
Abstract
Idiopathic pulmonary fibrosis (IPF) carries a high mortality rate and has a poor prognosis. The pathogenesis of pulmonary fibrosis (PF) is highly related to dysregulation of multiple RNAs. This study aims to identify and validate dysregulated RNAs that exhibited dynamic alterations in response to bleomycin (BLM)-induced PF. The results will provide therapeutic targets for patients suffering from IPF. Whole transcriptomic profiles of BLM-induced PF were obtained through high-throughput RNA sequencing. miRNA profiling was downloaded from GSE45789 database in the Gene Expression Omnibus (GEO). We identified the differentially expressed RNAs (DERNAs) that exhibited dynamic alterations in response to BLM-induced PF. Subsequently, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genome (KEGG) pathway enrichment analysis were conducted to discovery regulatory processes of PF. Weighted gene co-expression network analysis (WGCNA), protein–protein interaction (PPI) analysis, and co-expression analysis were performed to identify key genes and pathogenic pattern during the progression of PF. MiRanda, miRcode, and TargetScan were utilized to predict target relationships in the potential competing endogenous RNA (ceRNA) network. The results were verified by qRT-PCR analysis. In the context of BLM-induced PF, this study identified a total of 167 differentially expressed messenger RNAs (DEmRNAs), 115 differentially expressed long non-coding RNAs (DElncRNAs), 45 differentially expressed circular RNAs (DEcircRNAs), and 87 differentially expressed microRNAs (DEmiRNAs). These RNA molecules showed dynamic alterations in response to BLM-induced PF. These DEmRNAs exhibited a predominant association with the biological processes pertaining to the organization of extracellular matrix. A regulatory network was built in PF, encompassing 31 DEmRNAs, 18 DE lncRNAs, 13 DEcircRNAs, and 13 DEmiRNAs. Several DERNA molecules were subjected to validate using additional BLM-induced PF model. The outcomes of this validation process shown a strong correlation with the results obtained from RNA sequencing analysis. The GSE213001 dataset was utilized to validate the expression levels and diagnostic efficacy of four specific hub mRNAs (CCDC80, CLU, COL5A1, and COL6A3) in individuals diagnosed with PF. In this study, we identified and validated several RNA molecules that exhibited dynamic alternations in response to BLM-induced PF. These dysregulated RNAs participated in the pathogenesis of PF and can be used as therapeutic targets for early-stage IPF. Although more work must be done to confirm the results, our study may provide directions for future studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Idiopathic pulmonary fibrosis (IPF) is a heterogeneous respiratory disease with high morbidity and short survival time [1]. It is characterized by accumulation of extracellular matrix and inflammatory cell infiltration [2]. Several pathological processes have been indicated to explain the occurrence of pulmonary fibrosis (PF), including fibroblast-to-myofibroblast transition (FMT), resistance to apoptosis, immune cell infiltration, as well as epithelial-mesenchymal transition (EMT) [3]. However, understanding this pathological process is still in its infancy, and the detailed mechanism remains largely unknown. Due to delays in diagnosis and lack of effective treatments, the prognosis associated with this condition is poor.
Recently, some studies highlighted the importance of RNA dysregulation during the development of IPF. The dysregulation of mRNAs (GPR41, SPP1, CCL1, and FGF19) can promote pulmonary fibroblast activation and fibrosis to regulate different signaling pathways, such as Smad2/3, ERK1/2 phosphorylation and JNK phosphorylation [4,5,6,7]. Kraven et al. used blood gene expression signatures to predict mortality in IPF [8]. In addition, long non-coding RNAs (lncRNA) PCF can promote pulmonary fibrogenesis by regulating miR-334a/map3k11 axis [9]. LncRNA-MALAT1 was associated with abnormal activation of macrophages, which affects pulmonary pathogeneses [10]. It has been reported that circular RNAs (circRNAs) HIPK3 accelerates PF by regulating miR-30a-3p/FOXK2 expression [11]. Based on above-mentioned findings, it is obvious that non-coding RNAs (ncRNAs) can regulate the biological process of mRNAs and affect the pathogenesis of PF.
Several studies reported the dysregulation of mRNA-microRNA-lncRNA-circRNA interaction was highly correlative to the development of sclerosis-associated PF [12]. However, the key genes exhibiting dynamic alterations in response to bleomycin (BLM)-induced PF remain to be characterized. Thus, a comprehensive exploration of dysregulated RNAs could enhance our understanding of PF disease.
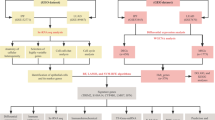
To identify the dysregulated RNAs in the development of PF, we performed whole transcriptome sequencing analysis on BLM-induced lung tissues and normal control tissues. According to the flow diagram of our study (Fig. 1), we identified differentially expressed RNAs (DERNAs) that showed dynamic alterations in PF development. Furthermore, the potential competing endogenous RNA (ceRNA) network, weighted gene co-expression network analysis (WGCNA), functional annotation, and protein–protein interaction (PPI) were conducted to identify key genes and pathogenic pattern during the progression of PF. Several DERNAs were subjected to validate using additional BLM-induce PF model through quantitative real-time PCR (qRT-PCR). Finally, the GSE213001 dataset was utilized to validate the expression levels and diagnostic efficacy of four specific hub mRNAs in individuals diagnosed with PF. These dysregulated RNAs participated in the pathogenesis of PF and can be used as therapeutic targets for early-stage IPF.

Flow diagram of the study design
Materials and Methods
Pulmonary Fibrosis Models
Male C57BL/6 mice (6–8 weeks old, 23–25 g weight) were housed in separate chambers with abundant food and water under the optimal conditions (20 ± 2 °C, 55 ± 5% humidity). All animals were randomly allocated to two groups: normal control (NC) group (saline vehicle, 50 μL) and BLM group (BLM was dissolved in saline vehicle, 3 mg/Kg, 50 μL). C57BL/6 mice were lightly anaesthetized with isoflurane gas (2%), and BLM or 0.9% NaCl was administered intratracheally for a single time. Mice were sacrificed at 3, 7, 14, or 21 days after BLM or 0.9% NaCl treatment. The left lung tissues were used for pathological staining. The right lung was collected for RNA sequencing and PCR tests.
Histological Examination
Left lung tissue was fixed with 4% paraformaldehyde at room temperature for 24 h, embedded in paraffin, and sliced into 5 μm sections, as described in previous article [13]. Hematoxylin and eosin (H&E) staining and Masson staining were used for histopathological analyses. And the severity of PF was evaluate depending on Ashcroft scoring system [14].
Whole-Transcriptome Sequencing
Total RNA of lung tissues was extracted and purified using RNAiso Plus (Takara, Japan). RNA quality and integrity were evaluated using a NanoDrop 2000c (Thermo, USA), an Agilent 2100 bioanalyzer (Agilent, USA), and denaturing agarose gel electrophoresis (GE Healthcare, USA). The high-throughput whole transcriptome sequencing was conducted by Geneplus Technology Co., Ltd. (Shenzhen, China).
MiRNA profiling was downloaded from GSE45789 database in the Gene Expression Omnibus (GEO).
The valid data were generated from the raw reads using an internal python script and mapped to the reference genome. The StringTie tool was used to determined transcripts of different RNAs. The identification of circRNAs was based on previous studies [15]. After quality filtering, miRNA sequences were mapped and counted using an internal script.
Identification of DERNAs
This study aims to find out the genes that showed the dynamic alterations during the progression of PF. The FPKM or TPM value of all DERNAs was calculated, and low expression RNAs were excluded. In general, we first screened differentially expressed genes (DEGs) from BLM-21d group and NC group using R software “DEseq2 (v1.32.0)” package. DERNAs were selected using |Log2 (FoldChange)|≥ 2 and P < 0.05 as the threshold criteria. According to the above data, we then identified the DERNAs from four groups (BLM-3d, BLM-7d, BLM-14d, and BLM-21d), which showed the dynamic alterations in the development of PF. Finally, volcano plots and heatmap revealed DERNAs between the two groups.
Functional Enrichment of DEmRNAs
Gene ontology (GO) enrichment analysis was utilized to assess regulatory processes. Kyoto Encyclopedia of Genes and Genome (KEGG) pathway enrichment analysis was conducted to enrich the signaling pathways. We selected the top 10 most significantly enriched term in GO analysis and KEGG pathway and imported these results into R package for visualization.
Construction of ceRNA network
The ceRNA interactions among DEmRNAs, DElncRNAs, DEcircRNAs, and DEmiRNAs were constructed and visualized using Cytoscape software (https://cytoscape.org). DEmiRNAs were acted as the central point of network. Pairing relationships between DEmiRNA and other kinds of RNA were obtained by multiple ways.
Weighted Gene Co-Expression Network (WGCNA) Analysis
The genes associated with PF phenotype were identified using WCCNA approach [16]. Firstly, we identified and analyzed distinct gene modules. Secondly, different gene modules were performed to establish the co-expression network. Finally, Pearson correlation was used to evaluate the relationship between module-eigengene and disease status.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was extracted from lung tissues of PF mice and performed Quantitative Real-Time PCR analyses to validate above data. M-MuLV reverse transcriptase (FAPON, China) was used to reverse transcribed RNA into cDNA. We used SYBR Green/ROX and primers to amplify and quantify gene expression (Applied Biosystems, USA). β-actin level was used to normalize the relative value of each target gene. miRNA expression profile was evaluated according to our previous study [17]. Gene expression of all genes was calculated by the 2−ΔΔCt method. All primers were shown in Additional file 1: Table S4.
Diagnostic Evaluation of Hub DEmRNAs
To improve the reliability of these results, the expression level and diagnostic efficacy of key DEmRNAs in an independent dataset (GSE213001) were validated. Based on the normalized results, we used GraphPad Prism 8.0 to analyze gene expression profiling. The receiver operating characteristics (ROC) curves was conducted to obtain the corresponding area under the curve (AUC), sensitivity, and specificity.
Statistical Analysis
Data were compared and analyzed using Graphpad Prism 8.0. All variables were displayed as the mean ± standard error of measurement (SEM). Unpaired Student’s t-test was employed for the comparison of two groups. Two-tailed P value < 0.05 was considered statistically significant.
Results
BLM-Induced Pulmonary Fibrosis Mice Model
This study established a BLM-induced PF in mice to identify the genes with dynamic alterations during the progression of PF. Injuries of inflammation, alveolar structure, and the severity of PF was evaluated by H&E and Masson staining. The H&E staining showed that BLM-exposed lungs had markedly morphologic changes, including thickening of alveolar epithelium, lung architecture destruction, and inflammatory cell infiltration in the alveolar space. BLM-induced PF group had a higher percentage of collagen fibers area. (Fig. 2A). The Ashcroft score was remarkably increased in BLM-induced group than NC group (Fig. 2B). In general, the mice treated with BLM showed a decreased trend in body weight, whereas the NC group maintained their bodyweight (Fig. 2C). These results indicated that BLM treatment can induce pulmonary fibrosis.

BLM-induced pulmonary fibrosis mice model. A The lung tissue was stained using H&E and Masson staining. B Ashcroft score of two groups. C Body weight change of two groups for different days. Data were expressed as mean ± SEM with n = 4 per group. A value of *p < 0.05, **p < 0.01, ***p < 0.001 was compared with the NC group. BLM bleomycin, NC normal control group. Scale bar: 500 μm
Differentially Expression Analysis
Lung tissues from both BLM-induced PF at four different time points (BLM-3d, BLM-7d, BLM-14d, and BLM-21d) and NC group were used to extract and purify total RNAs. Total RNAs were used to perform whole transcriptome sequencing. After quality filtering, clean reads were generated from the original reads, which were then mapped to mouse genome (Additional file: Table S1).
The reliability of sequencing results was evaluated through principal component analysis (PCA). BLM-induced PF samples could be clearly separated from the NC group, implying the reliability of the results. (Fig. 3A–C). Given the heterogeneity among different mice, we set strict screening standard |Log2(FoldChange)|≥ 2 and P value < 0.05 to identify DERNAs between BLM-21d group and NC group mice. Subsequently, we filtered the DERNAs with dynamic alterations during the development of PF at four points (BLM-3d, BLM-7d, BLM-14d, and BLM-21d). Finally, a total of 167 differentially expressed messenger RNAs (DEmRNAs), 115 differentially expressed long non-coding RNAs (DElncRNAs), and 45 differentially expressed circular RNAs (DEcircRNAs) were identified. A volcano plot was performed to compare DEGs between BLM-induced PF group and NC group (Fig. 3D–F). DERNAs were analyzed by the clustering heatmaps. (Fig. 3G–I).

Differentially expression analysis. Principal component analysis (PCA) for mRNAs (A), lncRNAs (B), and circRNAs (C). Volcano plot for DEmRNAs (D), DElncRNAs (E), and DEcircRNAs (F). The heatmap for DEmRNAs (G), DElncRNAs (H), and DEcircRNAs (I). BLM-21d bleomycin-21d group, NC normal control group
Functional Enrichment of DEmRNAs
DEmRNAs were used to explore the biological processes in BLM-induced PF. GO analysis and KEGG pathway analysis found that the enriched biological processes were primarily associated with extracellular matrix organization. (Fig. 4A, B; Additional file1: Table S2-3). These results suggested that extracellular matrix changes may serve as pathological factors of the occurrence of BLM-induced PF.

GO ontology (A) and KEGG pathway enrichment (B) of DEmRANs
Construction of a Potential ceRNA Regulatory Network
As is known to all, miRNAs can interact with other kinds of RNAs. To investigate the regulatory relationship of mRNA and ncRNAs related to PF, a ceRNA network was constructed, including 31 DEmRNAs, 18 DElncRNAs, 13 DEcircRNAs, and 13 DEmiRNAs (Table 1). According to the above screening criteria, the 75 hub genes within the ceRNA regulatory network exhibited dynamic alterations during PF development (Fig. 5B–D). The results of the ceRNA interactions demonstrated that some miRNAs could interact with either lncRNAs or circRNAs, while only a few miRNAs interact with both lncRNAs and circRNAs. Cytoscape software was used to visualize the interacting relationships that responds in the BLM-induced PF (Fig. 5A). An insightful understanding of the pathogenesis of PF can be gained through the interactions between mRNAs and ncRNAs in this network.

Competitive endogenous RNA (ceRNA) network constructed with DERNAs (A). The 31 mRNAs (B), 18 lncRNAs (C), and 13 circRNAs (D) associated with ceRNA regulatory network showed the dynamic alterations in response to BLM-induced PF
Otherwise, we established a PPI network and co-expression pattern of DEmRNAs. (Fig. 6A). There are 48 co-expression gene pairs in the ceRNA network, including Col1a1-Col1a2, Col5a1-Mmp2, CCDC80-Col1a2, and so on (Fig. 6B).

The PPI analysis of DEmRANs (A). Co-expression analysis of DEmRANs (B)
Construction of Weight Gene Co-expression Network Analysis (WGCNA)
In this study, gene modules that could be involved in PF phenotype were identified. Using the dynamic tree cut method, we identified distinct gene modules in our dataset. Hierarchical clustering dendrograms of different gene modules were utilized to visualize the relationship between genes in our dataset (Fig. 7A). These gene modules were then further analyzed to identify their relationship with the PF phenotype. A heatmap was generated to display the relationship between all the gene modules and PF disease (Fig. 7B), which suggested that certain gene modules are positively or negatively correlated with PF, indicating that these gene modules could potentially be associated with the pathogenesis of PF.

PF-related genes were screened by WGCNA analysis. A Gene dendrogram and module colors of WGCNA. B Analysis of correlations between the module eigengenes and PF disease, p values are shown. C The eigengene dendrogram and heatmap identifying groups of correlated eigengenes. D Scatter plot analysis of module eigengenes in turquoise module. E Scatter plot analysis of module eigengenes in blue module. F Scatter plot analysis of module eigengenes in red module. WGCNA, weighted gene co-expression network analysis
We then conducted module-eigengene analysis to explore their relationships. The results revealed that different gene modules in our dataset were well separated and could be used to distinguish between different stages of PF (Fig. 7C). This analysis allowed us to identify key modules that could associate with the development of PF. The relationships between gene significance and module membership in different gene modules was explored. Modules membership in turquoise, blue, and red showed remarkably significant relationship with gene significance, which could serve as potential biomarkers for the disease. (Fig. 7D–F).
Validation of the Expression of Key Genes
The expression of key genes that exhibited dynamic alterations were confirmed using qRT-PCR analyses. In accordance with the results of whole transcriptome sequencing in PF lung tissues, the expression of CCDC80, CLU, COL5A1, COL6A3, and ATP6V1C2 were significantly different between BLM-induced PF group and NC group (Fig. 8A–E). In addition, the expression of miR-223-5p, miR-21-5p, miR-218, and miR-214-3p were significantly increased in BLM-induced group (Fig. 8K–N). Moreover, miR-92c, LINC7172, LINC5923, LINC6803, LINC6849 and circ_712183 were also confirmed to be downregulated in BLM-induced lung samples (Fig. 8F–J, O). The key genes that exhibited dynamic alterations could provide therapeutic targets for IPF patients and promote research on the pathogenesis of PF.

Validation of the expression of key genes. Expression levels of CCDC80 (A), CLU (B), COL5A1 (C), COL6A3 (D), ATP6V1C2 (E), LINC7172 (F), LINC5923 (G), LINC6803 (H), LINC6849 (I), circ_712183 (J), miR-223-5p (K), miR-21-5p (L), miR-218 (M), miR-214-3p (N), and miR-92c (O). Data were expressed as mean ± SEM with n = 4 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. BLM bleomycin, NC normal control group
Evaluation of Hub DEmRNAs in the Gene Expression Level and Diagnostic Effectiveness
To further explore the clinical value of genes during the progression of PF, we analyzed the GSE213001 dataset, which included 62 patients with IPF, 36 patients with non-IPF interstitial lung disease (non-IPF ILD), and 41 non-disease control donors. In contrast to non-diseased control donors, four DEmRNAs (CCDC80, CLU, COL5A1, and COL6A3) were significantly upregulated in patients with IPF and non-IPF ILD (Fig. 9A–D). ROC curves were employed to assess the sensitivity and specificity of four specific genes in differential lung tissues from PF patients and non-diseased control donors yielded the following AUCs: CCDC80, 0.9308 (95% confidence interval (CI), 0.8761–0.9854), CLU, 0.8379 (95% CI, 0.7548–0.9211), COL5A1, 0.9099 (95% CI, 0.8509–0.9690), and COL6A3, 0.9288 (95% CI, 0.8735–0.9841) (Fig. 9E–H). AUC values > 0.8 for four hub genes suggested their relatively significant diagnostic efficacy of PF.

