
Abstract
Idiopathic pulmonary fibrosis (IPF) is an interstitial lung disease characterized by chronic progressive pulmonary fibrosis and a poor prognosis. Genetic studies, including transcriptomic and proteomics, have provided new insight into revealing mechanisms of IPF. Herein we provided a novel strategy to identify biomarkers by integrative analysis of transcriptomic and proteomic profiles of IPF patients. We examined the landscape of IPF patients' gene expression in the transcription and translation phases and investigated the expression and functions of two new potential biomarkers. Differentially expressed (DE) mRNAs were mainly enriched in pathways associated with immune system activities and inflammatory responses, while DE proteins are related to extracellular matrix production and wound repair. The upregulated genes in both phases are associated with wound repair and cell differentiation, while the downregulated genes in both phases are associated with reduced immune activities and the damage of the alveolar tissues. On this basis, we identified thirteen potential marker genes. Among them, we validated the expression changes of butyrophilin-like 9 (BTNL9) and plasmolipin (PLLP) and investigated their functional pathways in the IPF mechanism. Both genes are downregulated in the tissues of IPF patients and Bleomycin-induced mice, and co-expression analysis indicates that they have a protective effect by inhibiting extracellular matrix production and promoting wound repair in alveolar epithelial cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
IPF is a type of interstitial lung disease with a poor prognosis and is associated with immune and inflammatory responses. It is characterized by persistent, progressive pulmonary fibrosis. Over 80% of IPF patients have an average survival period of 3–5 years after diagnosis. IPF primarily affects people over the age of 50, and its prevalence rises with age. There is a strong correlation between smoking or smoking history and the incidence of IPF. Despite extensive research, the etiology and pathogenesis of IPF are still not fully understood. Many hypothetical mechanisms, such as immune-mediated inflammation and malfunctioned alveolar epithelial cell (AEC) repairment, have been proposed in recent decades to contribute to the progression of IPF [1]. In the last decades, genome-wide sequencing has made achievements in identifying disease susceptible motifs in familial and sporadic pulmonary fibrosis. Mutations in several genes, including surfactant proteins C (SFTPC) [2], surfactant proteins A2 (SFTPA2) [3], and mucin 5B (MUC5B) [4], have been identified as a driving factor for IPF development, and transcriptomic and proteomic studies have identified pathways and biological processes that may be involved in the IPF mechanism [5].
In addition to traditional genome analysis, research on the IPF transcriptome and proteome has provided new insight into the mechanism of IPF. Proteome analysis using two-dimensional gel electrophoresis and MALDI–TOF–MS on IPF lung tissues as early as 2011 identified up- and down-regulated proteins in IPF, such as heat–shock protein 27 (Hsp27) [6]. Along with the development of novel experiment technologies, proteomes studies on IPF have been conducted using technologies, such as two-dimensional reversed-phase liquid chromatography and ion-mobility-assisted data-independent acquisition (HDMSE) [7], SOMAscan [8], and iTRAQ-based LC–MS/MS [9, 10]. The biological specimen used for the proteomics studies include lung tissues [6, 10], bronchoalveolar lavage fluid (BALF) [7], and peripheral blood [8, 9, 11]. These proteomics studies have detected differentially expressed (DE) proteins and identified biomarkers for IPF, such as matrix metallopeptidase 7 (MMP7), alpha Heremans–Schmid glycoprotein (AHSG), and vascular endothelial growth factor receptor (VEGFR).
Although the application of whole transcriptomic technology in IPF is later than that of proteomics, several transcriptomic studies have shed light on understanding the mechanism of IPF. In 2018, transcriptomic analysis of IPF lung tissues revealed transcriptomic changes in normal-appearing and scarred areas [12]. In 2019, Sheu et al. investigated the expression changes associated with Nintedanib treatment in IPF Fibroblasts and identified down-regulated genes and associated pathways [13]. Sheu’s team also identified dysregulated genes in IPF fibroblasts the same year [14].
As research on the proteomics and transcriptomics of IPF progresses, there emerged a need to integrate and analyze the comprehensive characterization of IPF gene expression by combining multiple omics. As the first attempt, Konigsberg et al. identified molecular signatures and their signaling pathways by combining transcriptome, DNA methylome, and proteome of lung tissues from IPF patients [15]. To make further use of multi-omics analysis and identify novel IPF biomarkers, we designed the present experiment to jointly sequence and analyze the transcriptomes and proteomes of lung tissue samples from end-stage IPF patients. Our findings demonstrated the differences and correlations between the characteristics of gene expression during the transcription and translation phases, as well as an overview of the non-coding RNA regulative network. We revealed the featuring pathological processes that occur during transcription and protein translation and identified butyrophilin-like 9 (BTNL9) and plasmolipin (PLLP) as promising new IPF-associated biomarkers. Our research and efforts point in a new direction and might provide guidance for future studies that aim to unravel the mystery of the IPF mechanism.
Materials and methods
Participant description
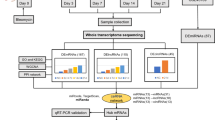
The overall study population consists of IPF lung tissues from nine end-stage IPF patients who underwent lung transplantation surgery at the First Affiliated Hospital of Guangzhou Medical University, Guangdong Province, China, and the healthy lung tissues from nine lung donors. Six IPF tissues and five healthy tissues were collected for the multi-omics experiments at the first stage, and the other three IPF tissues and four healthy tissues were further collected for the validation experiment (qPCR, and Western blot). This study was approved by the ethics committee of The First Affiliated Hospital of Guangzhou Medical University (Reference number: 2018-92). Signed informed consent was obtained from each patient. All IPF patients were diagnosed following the criteria suggested by the ATS/ERS/JRS/ALAT Clinical Practice Guideline [16] and the “Chinese Expert Consensus on Diagnosis and Treatment of Idiopathic Pulmonary Fibrosis” [17]: (1) Exclusion of other known causes of ILD (e.g., domestic and occupational environmental exposures, connective tissue disease, drug toxicity); (2) the presence of a UIP pattern on the high resolution computed tomography (HRCT); (3) for patients who had undergone surgical lung biopsy, the diagnosis is made by the present of both histopathology patterns and HRCT patterns. In this study, the diagnosis of all the subjects was further confirmed by histology for each IPF patient.
Nine IPF patients include seven males and two females, with an average age of 61.2 years. No patient had a family history of IPF. The medical history of symptom onset is 5.5 years on average. