglia kill amyloid-beta(1–42) damaged neurons by a CD14-dependent process. Neuroreport 15:1427–1430
Bauer MK, Lieb K, Schulze-Osthoff K, Berger M, Gebicke-Haerter PJ, Bauer J, Fiebich BL (1997) Expression and regulation of cyclooxygenase-2 in rat microglia. Eur J Biochem 243:726–731
Beard CM, Waring SC, O’Brien PC, Kurland LT, Kokmen E (1998) Nonsteroidal anti-inflammatory drug use and Alzheimer’s disease: a case-control study in Rochester, Minnesota, 1980 through 1984. Mayo Clin Proc 73:951–955
Beglopoulos V, Sun X, Saura CA, Lemere CA, Kim RD, Shen J (2004) Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem 279:46907–46914
Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118:103–113
Benveniste EN, Nguyen VT, O’Keefe GM (2001) Immunological aspects of microglia: relevance to Alzheimer’s disease. Neurochem Int 39:381–391
Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR (1999) Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging 20:581–589
Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A (2001a) CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4:702–710
Bezzi P, Domercq M, Vesce S, Volterra A (2001b) Neuron-astrocyte cross-talk during synaptic transmission: physiological and neuropathological implications. Prog Brain Res 132:255–265
Bilak M, Wu L, Wang Q, Haughey N, Conant K, St Hillaire C, Andreasson K (2004) PGE2 receptors rescue motor neurons in a model of amyotrophic lateral sclerosis. Ann Neurol 56:240–248
Blasko I, Marx F, Steiner E, Hartmann T, Grubeck-Loebenstein B (1999) TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J 13:63–68
Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B (2000) Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1–40 and Abeta1–42 by human astrocytes. Neurobiol Dis 7:682–689
Blasko I, Beer R, Bigl M, Apelt J, Franz G, Rudzki D, Ransmayr G, Kampfl A, Schliebs R (2004) Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease beta-secretase (BACE-1). J Neural Transm 111:523–536
Boje KM, Arora PK (1992) Microglial-produced nitric-oxide and reactive nitrogen-oxides mediate neuronal cell-death. Brain Res 587:250–256
Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 28:4283–4292
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405:458–462
Botchkina GI, Meistrell ME III, Botchkina IL, Tracey KJ (1997) Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med 3:765–781
Botto M (1998) C1q knock-out mice for the study of complement deficiency in autoimmune disease. Exp Clin Immunogenet 15:231–234
Braak H, Braak E (1998) Evolution of neuronal changes in the course of Alzheimer’s disease. J Neural Transm Suppl 53(127–40):127–140
Braida D, Sacerdote P, Panerai AE, Bianchi M, Aloisi AM, Iosue S, Sala M (2004) Cognitive function in young and adult IL (interleukin)-6 deficient mice. Behav Brain Res 153:423–429
Breder CD, Tsujimoto M, Terano Y, Scott DW, Saper CB (1993) Distribution and characterization of tumor necrosis factor-alpha-like immunoreactivity in the murine central nervous system. J Comp Neurol 337:543–567
Bredt DS, Snyder SH (1990) Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci USA 87:682–685
Breidert T, Callebert J, Heneka MT, Landreth G, Launay JM, Hirsch EC (2002) Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. J Neurochem 82:615–624
Breitner JC (1996) Inflammatory processes and antiinflammatory drugs in Alzheimer’s disease: a current appraisal. Neurobiol Aging 17:789–794
Breitner JC, Zandi PP (2001) Do nonsteroidal antiinflammatory drugs reduce the risk of Alzheimer’s disease? N Engl J Med 345:1567–1568
Breitner JC, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, Pericak-Vance MA, Saunders AM (1995) Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging 16:523–530
Brown GC, Bal-Price A (2003) Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 27:325–355
Butovsky O, Talpalar AE, Ben Yaakov K, Schwartz M (2005) Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci 29:381–393
Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Alvarez D, Al Dalain S, Martinez G, Leon OS, Springer JE (2003) Assessment of the relative contribution of COX-1 and COX-2 isoforms to ischemia-induced oxidative damage and neurodegeneration following transient global cerebral ischemia. J Neurochem 86:545–555
Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang DR, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9:917–924
Castell JV, Andus T, Kunz D, Heinrich PC (1989) Interleukin-6. The major regulator of acute-phase protein synthesis in man and rat. Ann N Y Acad Sci 557:87–99 (discussion 100-1)
Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, Zubair AC, Dickson D, Golde TE, Das P (2010) Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J 24:548–559
Chao CC, Hu S, Sheng WS, Bu D, Bukrinsky MI, Peterson PK (1996) Cytokine-stimulated astrocytes damage human neurons via a nitric oxide mechanism. Glia 16:276–284
Charo IF, Ransohoff RM (2006) Mechanisms of disease—the many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 354:610–621
Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM (2001) PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med 7:48–52
Chen KQ, Iribarren P, Hu JY, Chen JH, Gong WH, Cho EH, Lockett S, Dunlop NM, Wang JM (2006) Activation of toll-like receptor 2 on microglia promotes cell uptake of Alzheimer disease-associated amyloid beta peptide. J Biol Chem 281:3651–3659
Chong Y (1997) Effect of a carboxy-terminal fragment of the Alzheimer’s amyloid precursor protein on expression of proinflammatory cytokines in rat glial cells. Life Sci 61:2323–2333
Ciesielski-Treska J, Ulrich G, Aunis D, Bader MF (1998a) Chromogranin A triggers the expression of neurotoxic phenotype in microglia and induces apoptosis in neuronal/microglial cocultures. Eur J Neurosci 10:164
Ciesielski-Treska J, Ulrich G, Taupenot L, Chasserot-Golaz S, Corti A, Aunis D, Bader MF (1998b) Chromogranin A induces a neurotoxic phenotype in brain microglial cells. J Biol Chem 273:14339–14346
Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE (2000) Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci 20:558–567
Combs CK, Karlo JC, Kao SC, Landreth GE (2001) beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci 21:1179–1188
Corradin SB, Mauel J, Donini SD, Quattrocchi E, Ricciardi-Castagnoli P (1993) Inducible nitric oxide synthase activity of cloned murine microglial cells. Glia 7:255–262
Cunard R, Eto Y, Muljadi JT, Glass CK, Kelly CJ, Ricote M (2004) Repression of IFN-gamma expression by peroxisome proliferator-activated receptor gamma. J Immunol 172:7530–7536
D’Andrea MR, Cole GM, Ard MD (2004) The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging 25:675–683
Davis S, Laroche S (2003) What can rodent models tell us about cognitive decline in Alzheimer’s disease? Mol Neurobiol 27:249–276
Dawson VL, Brahmbhatt HP, Mong JA, Dawson TM (1994) Expression of inducible nitric oxide synthase causes delayed neurotoxicity in primary mixed neuronal-glial cortical cultures. Neuropharmacology 33:1425–1430
Daynes RA, Jones DC (2002) Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol 2:748–759
DeGiorgio LA, Shimizu Y, Chun HS, Cho BP, Sugama S, Joh TH, Volpe BT (2002) APP knockout attenuates microglial activation and enhances neuron survival in substantia nigra compacta after axotomy. Glia 38:174–178
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem 88:494–501
del Zoppo G, Ginis I, Hallenbeck JM, Iadecola C, Wang X, Feuerstein GZ (2000) Inflammation and stroke: putative role for cytokines, adhesion molecules and iNOS in brain response to ischemia. Brain Pathol 10:95–112
Demerle-Pallardy C, Lonchampt MO, Chabrier PE, Braquet P (1993) Nitric oxide synthase induction in glial cells: effect on neuronal survival. Life Sci 52:1883–1890
Dewitt DA, Perry G, Cohen M, Doller C, Silver J (1998) Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol 149:329–340
Diab A, Hussain RZ, Lovett-Racke AE, Chavis JA, Drew PD, Racke MK (2004) Ligands for the peroxisome proliferator-activated receptor-gamma and the retinoid X receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. J Neuroimmunol 148:116–126
Dickson DW, Lee SC, Mattiace LA, Yen SH, Brosnan C (1993) Microglia and cytokines in neurological disease, with special reference to AIDS and Alzheimer’s disease. Glia 7:75–83
Doi Y, Mizuno T, Maki Y, Jin S, Mizoguchi H, Ikeyama M, Doi M, Michikawa M, Takeuchi H, Suzumura A (2009) Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid beta neurotoxicity in in vitro and in vivo models of Alzheimer’s disease. Am J Pathol 175:2121–2132
Du YS, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW, Rajavashisth T, Chen X, Godman GC, Stern D, Schmidt AM (1997) Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci USA 94:5296–5301
Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LBA, Lipsky PE (1998) Cyclooxygenase in biology and disease. FASEB J 12:1063–1073
Eikelenboom P, van Gool WA (2004) Neuroinflammatory perspectives on the two faces of Alzheimer’s disease. J Neural Transm 111:281–294
Eikelenboom P, Veerhuis R (1996) The role of complement and activated microglia in the pathogenesis of Alzheimer’s disease. Neurobiol Aging 17:673–680
Eikelenboom P, Hack CE, Rozemuller JM, Stam FC (1989) Complement activation in amyloid plaques in Alzheimers dementia. Virchows Arch B Cell Pathol Incl Mol Pathol 56:259–262
Eikelenboom P, Zhan SS, van Gool WA, Allsop D (1994) Inflammatory mechanisms in Alzheimer’s disease. Trends Pharmacol Sci 15:447–450
Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE (2003) NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest 112:440–449
Etminan M, Gill S, Samii A (2003) Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ 327:128
Fassbender K, Walter S, Kühl S, Landmann R, Ishii K, Bertsch T, Stalder AK, Muehlhauser F, Liu Y, Ulmer AJ, Rivest S, Lentschat A, Gulbins E, Jucker M, Staufenbiel M, Brechtel K, Walter J, Multhaup G, Penke B, Adachi Y, Hartmann T, Beyreuther K (2004) The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J 18:203–205
Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, Landreth GE, Pershadsingh HA, Weinberg G, Heneka MT (2002a) Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol 51:694–702
Feinstein DL, Heneka MT, Gavrilyuk V, Dello RC, Weinberg G, Galea E (2002b) Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int 41:357–365
Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, Russo CD (2005) Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol 70:177–188
Feuerstein GZ, Liu T, Barone FC (1994) Cytokines, inflammation, and brain injury: role of tumor necrosis factor-alpha. Cerebrovasc Brain Metab Rev 6:341–360
Fiebich BL, Schleicher S, Spleiss O, Czygan M, Hull M (2001) Mechanisms of prostaglandin E2-induced interleukin-6 release in astrocytes: possible involvement of EP4-like receptors, p38 mitogen-activated protein kinase and protein kinase C. J Neurochem 79:950–958
Fishel MA, Watson GS, Montine TJ, Wang Q, Green PS, Kulstad JJ, Cook DG, Peskind ER, Baker LD, Goldgaber D, Nie W, Asthana S, Plymate SR, Schwartz MW, Craft S (2005) Hyperinsulinemia provokes synchronous increases in central inflammation and {beta}-amyloid in normal adults. Arch Neurol 62:1539–1544
Floden AM, Combs CK (2006) beta-Amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J Neurosci 26:4644–4648
Fonseca MI, Zhou J, Botto M, Tenner AJ (2004) Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci 24:6457–6465
Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, Laferla FM, Taylor SM, Woodruff TM, Tenner AJ (2009) Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol 183:1375–1383
Forno L (1966) Pathology of Parkinsonism: a preliminary report of 24 cases. J Neurosurg (Supplement, Part II):266–271
Frank S, Copanaki E, Burbach GJ, Muller UC, Deller T (2009) Differential regulation of toll-like receptor mRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neurosci Lett 453:41–44
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152:307–317
Freedman R, Foote SL, Bloom FE (1975) Histochemical characterization of a neocortical projection of the nucleus locus coeruleus in the squirrel monkey. J Comp Neurol 164:209–231
Frei K, Nohava K, Malipiero UV, Schwerdel C, Fontana A (1992) Production of macrophage colony-stimulating factor by astrocytes and brain macrophages. J Neuroimmunol 40:189–196
Fritschy JM, Grzanna R (1989) Immunohistochemical analysis of the neurotoxic effects of DSP-4 identifies two populations of noradrenergic axon terminals. Neuroscience 30:181–197
Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, Laferla FM, Kretzschmar H, Herms J (2010) Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci 12:1358–1360
Galea E, Feinstein DL, Reis DJ (1992) Induction of calcium-independent nitric oxide synthase activity in primary rat glial cultures. Proc Natl Acad Sci USA 89:10945–10949
Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373:523–527
Gasque P, Fontaine M, Morgan BP (1995) Complement expression in human brain. Biosynthesis of terminal pathway components and regulators in human glial cells and cell lines. J Immunol 154:4726–4733
Gavrilyuk V, Dello RC, Heneka MT, Pelligrino D, Weinberg G, Feinstein DL (2002) Norepinephrine increases I kappa B alpha expression in astrocytes. J Biol Chem 277:29662–29668
German DC, Manaye KF, White CL III, Woodward DJ, McIntire DD, Smith WK, Kalaria RN, Mann DM (1992) Disease-specific patterns of locus coeruleus cell loss. Ann Neurol 32:667–676
Glabinski AR, Ransohoff RM (1999) Chemokines and chemokine receptors in CNS pathology. J Neurovirol 5:3–12
Gong C, Qin Z, Betz AL, Liu XH, Yang GY (1998) Cellular localization of tumor necrosis factor alpha following focal cerebral ischemia in mice. Brain Res 801:1–8
Graham AJ, Ray MA, Perry EK, Jaros E, Perry RH, Volsen SG, Bose S, Evans N, Lindstrom J, Court JA (2003) Differential nicotinic acetylcholine receptor subunit expression in the human hippocampus. J Chem Neuroanat 25:97–113
Grammas P, Ovase R (2001) Inflammatory factors are elevated in brain microvessels in Alzheimer’s disease. Neurobiol Aging 22:837–842
Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL, Jucker M (2009) Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci 12:1361–1363
Griffin WST (2000) IL-1 and the cytokine cycle in Alzheimer’s disease. J Neurochem 74:S52
Griffin WS, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, Roberts GW, Mrak RE (1998) Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol 8:65–72
Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM (2007) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging 28:327–335
Guo JT, Yu J, Grass D, de Beer FC, Kindy MS (2002) Inflammation-dependent cerebral deposition of serum amyloid a protein in a mouse model of amyloidosis. J Neurosci 22:5900–5909
Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB (1992) Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature 359:322–325
Hanisch UK (2002) Microglia as a source and target of cytokines. Glia 40:140–155
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12:383–388
Hartlage-Rubsamen M, Zeitschel U, Apelt J, Gartner U, Franke H, Stahl T, Gunther A, Schliebs R, Penkowa M, Bigl V, Rossner S (2003) Astrocytic expression of the Alzheimer’s disease beta-secretase (BACE1) is stimulus-dependent. Glia 41:169–179
He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y (2007) Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer’s mice. J Cell Biol 178:829–841
Hein AM, Stasko MR, Matousek SB, Scott-McKean JJ, Maier SF, Olschowka JA, Costa AC, O'Banion MK (2010) Sustained hippocampal IL-1beta overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun 24(2):243–253
Heneka MT, Feinstein DL (2001) Expression and function of inducible nitric oxide synthase in neurons. J Neuroimmunol 114:8–18
Heneka MT, O’Banion MK (2007) Inflammatory processes in Alzheimer’s disease. J Neuroimmunol 184:69–91
Heneka MT, Loschmann PA, Gleichmann M, Weller M, Schulz JB, Wullner U, Klockgether T (1998) Induction of nitric oxide synthase and nitric oxide-mediated apoptosis in neuronal PC12 cells after stimulation with tumor necrosis factor-alpha/lipopolysaccharide. J Neurochem 71:88–94
Heneka MT, Feinstein DL, Galea E, Gleichmann M, Wullner U, Klockgether T (1999) Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J Neuroimmunol 100:156–168
Heneka MT, Klockgether T, Feinstein DL (2000) Peroxisome proliferator-activated receptor-gamma ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J Neurosci 20:6862–6867
Heneka MT, Wiesinger H, Dumitrescu-Ozimek L, Riederer P, Feinstein DL, Klockgether T (2001) Neuronal and glial coexpression of argininosuccinate synthetase and inducible nitric oxide synthase in Alzheimer disease. J Neuropathol Exp Neurol 60:906–916
Heneka MT, Galea E, Gavriluyk V, Dumitrescu-Ozimek L, Daeschner J, O’Banion MK, Weinberg G, Klockgether T, Feinstein DL (2002) Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: implications for Alzheimer’s disease. J Neurosci 22:2434–2442
Heneka MT, Gavrilyuk V, Landreth GE, O’Banion MK, Weinberg G, Feinstein DL (2003) Noradrenergic depletion increases inflammatory responses in brain: effects on IkappaB and HSP70 expression. J Neurochem 85:387–398
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven F (2005a) Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation 2:22
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O’Banion K, Klockgether T, Van Leuven F, Landreth GE (2005b) Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain 128:1442–1453
Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M (2006) Locus Ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci 26:1343–1354
Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK, Kummer MP (2010) Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci USA 107:6058–6063
Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA (1994) A model for beta-amyloid aggregation and neurotoxicity based on free-radical generation by the peptide—relevance to Alzheimer-disease. Proc Natl Acad Sci USA 91:3270–3274
Hess DC, Abe T, Hill WD, Studdard AM, Carothers J, Masuya M, Fleming PA, Drake CJ, Ogawa M (2004) Hematopoietic origin of microglial and perivascular cells in brain. Exp Neurol 186:134–144
Hesselgesser J, Horuk R (1999) Chemokine and chemokine receptor expression in the central nervous system. J Neurovirol 5:13–26
Hewett SJ, Csernansky CA, Choi DW (1994) Selective potentiation of NMDA-induced neuronal injury following induction of astrocytic iNOS. Neuron 13:487–494
Heyser CJ, Masliah E, Samimi A, Campbell IL, Gold LH (1997) Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc Natl Acad Sci USA 94:1500–1505
Hickman SE, Allison EK, El Khoury J (2008) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci 28:8354–8360
Ho L, Purohit D, Haroutunian V, Luterman JD, Willis F, Naslund J, Buxbaum JD, Mohs RC, Aisen PS, Pasinetti GM (2001) Neuronal cyclooxygenase 2 expression in the hippocampal formation as a function of the clinical progression of Alzheimer disease. Arch Neurol 58:487–492
Ho GJ, Drego R, Hakimian E, Masliah E (2005) Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr Drug Targets Inflamm Allergy 4:247–256
Hoozemans JJM, Rozemuller AJM, Janssen I, De Groot CJA, Veerhuis R, Eikelenboom P (2001) Cyclooxygenase expression in microglia and neurons in Alzheimer’s disease and control brain. Acta Neuropathol 101:2–8
Hoozemans J, Veerhuis R, Rozemuller A, Arendt T, Eikelenboom P (2004) Neuronal immunoreactivity for COX-2 and phosphorylation of prb precede P38 MAPK activation and neurofibrillary changes in ad temporal cortex. Neurobiol Aging 25:S416
Hori K, Burd PR, Furuke K, Kutza J, Weih KA, Clouse KA (1999) Human immunodeficiency virus-1-infected macrophages induce inducible nitric oxide synthase and nitric oxide (NO) production in astrocytes: astrocytic NO as a possible mediator of neural damage in acquired immunodeficiency syndrome. Blood 93:1843–1850
Horuk R, Martin AW, Wang ZX, Schweitzer L, Gerassimides A, Guo HH, Lu ZH, Hesselgesser J, Perez HD, Kim J, Parker J, Hadley TJ, Peiper SC (1997) Expression of chemokine receptors by subsets of neurons in the central nervous system. J Immunol 158:2882–2890
Hoshino T, Namba T, Takehara M, Nakaya T, Sugimoto Y, Araki W, Narumiya S, Suzuki T, Mizushima T (2009) Prostaglandin E2 stimulates the production of amyloid-beta peptides through internalization of the EP4 receptor. J Biol Chem 284:18493–18502
In t’Veld, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med 345:1515–1521
Iribarren P, Chen KQ, Hu JY, Gong WH, Cho EH, Lockett S, Uranchimeg B, Wang JM (2005) CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid beta 1–42 peptide by up-regulating the expression of the G-protein-coupled receptor mFPR2. FASEB J 19:2032–2034
Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T (1997) Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin Neurosci 51:135–138
Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D (1989) Relationship of Microglia and Astrocytes to Amyloid Deposits of Alzheimer-Disease. J Neuroimmunol 24:173–182
Iversen LL, Rossor MN, Reynolds GP, Hills R, Roth M, Mountjoy CQ, Foote SL, Morrison JH, Bloom FE (1983) Loss of pigmented dopamine-beta-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer’s type. Neurosci Lett 39:95–100
Jana M, Palencia CA, Pahan K (2008) Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer’s disease. J Immunol 181:7254–7262
Janelsins MC, Mastrangelo MA, Park KM, Sudol KL, Narrow WC, Oddo S, Laferla FM, Callahan LM, Federoff HJ, Bowers WJ (2008) Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol 173:1768–1782
Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J (2008) Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 28:11650–11661
Kalaria RN, Andorn AC, Tabaton M, Whitehouse PJ, Harik SI, Unnerstall JR (1989) Adrenergic receptors in aging and Alzheimer’s disease: increased beta 2-receptors in prefrontal cortex and hippocampus. J Neurochem 53:1772–1781
Kamboh MI, Sanghera DK, Ferrell RE, DeKosky ST (1995) APOE*4-associated Alzheimer’s disease risk is modified by alpha 1-antichymotrypsin polymorphism. Nat Genet 10:486–488
Kelley KA, Ho L, Winger D, Freire-Moar J, Borelli CB, Aisen PS, Pasinetti GM (1999) Potentiation of excitotoxicity in transgenic mice overexpressing neuronal cyclooxygenase-2. Am J Pathol 155:995–1004
Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S (2004) Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol 5:104–112
Kiaei M, Kipiani K, Chen JY, Calingasan NY, Beal MF (2005) Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol 191:331–336
Kitamura Y, Shimohama S, Koike H, Kakimura J, Matsuoka Y, Nomura Y, Gebicke-Haerter PJ, Taniguchi T (1999) Increased expression of cyclooxygenases and peroxisome proliferator-activated receptor-gamma in Alzheimer’s disease brains. Biochem Biophys Res Commun 254:582–586
Kitazawa M, Oddo S, Yamasaki TR, Green KN, Laferla FM (2005) Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci 25:8843–8853
Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, Umesono K, Evans RM (1994) Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA 91:7355–7359
Koenigsknecht J, Landreth G (2004) Microglial phagocytosis of fibrillar beta-amyloid through a beta1 integrin-dependent mechanism. J Neurosci 24:9838–9846
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318
Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ (2009) Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323:1211–1215
Landreth GE, Heneka MT (2001) Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol Aging 22:937–944
Lee SC, Zhao ML, Hirano A, Dickson DW (1999) Inducible nitric oxide synthase immunoreactivity in the Alzheimer disease hippocampus: association with Hirano bodies, neurofibrillary tangles, and senile plaques. J Neuropathol Exp Neurol 58:1163–1169
Lee VM, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24:1121–1159
Lee YB, Nagai A, Kim SU (2002) Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res 69:94–103
Lee EO, Shin YJ, Chong YH (2004) Mechanisms involved in prostaglandin E2-mediated neuroprotection against TNF-alpha: possible involvement of multiple signal transduction and beta-catenin/T-cell factor. J Neuroimmunol 155:21–31
Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA (1997) Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem 272:3406–3410
Li M, Pascual G, Glass CK (2000) Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol 20:4699–4707
Li YK, Liu L, Barger SW, Griffin WST (2003) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 23:1605–1611
Liang XB, Wang Q, Hand T, Wu LJ, Breyer RM, Montine TJ, Andreasson K (2005) Deletion of the prostaglandin E-2 EP2 receptor reduces oxidative damage and amyloid burden in a model of Alzheimer’s disease. J Neurosci 25:10180–10187
Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM (2000) Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci 20:5709–5714
Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao-Ashec K, Frautschy SA, Cole GM (2001) Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging 22:983–991
Lindberg C, Selenica MLB, Westlind-Danielsson A, Schultzberg M (2005) beta-Amyloid protein structure determines the nature of cytokine release from rat microglia. J Mol Neurosci 27:1–12
Liu B, Hong JS (2003) Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther 304:1–7
Liu Y, Walter S, Stagi M, Cherny D, Letiembre M, Schulz-Schaeffer W, Heine H, Penke B, Neumann H, Fassbender K (2005) LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 128:1778–1789
Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, Frosch MP, Irizarry M, Hyman BT (2004) Nonsteroidal anti-inflammatory drugs lower Abeta42 and change presenilin 1 conformation. Nat Med 10:1065–1066
Lotz M, Ebert S, Esselmann H, Iliev AI, Prinz M, Wiazewicz N, Wiltfang J, Gerber J, Nau R (2005) Amyloid beta peptide 1–40 enhances the action of Toll-like receptor-2 and -4 agonists but antagonizes Toll-like receptor-9-induced inflammation in primary mouse microglial cell cultures. J Neurochem 94:289–298
Lue LF, Rydel R, Brigham EF, Yang LB, Hampel H, Murphy GM, Brachova L, Yan SD, Walker DG, Shen Y, Rogers J (2001) Inflammatory repertoire of Alzheimer’s disease and nondemented elderly microglia in vitro. Glia 35:72–79
Lukiw WJ, Bazan NG (1997) Cyclooxygenase 2 RNA message abundance, stability, and hypervariability in sporadic Alzheimer neocortex. J Neurosci Res 50:937–945
Lukiw WJ, Zhao Y, Cui JG (2008) An NF-kappaB-sensitive micro RNA-146a-mediated inflammatory circuit in Alzheimer disease and in stressed human brain cells. J Biol Chem 283:31315–31322
Luster AD (1998) Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med 338:436–445
Ma J, Yee A, Brewer HB Jr, Das S, Potter H (1994) Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 372:92–94
Mackenzie IR (2001) Postmortem studies of the effect of anti-inflammatory drugs on Alzheimer-type pathology and associated inflammation. Neurobiol Aging 22:819–822
Mackenzie IR, Munoz DG (1998) Nonsteroidal anti-inflammatory drug use and Alzheimer-type pathology in aging. Neurology 50:986–990
Magnuson DS, Staines WA, Marshall KC (1993) Electrophysiological changes accompanying DSP-4 lesions of rat locus coeruleus neurons. Brain Res 628:317–320
Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, Koistinaho J (2005) Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis 18:134–142
Mann DM, Lincoln J, Yates PO, Stamp JE, Toper S (1980) Changes in the monoamine containing neurones of the human CNS in senile dementia. Br J Psychiatry 136:533–541
Mann DM, Yates PO, Hawkes J (1982) The noradrenergic system in Alzheimer and multi-infarct dementias. J Neurol Neurosurg Psychiatry 45:113–119
Marcyniuk B, Mann DM, Yates PO (1986) The topography of cell loss from locus caeruleus in Alzheimer’s disease. J Neurol Sci 76:335–345
Marien MR, Colpaert FC, Rosenquist AC (2004) Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res Brain Res Rev 45:38–78
Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O’Banion MK, Tenner AJ, Lemere CA, Duff K (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am J Pathol 158:1345–1354
Mavridis M, Degryse AD, Lategan AJ, Marien MR, Colpaert FC (1991) Effects of locus coeruleus lesions on parkinsonian signs, striatal dopamine and substantia nigra cell loss after 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine in monkeys: a possible role for the locus coeruleus in the progression of Parkinson’s disease. Neuroscience 41:507–523
McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, Breyer RM, Andreasson K (2004) Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia. J Neurosci 24:257–268
McCusker SM, Curran MD, Dynan KB, McCullagh CD, Urquhart DD, Middleton D, Patterson CC, McIlroy SP, Passmore AP (2001) Association between polymorphism in regulatory region of gene encoding tumour necrosis factor alpha and risk of Alzheimer’s disease and vascular dementia: a case-control study. Lancet 357:436–439
McGeer PL, McGeer EG (1995) The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 21:195–218
McGeer EG, McGeer PL (1999) Brain inflammation in Alzheimer disease and the therapeutic implications. Curr Pharm Des 5:821–836
McGeer PL, McGeer EG (2002) The possible role of complement activation in Alzheimer disease. Trends Mol Med 8:519–523
McGeer PL, Schulzer M, McGeer EG (1996) Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology 47:425–432
Meda L, Baron P, Prat E, Scarpini E, Scarlato G, Cassatella MA, Rossi F (1999) Proinflammatory profile of cytokine production by human monocytes and murine microglia stimulated with beta-amyloid[25–35]. J Neuroimmunol 93:45–52
Mehlhorn G, Hollborn M, Schliebs R (2000) Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int J Dev Neurosci 18:423–431
Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT (2008) Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451:720–724
Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, Prinz M (2007) Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci 10:1544–1553
Minghetti L, Polazzi E, Nicolini A, Creminon C, Levi G (1997) Up-regulation of cyclooxygenase-2 expression in cultured microglia by prostaglandin E2, cyclic AMP and non-steroidal anti-inflammatory drugs. Eur J Neurosci 9:934–940
Moncada C, Lekieffre D, Arvin B, Meldrum B (1992) Effect of NO synthase inhibition on NMDA- and ischaemia-induced hippocampal lesions. Neuroreport 3:530–532
Montine TJ, Sidell KR, Crews BC, Markesbery WR, Marnett LJ, Roberts LJ, Morrow JD (1999) Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology 53:1495–1498
Moore AH, Olschowka JA, Williams JP, Okunieff P, O’Banion MK (2005) Regulation of prostaglandin E-2 synthesis after brain irradiation. Int J Radiat Oncol Biol Phys 62:267–272
Moore AH, Wu M, Shaftel SS, Graham KA, O’Banion MK (2009) Sustained expression of interleukin-1beta in mouse hippocampus impairs spatial memory. Neuroscience 164(4):1484–1495
Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, Frautschy SA, Cole GM (2005) Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer’s models. Neuropsychopharmacology 30:1111–1120
Morimoto K, Murasugi T, Oda T (2002) Acute neuroinflammation exacerbates excitotoxicity in rat hippocampus in vivo. Exp Neurol 177:95–104
Mrak RE, Griffin WS (2001) Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging 22:903–908
Murphy PG, Borthwick LS, Johnston RS, Kuchel G, Richardson PM (1999) Nature of the retrograde signal from injured nerves that induces interleukin-6 mRNA in neurons. J Neurosci 19:3791–3800
Murray CA, Lynch MA (1998) Evidence that increased hippocampal expression of the cytokine interleukin-1 beta is a common trigger for age- and stress-induced impairments in long-term potentiation. J Neurosci 18:2974–2981
Nagatsu T, Sawada M (2005) Inflammatory process in Parkinson’s disease: role for cytokines. Curr Pharm Des 11:999–1016
Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971:197–209
Narumiya S, Sugimoto Y, Ushikubi F (1999) Prostanoid receptors: structures, properties, and functions. Physiol Rev 79:1193–1226
Natarajan C, Bright JJ (2002) Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun 3:59–70
Nicoll JA, Mrak RE, Graham DI, Stewart J, Wilcock G, MacGowan S, Esiri MM, Murray LS, Dewar D, Love S, Moss T, Griffin WS (2000) Association of interleukin-1 gene polymorphisms with Alzheimer’s disease. Ann Neurol 47:365–368
Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A, Tashiro K, Onoe K (2001) Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J Neuroimmunol 116:40–48
Nishino K, Lin CS, Morse JK, Davis JN (1991) DSP4 treatment worsens hippocampal pyramidal cell damage after transient ischemia. Neuroscience 43:361–367
O’Banion MK (1999) Cyclooxygenase-2: Molecular biology, pharmacology, and neurobiology. Crit Rev Neurobiol 13:45–82
O’Banion MK, Miller JC, Chang JW, Kaplan MD, Coleman PD (1996) Interleukin-1 beta induces prostaglandin G/H synthase-2 (cyclooxygenase-2) in primary murine astrocyte cultures. J Neurochem 66:2532–2540
O’Banion MK, Chang JW, Coleman PD (1997) Decreased expression of prostaglandin G/H synthase-2 (PGHS-2) in Alzheimer’s disease brain. Adv Exp Med Biol 407:171–177
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, Laferla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular A beta and synaptic dysfunction. Neuron 39:409–421
Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, Laferla FM (2006) Temporal profile of amyloid-beta (A beta) oligomerization in an in vivo model of Alzheimer disease—A link between A beta and tau pathology. J Biol Chem 281:1599–1604
Oka A, Takashima S (1997) Induction of cyclo-oxygenase 2 in brains of patients with Down’s syndrome and dementia of Alzheimer type: specific localization in affected neurones and axons. Neuroreport 8:1161–1164
Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV (2009) Toll-like receptors in neurodegeneration. Brain Res Rev 59:278–292
Olpe HR, Laszlo J, Dooley DJ, Heid J, Steinmann MW (1983) Decreased activity of locus coeruleus neurons in the rat after DSP-4 treatment. Neurosci Lett 40:81–84
Orzylowska O, Oderfeld-Nowak B, Zaremba M, Januszewski S, Mossakowski M (1999) Prolonged and concomitant induction of astroglial immunoreactivity of interleukin-1beta and interleukin-6 in the rat hippocampus after transient global ischemia. Neurosci Lett 263:72–76
Osaka H, Mukherjee P, Aisen PS, Pasinetti GM (1999) Complement-derived anaphylatoxin C5a protects against glutamate-mediated neurotoxicity. J Cell Biochem 73:303–311
Owens T, Babcock AA, Millward JM, Toft-Hansen H (2005) Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Res Brain Res Rev 48:178–184
Papassotiropoulos A, Bagli M, Jessen F, Bayer TA, Maier W, Rao ML, Heun R (1999) A genetic variation of the inflammatory cytokine interleukin-6 delays the initial onset and reduces the risk for sporadic Alzheimer’s disease. Ann Neurol 45:666–668
Paresce DM, Ghosh RN, Maxfield FR (1996) Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 17:553–565
Pasinetti GM, Tocco G, Sakhi S, Musleh WD, DeSimoni MG, Mascarucci P, Schreiber S, Baudry M, Finch CE (1996) Hereditary deficiencies in complement C5 are associated with intensified neurodegenerative responses that implicate new roles for the C-system in neuronal and astrocytic functions. Neurobiol Dis 3:197–204
Patsouris D, Muller M, Kersten S (2004) Peroxisome proliferator activated receptor ligands for the treatment of insulin resistance. Curr Opin Investig Drugs 5:1045–1050
Pavlov VA, Tracey KJ (2005) The cholinergic anti-inflammatory pathway. Brain Behav Immun 19:493–499
Perry RT, Collins JS, Harrell LE, Acton RT, Go RC (2001a) Investigation of association of 13 polymorphisms in eight genes in southeastern African American Alzheimer disease patients as compared to age-matched controls. Am J Med Genet 105:332–342
Perry RT, Collins JS, Wiener H, Acton R, Go RC (2001b) The role of TNF and its receptors in Alzheimer’s disease. Neurobiol Aging 22:873–883
Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G (1995) Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc Natl Acad Sci USA 92:11294–11298
Qiu WQ, Walsh DM, Ye Z, Vekrellis K, Zhang J, Podlisny MB, Rosner MR, Safavi A, Hersh LB, Selkoe DJ (1998) Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem 273:32730–32738
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362:329–344
Rangon CM, Haik S, Faucheux BA, Metz-Boutigue MH, Fierville F, Fuchs JP, Hauw JJ, Aunis D (2003) Different chromogranin immunoreactivity between prion and a-beta amyloid plaque. Neuroreport 14:755–758
Ransohoff RM (2009) Chemokines and chemokine receptors: standing at the crossroads of immunobiology and neurobiology. Immunity 31:711–721
Ray I, Chauhan A, Wisniewski HM, Wegiel J, Kim KS, Chauhan VP (1998) Binding of amyloid beta-protein to intracellular brain proteins in rat and human. Neurochem Res 23:1277–1282
Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE (2009) CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci 29:11982–11992
Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC (2004) Rofecoxib—no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 62:66–71
Rich JB, Rasmusson DX, Folstein MF, Carson KA, Kawas C, Brandt J (1995) Nonsteroidal anti-inflammatory drugs in Alzheimer’s disease. Neurology 45:51–55
Richard KL, Filali M, Prefontaine P, Rivest S (2008) Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1–42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. J Neurosci 28:5784–5793
Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391:79–82
Ristow M (2004) Neurodegenerative disorders associated with diabetes mellitus. J Mol Med 82:510–529
Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A (2009) Astroglia in dementia and Alzheimer’s disease. Cell Death Diff 16:378–385
Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, Lieberburg I (1992) Complement activation by beta-amyloid in Alzheimer-disease. Proc Natl Acad Sci USA 89:10016–10020
Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P (1993) Clinical trial of indomethacin in Alzheimer’s disease. Neurology 43:1609–1611
Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN (1999) Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J Biol Chem 274:6421–6431
Rossi F, Bianchini E (1996) Synergistic induction of nitric oxide by beta-amyloid and cytokines in astrocytes. Biochem Biophys Res Commun 225:474–478
Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG (2000) Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of I kappa B kinase. Nature 403:103–108
Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR (2005) Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J Neurochem 92:226–234
Rota E, Bellone G, Rocca P, Bergamasco B, Emanuelli G, Ferrero P (2006) Increased intrathecal TGF-beta 1, but not IL-12, IFN-gamma and IL-10 levels in Alzheimer’s disease patients. Neurol Sci 27:33–39
Rozemuller JM, Eikelenboom P, Stam FC, Beyreuther K, Masters CL (1989) A4-protein in Alzheimers-disease—primary and secondary cellular events in extracellular amyloid deposition. J Neuropath Exp Neurol 48:674–691
Sanz JM, Chiozzi P, Ferrari D, Colaianna M, Idzko M, Falzoni S, Fellin R, Trabace L, Di Virgilio F (2009) Activation of Microglia by Amyloid beta Requires P2X(7) Receptor Expression. J Immunol 182:4378–4385
Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, Van Leuven F, Heneka MT (2003) Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci 23:9796–9804
Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, Van Leuven F, Heneka MT (2006) Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPAR gamma. Proc Natl Acad Sci USA 103:443–448
Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, Mehta PD, Spinner DS, Wisniewski T (2009) Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci 29:1846–1854
Schubert P, Morino T, Miyazaki H, Ogata T, Nakamura Y, Marchini C, Ferroni S (2000) Cascading glia reactions: a common pathomechanism and its differentiated control by cyclic nucleotide signaling. Ann N Y Acad Sci 903:24–33
Schutz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schurmann B, Zimmer A, Heneka MT (2005) The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci 25:7805–7812
Selkoe DJ (2005) Defining molecular targets to prevent Alzheimer disease. Arch Neurol 62:192–195
Selmaj KW, Farooq M, Norton WT, Raine CS, Brosnan CF (1990) Proliferation of astrocytes in vitro in response to cytokines. A primary role for tumor necrosis factor. J Immunol 144:129–135
Semmler A, Okulla T, Sastre M, Dumitrescu-Ozimek L, Heneka MT (2005) Systemic inflammation induces apoptosis with variable vulnerability of different brain regions. J Chem Neuroanat 30:144–157
Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C (1992) Isolation and quantification of soluble Alzheimer’s beta-peptide from biological fluids. Nature 359:325–327
Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK (2007) Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest 117:1595–1604
Shen Y, Meri S (2003) Yin and Yang: complement activation and regulation in Alzheimer’s disease. Prog Neurobiol 70:463–472
Sheng JG, Zhu SG, Jones RA, Griffin WST, Mrak RE (2000) Interleukin-1 promotes expression and phosphorylation of neurofilament and tau proteins in vivo. Ex Neurol 163:388–391
Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE (2003) Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis 14:133–145
Shie FS, Breyer RM, Montine TJ (2005a) Microglia lacking E Prostanoid Receptor subtype 2 have enhanced Abeta phagocytosis yet lack Abeta-activated neurotoxicity. Am J Pathol 166:1163–1172
Shie FS, Montine KS, Breyer RM, Montine TJ (2005b) Microglial EP2 is critical to neurotoxicity from activated cerebral innate immunity. Glia 52:70–77