Evaluation of four specific DEmRNAs in the gene expression level and diagnostic efficacy. Expression levels of CCDC80 (A), CLU (B), COL5A1 (C), and COL6A3 (D) in patients with PF and non-diseased control donors. ROC curves and AUC statistics found the clinical significance of CCDC80 (E), CLU (F), COL5A1 (G), and COL6A3 (H) for diagnosing PF. A value of *p < 0.05, **p < 0.01, ***p < 0.001 was compared with non-diseased control donors. NDC non-diseased control donors, IPF idiopathic pulmonary fibrosis, Non-IPF ILD non-IPF interstitial lung disease
Discussion
IPF is one of the most aggressive forms of ILD, threatening millions of patients worldwide [18]. Although evidence has been proposed to explain the development of IPF, the prognosis of the disease has not been improved. Hence, it is essential to investigate the underlying pathogenesis and recognize novel genes with a high degree of sensitivity and specificity. At present study, a BLM-induced PF model was established to perform whole transcriptome sequencing and explore the key genes that exhibited dynamic alterations. And we applied related experiments to validate interaction of RNAs and their regulatory pattern in PF, which could provide therapeutic targets for IPF patients and promote research on the pathogenesis of PF.
Apart from aberrant expression of mRNAs, dysregulation of ncRNAs is also important for the progression of PF. In this study, we identified several DERNAs, including 31 DEmRNAs, 18 DE lncRNAs, 13 DE circRNAs, and 13 DEmiRNAs, which showed dynamic alterations in response to BLM-induced PF. In fact, DEmRNAs, such as COL1A1 [19], MMP2 [20], IGF1 [21], FSTL1 [22], and SFN [23] have been found to be associated with the occurrence of PF via different signaling pathways in the previous studies. Several studies have reported that inhibiting the expression of collagen-related genes during the progression of PF may be a value approach to treat PF [24, 25]. Hamaguchi et al. indicated that the concentration of serum IGF1 were increased in patients suffering from severe pulmonary fibrosis [26]. Moreover, among the 13 DEmiRNAs, miR-210-3p [27], miR-21-5p [28], miR-17-5p [29], miR-15b [30], and miR-214-3p [31] has been reported as effective targets for the treatment of PF. Furthermore, 18 DElncRNAs and 13 novel DEcircRNAs have been identified to be related to the occurrence of BLM-induced PF. We found dysregulated RNAs could interact with each other and affect many signaling pathways.
Salmena et al. recently proposed the hypothesis of ceRNA network, indicating that RNAs can interact with others and participate in the regulation of physiological processes [32]. A network comprises different types of RNAs, such as mRNAs and ncRNAs [33]. According to the analysis of theory, lncRNAs and circRNAs could act as natural miRNA sponges, reducing the stability of target mRNAs or inhibiting their translation [34]. However, the research on ceRNA regulation has primarily focused on tumorigenesis, with only a few studies exploring the ceRNA network in PF [35, 36].
It is widely known that ncRNAs are specifically expressed in certain diseases and are widely participated in many biological processes. miRNAs, a key component of ncRNAs, play a vital function in silencing or degrading the target mRNAs [37]. In this study, we predicted the network between DEmiRNAs and DEmRNAs/DElncRNAs/DEcircRNAs using DEmiRNAs as hub nodes. We identified 13 miRNAs that interacted with 18 lncRNAs and 13 circRNAs and affected the function of 31 mRNAs. Jia et al. reported that lncRNA-HOTAIR regulates the expression of MMP2 by binding to miR-17-5p in paraquat-induced PF [29]. Our data revealed that a novel lncRNA (LINC5980) may regulate the expression of MMP2 via specifically sponge miR-17-5p. Another research indicated that lncRNA-DNM3OS influence SMAD and non-SMAD components of TGF-β signaling by interacting with miRNA-214-3p. We discovered that an additional 6 lncRNAs could act as miR-761 sponges to affect the level of 9 mRNAs, as showed in Table 1. Furthermore, silencing lncRNA-NEAT1 reduces SMAD3 expression via the miR-455-3p axis, thereby affecting its function [38]. Our study predicted that lncRNA-NEAT1 could actively sponge miR-218 and miR-221-3p to affect the expression of TNC, IGF1, and THBS1.
Furthermore, accumulating evidence shows that circRNAs participated in various biological functions through different mechanisms of PF. CircRNAs participate in gene regulation in post-transcriptional manner by isolating miRNAs and inhibiting their target mRNAs [39, 40]. circRNA-TADA2A alleviated IPF via miR-526b/Cav1 axis [41]. Another study demonstrated that circRNA-HECTD was associated with SiO2-induced PF [42]. In this study, DEcircRNAs were also identified, including five downregulated circRNAs (circRNA_712183, circRNA_595255, circRNA_926249, circRNA_301367, and circRNA_800023) and nine upregulated circRNAs, which directly or indirectly influence the expression of mRNAs by targeting different miRNAs. Our results indicate that DEcircRNAs are indeed involved in many aspects of the PF progression.
We conducted GO ontology and KEGG analysis to explore the biologic functions of DEmRNAs in BLM-induced PF. The results of these analyses were primarily enriched in extracellular matrix organization. Lin et al. reported that triptolide attenuates PF by preventing fibrotic extracellular matrix remodeling mediated by MMPs/LOX/integrin [43]. A recent study has highlighted the role of extracellular matrix in IPF [44]. These findings suggest that accumulation of extracellular matrix may be the primary drives of irreversible pulmonary structure remodeling and fibrosis formation and may identify new biology mechanisms and potential therapeutic targets in PF development.