Shimizu E, Kawahara K, Kajizono M, Sawada M, Nakayama H (2008) IL-4-induced selective clearance of oligomeric beta-amyloid peptide(1–42) by rat primary type 2 microglia. J Immunol 181:6503–6513
Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B (1992) Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258:126–129
Simard AR, Rivest S (2004) Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J 18:998–1000
Simard AR, Rivest S (2007) Neuroprotective effects of resident microglia following acute brain injury. J Comp Neurol 504:716–729
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49:489–502
Simmons ML, Murphy S (1992) Induction of nitric oxide synthase in glial cells. J Neurochem 59:897–905
Skaper SD, Facci L, Leon A (1995) Inflammatory mediator stimulation of astrocytes and meningeal fibroblasts induces neuronal degeneration via the nitridergic pathway. J Neurochem 64:266–276
Slawik H, Volk B, Fiebich B, Hull M (2004) Microglial expression of prostaglandin EP3 receptor in excitotoxic lesions in the rat striatum. Neurochem Int 45:653–660
Sly LM, Krzesicki RF, Brashler JR, Buhl AE, McKinley DD, Carter DB, Chin JE (2001) Endogenous brain cytokine mRNA and inflammatory responses to lipopolysaccharide are elevated in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res Bull 56:581–588
Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G (1997) Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci 17:2653–2657
Smithswintosky VL, Pettigrew LC, Craddock SD, Culwell AR, Rydel RE, Mattson MP (1994) Secreted forms of beta-amyloid precursor protein protect against ischemic brain injury. J Neurochem 63:781–784
Smits HA, Rijsmus A, van Loon JH, Wat JWY, Verhoef J, Boven LA, Nottet HSLM (2002) Amyloid-beta-induced chemokine production in primary human macrophages and astrocytes. J Neuroimmunol 127:160–168
Snyder SW, Wang GT, Barrett L, Ladror US, Casuto D, Lee CM, Krafft GA, Holzman RB, Holzman TF (1994) Complement C1Q does not bind monomeric beta-amyloid. Exp Neurol 128:136–142
Stalder AK, Carson MJ, Pagenstecher A, Asensio VC, Kincaid C, Benedict M, Powell HC, Masliah E, Campbell IL (1998) Late-onset chronic inflammatory encephalopathy in immune-competent and severe combined immune-deficient (SCID) mice with astrocyte-targeted expression of tumor necrosis factor. Am J Pathol 153:767–783
Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M (2005) Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci 25:11125–11132
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178
Stewart WF, Kawas C, Corrada M, Metter EJ (1997) Risk of Alzheimer’s disease and duration of NSAID use. Neurology 48:626–632
Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, Khoury JE, Golenbock DT, Moore KJ (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol 11:155–161
Strieter RM, Polverini PJ, Kunkel SL, Arenberg DA, Burdick MD, Kasper J, Dzuiba J, Van Damme J, Walz A, Marriott D (1995) The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J Biol Chem 270:27348–27357
Stuehr DJ, Marletta MA (1985) Mammalian nitrate biosynthesis: mouse macrophages produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide. Proc Natl Acad Sci USA 82:7738–7742
Sung S, Yang H, Uryu K, Lee EB, Zhao L, Shineman D, Trojanowski JQ, Lee VM, Pratico D (2004) Modulation of nuclear factor-kappa B activity by indomethacin influences A beta levels but not A beta precursor protein metabolism in a model of Alzheimer’s disease. Am J Pathol 165:2197–2206
Suzuki S, Tanaka K, Nagata E, Ito D, Dembo T, Fukuuchi Y (1999) Cerebral neurons express interleukin-6 after transient forebrain ischemia in gerbils. Neurosci Lett 262:117–120
Szczepanik AM, Funes S, Petko W, Ringheim GE (2001a) IL-4, IL-10 and IL-13 modulate A beta(1–42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J Neuroimmunol 113:49–62
Szczepanik AM, Rampe D, Ringheim GE (2001b) Amyloid-beta peptide fragments p3 and p4 induce pro-inflammatory cytokine and chemokine production in vitro and in vivo. J Neurochem 77:304–317
Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, Goodman SN (2004) Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology 23:159–169
Takadera T, Shiraishi Y, Ohyashiki T (2004) Prostaglandin E2 induced caspase-dependent apoptosis possibly through activation of EP2 receptors in cultured hippocampal neurons. Neurochem Int 45:713–719
Tamagno E, Bardini P, Obbili A, Vitali A, Borghi R, Zaccheo D, Pronzato MA, Danni O, Smith MA, Perry G, Tabaton M (2002) Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis 10:279–288
Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M (1999) Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science 286:2352–2355
Tancredi V, D’Arcangelo G, Grassi F, Tarroni P, Palmieri G, Santoni A, Eusebi F (1992) Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci Lett 146:176–178
Tancredi V, D’Antuono M, Cafe C, Giovedi S, Bue MC, D’Arcangelo G, Onofri F, Benfenati F (2000) The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase ERK. J Neurochem 75:634–643
Tanzi RE, Bertram L (2005) Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120:545–555
Taupenot L, CiesielskiTreska J, Ulrich G, ChasserotGolaz S, Aunis D, Bader MF (1996) Chromogranin a triggers a phenotypic transformation and the generation of nitric oxide in brain microglial cells. Neuroscience 72:377–389
Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ (2000) A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med 192:359–366
Tchelingerian JL, Vignais L, Jacque C (1994) TNF alpha gene expression is induced in neurones after a hippocampal lesion. Neuroreport 5:585–588
Teaktong T, Graham A, Court J, Perry R, Jaros E, Johnson M, Hall R, Perry E (2003) Alzheimer’s disease is associated with a selective increase in alpha 7 nicotinic acetylcholine receptor immunoreactivity in astrocytes. Glia 41:207–211
Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, Assaid C, Nessly ML, Norman BA, Baranak CC, Reines SA (2005) A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 30:1204–1215
Thery C, Stanley ER, Mallat M (1992) Interleukin-1 and tumor-necrosis-factor-alpha stimulate the production of colony-stimulating factor-I by murine astrocytes. J Neurochem 59:1183–1186
Thiemermann C (1994) The role of the l-arginine: nitric oxide pathway in circulatory shock. Adv Pharmacol 28:45–79
Thomas T, Nadackal TG, Thomas K (2001) Aspirin and non-steroidal anti-inflammatory drugs inhibit amyloid-beta aggregation. Neuroreport 12:3263–3267
Tocco G, Freire-Moar J, Schreiber SS, Sakhi SH, Aisen PS, Pasinetti GM (1997) Maturational regulation and regional induction of cyclooxygenase-2 in rat brain: implications for Alzheimer’s disease. Exp Neurol 144:339–349
Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM (1994) mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8:1224–1234