PPI analysis was used to identify the key genes, including Col1a1, Col5a1, Col6a3, Col12a1, Mmp2, Fstl1, CCDC80 and Ltbp2, which could be promising biomarkers for the prevention and therapy of IPF patients. Co-expression further confirmed the significance of these genes. It has been previously shown that silencing the expression of Ltbp2 prevented PF via NF-κB signaling [45]. A recent study has demonstrated that Fstl1 affects the function of cells in the development of PF via p38 and JNK signaling pathway [46]. In our study, we found that the expression of CCDC80, CLU, Col5a1, and Col6a3 were remarkedly increased in patients with IPF and non-IPF ILD, which had high diagnostic value. However, due to the complexity of the regulatory network, it is necessary to explore it in depth. Overall, our results provide a meaningful direction for future research.
Nevertheless, there are some limitations in this study. First, the number of animal samples used to identify DERNAs was small. Second, we only validate the expression among the DERNAs in animal models, but additional experiments are needed to elucidate the underlying mechanisms in detail. Finally, the potential ceRNA regulatory network was based solely on bioinformatics analysis and required further experimental validation.
In conclusion, we identified DERNAs between BLM-induced PF group and NC group, which were performed to establish the ceRNA network. We validated the expression of some DERNAs that exhibited dynamic alterations during the progression of PF using qRT-PCR. Moreover, we also evaluate the diagnostic efficacy of key mRNAs. This study offers several potential biomarkers to illuminate the progression of PF, which may serve as therapeutic targets for IPF patients and facilitate research on the pathogenesis of PF.
Data Availability
NGS data are available from the NCBI Sequence Read Archive database (BioProject: PRJNA962191).
Abbreviations
- IPF:
-
Idiopathic pulmonary fibrosis
- ILD:
-
Interstitial lung disease
- GEO:
-
Gene Expression Omnibus
- DERNAs:
-
Differentially expressed RNAs
- CeRNA:
-
Competiting endogenous RNA
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genome
- PPI:
-
Protein–protein interaction
- WGCNA:
-
Weighted gene co-expression network analysis
- ncRNAs:
-
Non-coding RNAs
- PCA:
-
Principal component analysis
- FMT:
-
Fibroblast-to-myofibroblast transition
- EMT:
-
Epithelial-mesenchymal transition
- BLM:
-
Bleomycin
- qRT-PCR:
-
Quantitative Real-Time PCR
- DEGs:
-
Differentially expressed genes
- ROC:
-
Receive operating characteristic
- AUCs:
-
The area under the curve
References
Raghu, G., Remy-Jardin, M., Richeldi, L., et al. (2022). Idiopathic pulmonary fibrosis (an Update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. American Journal of Respiratory and Critical Care Medicine, 205(9), e18–e47.
Spagnolo, P., Kropski, J. A., Jones, M. G., et al. (2021). Idiopathic pulmonary fibrosis: Disease mechanisms and drug development. Pharmacology & therpeutics, 222, 107798.
Thannickal, V. J., Toews, G. B., White, E. S., Lynch, J. P., 3rd., & Martinez, F. J. (2004). Mechanisms of pulmonary fibrosis. Annual Review of Medicine, 55, 395–417.
Ren, Z., Pan, X., Li, J., et al. (2023). G protein coupled receptor 41 regulates fibroblast activation in pulmonary fibrosis via Gα(i/o) and downstream Smad2/3 and ERK1/2 phosphorylation. Pharmacological Research, 191, 106754.
Morse, C., Tabib, T., Sembrat, J., et al. (2019). Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. The European Respiratory Journal, 54(2), 1802441.
Liu, S. S., Liu, C., Lv, X. X., et al. (2021). The chemokine CCL1 triggers an AMFR-SPRY1 pathway that promotes differentiation of lung fibroblasts into myofibroblasts and drives pulmonary fibrosis. Immunity, 54(9), 2042–56.e8.
Justet, A., Ghanem, M., Boghanim, T., et al. (2022). FGF19 is downregulated in idiopathic pulmonary fibrosis and inhibits lung fibrosis in mice. American Journal of Respiratory Cell and Molecular Biology, 67(2), 173–187.
Kraven, L. M., Taylor, A. R., Molyneaux, P. L., et al. (2023). Cluster analysis of transcriptomic datasets to identify endotypes of idiopathic pulmonary fibrosis. Thorax, 78(6), 551–558.
Liu, H., Wang, B., Zhang, J., et al. (2017). A novel lnc-PCF promotes the proliferation of TGF-β1-activated epithelial cells by targeting miR-344a-5p to regulate map3k11 in pulmonary fibrosis. Cell Death & Disease, 8(10), e3137.
Cui, H., Banerjee, S., Guo, S., et al. (2019). Long noncoding RNA Malat1 regulates differential activation of macrophages and response to lung injury. JCI Insight. https://doi.org/10.1172/jci.insight.124522
Xu, Q., Cheng, D., Li, G., et al. (2021). CircHIPK3 regulates pulmonary fibrosis by facilitating glycolysis in miR-30a-3p/FOXK2-dependent manner. International Journal of Biological Sciences, 17(9), 2294–2307.
Wu, Q., Liu, Y., Xie, Y., Wei, S., & Liu, Y. (2021). Identification of potential ceRNA network and patterns of immune cell infiltration in systemic Sclerosis-associated interstitial lung disease. Frontiers in Cell and Developmental Biology, 9, 622021.
Liu, Q., Chen, X., Kan, M., et al. (2021). Gypenoside XLIX loaded nanoparticles targeting therapy for renal fibrosis and its mechanism. European Journal of Pharmacology, 910, 174501.
Ashcroft, T., Simpson, J. M., & Timbrell, V. (1988). Simple method of estimating severity of pulmonary fibrosis on a numerical scale. Journal of Clinical Pathology, 41(4), 467–470.
Shi, F., Xu, H., Liu, C., et al. (2021). Whole-transcriptome sequencing reveals a vernalization-related ceRNA regulatory network in chinese cabbage (Brassica campestris L. ssp pekinensis). BMC Genomics, 22(1), 819.
Langfelder, P., & Horvath, S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559.
Niu, Y., Zhang, L., Qiu, H., et al. (2015). An improved method for detecting circulating microRNAs with S-Poly(T) Plus real-time PCR. Scientific Reports, 5, 15100.
Selman, M., & Pardo, A. (2002). Idiopathic pulmonary fibrosis: An epithelial/fibroblastic cross-talk disorder. Respiratory Research, 3(1), 3.
Hormuzdi, S. G., Strandjord, T. P., Madtes, D. K., & Bornstein, P. (1999). Mice with a targeted intronic deletion in the Col1a1 gene respond to bleomycin-induced pulmonary fibrosis with increased expression of the mutant allele. Matrix Biology : Journal of the International Society for Matrix Biology, 18(3), 287–294.
Kunugi, S., Fukuda, Y., Ishizaki, M., & Yamanaka, N. (2001). Role of MMP-2 in alveolar epithelial cell repair after bleomycin administration in rabbits. Laboratory investigation: A Journal of Technical Methods and Pathology, 81(9), 1309–1318.
Hung, C. F., Rohani, M. G., Lee, S. S., Chen, P., & Schnapp, L. M. (2013). Role of IGF-1 pathway in lung fibroblast activation. Respiratory Research, 14(1), 102.
Dong, Y., Geng, Y., Li, L., et al. (2015). Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. The Journal of Experimental Medicine, 212(2), 235–252.
Son, E. S., Fei, X., Yoon, J. H., et al. (2021). Comparison of Pharmacokinetics and Anti-Pulmonary Fibrosis-Related Effects of Sulforaphane and Sulforaphane N-acetylcysteine. Pharmaceutics, 13(7), 958.
Parra, E. R., Teodoro, W. R., de Morais, J., et al. (2010). Increased mRNA expression of collagen V gene in pulmonary fibrosis of systemic sclerosis. European Journal of Clinical Investigation, 40(2), 110–120.
Jessen, H., Hoyer, N., Prior, T. S., et al. (2021). Turnover of type I and III collagen predicts progression of idiopathic pulmonary fibrosis. Respiratory Research, 22(1), 205.
Hamaguchi, Y., Fujimoto, M., Matsushita, T., Hasegawa, M., Takehara, K., & Sato, S. (2008). Elevated serum insulin-like growth factor (IGF-1) and IGF binding protein-3 levels in patients with systemic sclerosis: Possible role in development of fibrosis. The Journal of Rheumatology, 35(12), 2363–2371.
Snyder-Talkington, B. N., Dong, C., Sargent, L. M., et al. (2016). mRNAs and miRNAs in whole blood associated with lung hyperplasia, fibrosis, and bronchiolo-alveolar adenoma and adenocarcinoma after multi-walled carbon nanotube inhalation exposure in mice. Journal of Applied Toxicology : JAT, 36(1), 161–174.
Makiguchi, T., Yamada, M., Yoshioka, Y., et al. (2016). Serum extracellular vesicular miR-21-5p is a predictor of the prognosis in idiopathic pulmonary fibrosis. Respiratory Research, 17(1), 110.
Jia, R., Li, T., & Wang, N. (2021). Long noncoding RNA HOTAIR functions as ceRNA to regulate MMP2 in paraquat induced lung epithelial-mesenchymal transition. Toxicology, 461, 152891.
Jeon, S., Jin, H., Kim, J. M., et al. (2023). The miR-15b-Smurf2-HSP27 axis promotes pulmonary fibrosis. Journal of Biomedical Science, 30(1), 2.