Tugwood JD, Issemann I, Anderson RG, Bundell KR, McPheat WL, Green S (1992) The mouse peroxisome proliferator activated receptor recognizes a response element in the 5′ flanking sequence of the rat acyl CoA oxidase gene. EMBO J 11:433–439
Tuppo EE, Arias HR (2005) The role of inflammation in Alzheimer’s disease. Int J Biochem Cell Biol 37:289–305
Udan ML, Ajit D, Crouse NR, Nichols MR (2008) Toll-like receptors 2 and 4 mediate Abeta(1–42) activation of the innate immune response in a human monocytic cell line. J Neurochem 104:524–533
Vamecq J, Latruffe N (1999) Medical significance of peroxisome proliferator-activated receptors. Lancet 354:141–148
van Beek J, Elward K, Gasque P (2003) Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci 992:56–71
Varvel NH, Bhaskar K, Kounnas MZ, Wagner SL, Yang Y, Lamb BT, Herrup K (2009) NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J Clin Invest 119:3692–3702
Veerhuis R, Boshuizen RS, Morbin M, Mazzoleni G, Hoozemans JJM, Langedijk JPM, Tagliavini F, Langeveld JPM, Eikelenboom P (2005) Activation of human microglia by fibrillar prion protein-related peptides is enhanced by amyloid-associated factors SAP and C1q. Neurobiol Dis 19:273–282
Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, Weidner J, Mumford R, Webber R, Nathan C, Roberts AB, Lippa CF, Sporn MB (1996) Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer’s disease. J Exp Med 184:1425–1433
Wallace MN, Geddes JG, Farquhar DA, Masson MR (1997) Nitric oxide synthase in reactive astrocytes adjacent to beta-amyloid plaques. Exp Neurol 144:266–272
Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44:181–193
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002a) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416:535–539
Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ (2002b) Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans 30:552–557
Walter J, Fluhrer R, Hartung B, Willem M, Kaether C, Capell A, Lammich S, Multhaup G, Haass C (2001) Phosphorylation regulates intracellular trafficking of beta-secretase. J Biol Chem 276:14634–14641
Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K (2007) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem 20:947–956
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang HC, Yang H, Ulloa L, Al Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha 7 subunit is an essential regulator of inflammation. Nature 421:384–388
Wang Q, Rowan MJ, Anwyl R (2004) Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci 24:6049–6056
Weberpals M, Hermes M, Hermann S, Kummer MP, Terwel D, Semmler A, Berger M, Schafers M, Heneka MT (2009) NOS2 gene deficiency protects from sepsis-induced long-term cognitive deficits. J Neurosci 29:14177–14184
Webster S, Bonnell B, Rogers J (1997a) Charge-based binding of complement component C1q to the Alzheimer amyloid beta-peptide. Am J Pathol 150:1531–1536
Webster S, Lue LF, Brachova L, Tenner AJ, McGeer PL, Terai K, Walker DG, Bradt B, Cooper NR, Rogers J (1997b) Molecular and cellular characterization of the membrane attack complex, C5b–9, in Alzheimer’s disease. Neurobiol Aging 18:415–421
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH (2001) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414:212–216
Weldon DT, Rogers SD, Ghilardi JR, Finke MP, Cleary JP, O’Hare E, Esler WP, Maggio JE, Mantyh PW (1998) Fibrillar beta-amyloid induces microglial phagocytosis, expression of inducible nitric oxide synthase, and loss of a select population of neurons in the rat CNS in vivo. J Neurosci 18:2161–2173
Wilcock GK, Esiri MM, Bowen DM, Hughes AO (1988) The differential involvement of subcortical nuclei in senile dementia of Alzheimer’s type. J Neurol Neurosurg Psychiatry 51:842–849
Willenborg DO, Fordham SA, Staykova MA, Ramshaw IA, Cowden WB (1999a) IFN-gamma is critical to the control of murine autoimmune encephalomyelitis and regulates both in the periphery and in the target tissue: a possible role for nitric oxide. J Immunol 163:5278–5286
Willenborg DO, Staykova MA, Cowden WB (1999b) Our shifting understanding of the role of nitric oxide in autoimmune encephalomyelitis: a review. J Neuroimmunol 100:21–35
Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L (1997) Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature 389:603–606
Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E (2000) Chronic overproduction of transforming growth factor-beta 1 by astrocytes promotes Alzheimer’s disease-like microvascular degeneration in transgenic mice. Am J Pathol 156:139–150
Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci USA 99:10837–10842
Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat Med 9:453–457
Xia MQ, Hyman BT (1999) Chemokines/chemokine receptors in the central nervous system and Alzheimer’s disease. J Neurovirol 5:32–41
Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT (1998) Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am J Pathol 153:31–37
Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF (1993) Expression of a mitogen-inducible cyclooxygenase in brain neurons—regulation by synaptic activity and glucocorticoids. Neuron 11:371–386
Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P (1995) Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med 1:693–699
Yan SD, Stern D, Kane MD, Kuo YM, Lampert HC, Roher AE (1998) RAGE-A beta interactions in the pathophysiology of Alzheimer’s disease. Restor Neurol Neurosci 12:167–173
Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G (2003) Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J Neurosci 23:7504–7509
Yasojima K, Schwab C, McGeer EG, McGeer PL (1999a) Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res 830:226–236
Yasojima K, Schwab C, McGeer EG, McGeer PL (1999b) Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am J Pathol 154:927–936
Yates CM, Simpson J, Gordon A, Maloney AF, Allison Y, Ritchie IM, Urquhart A (1983) Catecholamines and cholinergic enzymes in pre-senile and senile Alzheimer-type dementia and Down’s syndrome. Brain Res 280:119–126
Yermakova A, O’Banion MK (2000) Cyclooxygenases in the central nervous system: implications for treatment of neurological disorders. Curr Pharm Des 6:1755–1776
Yermakova AV, O’Banion MK (2001) Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer’s disease. Neurobiol Aging 22:823–836
Yermakova AV, Rollins J, Callahan LM, Rogers J, O’Banion MK (1999) Cyclooxygenase-1 in human Alzheimer and control brain: quantitative analysis of expression by microglia and CA3 hippocampal neurons. J Neuropathol Exp Neurol 58:1135–1146
Yki-Jarvinen H (2004) Thiazolidinediones. N Engl J Med 351:1106–1118
Zarow C, Lyness SA, Mortimer JA, Chui HC (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol 60:337–341
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Heneka, M.T., O’Banion, M.K., Terwel, D. et al. Neuroinflammatory processes in Alzheimer’s disease. J Neural Transm 117, 919–947 (2010). https://doi.org/10.1007/s00702-010-0438-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-010-0438-z