Lei, X., He, N., Zhu, L., et al. (2021). Mesenchymal stem cell-derived extracellular vesicles attenuate radiation-induced lung injury via miRNA-214-3p. Antioxidants & Redox Signaling, 35(11), 849–862.
Salmena, L., Poliseno, L., Tay, Y., Kats, L., & Pandolfi pp. (2011). A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell, 146(3), 353–358.
Fatica, A., & Bozzoni, I. (2014). Long non-coding RNAs: New players in cell differentiation and development. Nature Reviews Genetics, 15(1), 7–21.
Tay, Y., Rinn, J., & Pandolfi pp. (2014). The multilayered complexity of ceRNA crosstalk and competition. Nature, 505(7483), 344–352.
Tay, Y., Kats, L., Salmena, L., et al. (2011). Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell, 147(2), 344–357.
Taulli, R., Loretelli, C., & Pandolfi pp. (2013). From pseudo-ceRNAs to circ-ceRNAs: a tale of cross-talk and competition. Nature Structural & Molecular Biology, 20(5), 541–543.
Guil, S., & Esteller, M. (2015). RNA-RNA interactions in gene regulation: The coding and noncoding players. Trends in Biochemical Sciences, 40(5), 248–256.
Liu, Y., Lu, F. A., Wang, L., Wang, Y. F., & Wu, C. F. (2021). Long non-coding RNA NEAT1 promotes pulmonary fibrosis by regulating the microRNA-455-3p/SMAD3 axis. Molecular Medicine Reports. https://doi.org/10.3892/mmr.2021.11857
Thomson, D. W., & Dinger, M. E. (2016). Endogenous microRNA sponges: Evidence and controversy. Nature Reviews Genetics, 17(5), 272–283.
Denzler, R., Agarwal, V., Stefano, J., Bartel, D. P., & Stoffel, M. (2014). Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Molecular cell, 54(5), 766–776.
Li, J., Li, P., Zhang, G., Qin, P., Zhang, D., & Zhao, W. (2020). CircRNA TADA2A relieves idiopathic pulmonary fibrosis by inhibiting proliferation and activation of fibroblasts. Cell Death & Disease, 11(7), 553.
Fang, S., Guo, H., Cheng, Y., et al. (2018). circHECTD1 promotes the silica-induced pulmonary endothelial-mesenchymal transition via HECTD1. Cell Death & Disease, 9(3), 396.
Lin, W., Song, Y., Li, T., et al. (2023). Triptolide attenuates pulmonary fibrosis by inhibiting fibrotic extracellular matrix remodeling mediated by MMPs/LOX/integrin. Biomedicine & Pharmacotherapy Biomedecine & Pharmacotherapie, 166, 115394.
Zheng, S., Zhang, Y., Hou, Y., et al. (2023). Underlying Molecular Mechanism and Construction of a miRNA-Gene Network in Idiopathic Pulmonary Fibrosis by Bioinformatics. International Journal of Molecular Sciences, 24(17), 13305.
Zou, M., Zou, J., Hu, X., Zheng, W., Zhang, M., & Cheng, Z. (2021). Latent transforming growth Factor-β binding protein-2 regulates lung fibroblast-to-Myofibroblast differentiation in pulmonary fibrosis via NF-κB Signaling. Frontiers in Pharmacology, 12, 788714.
Jin, Y. K., Li, X. H., Wang, W., et al. (2018). Follistatin-like 1 promotes Bleomycin-induced pulmonary fibrosis through the transforming growth factor Beta 1/mitogen-activated protein kinase signaling pathway. Chinese Medical Journal, 131(16), 1917–1925.
Acknowledgements
Not applicable.
Funding
This work was supported by The National Natural Science Foundation of China (89202586), Joint Application Foundation Project of Kunming Medical University and Yunnan Provincial Science and Technology Department (202101AY070001-271 and 202001AY070001-289), and Open Project of Yunnan Provincial Clinical Medical Center for Respiratory System Diseases (2020LCZXKF-HX08, 2022LCZXKF-HX04).
Author information
Authors and Affiliations
Contributions
YHZ and DMG: designed the study. DYL and JZ: constructed the BLM-induced PF mice model. JW, SJL, LQ, GHF and KW: performed comprehensive bioinformatics analysis. DYL, DXS, and ZMH: performed the qRT-PCR validation. DYL: wrote the article. JW, DMG and YHZ: revised the article. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All procedures comply with National Institutes of Health guidelines (NIH Publication, 8th edition, 2011), and were approved by the Institutional Animal Care and Use Committee (IACUC) of The First People’s Hospital of Yunnan Province.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, D., Wang, J., Zeng, J. et al. Identification and Validation of Genes Exhibiting Dynamic Alterations in Response to Bleomycin-Induced Pulmonary Fibrosis. Mol Biotechnol (2023). https://doi.org/10.1007/s12033-023-00943-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12033-023-00943-4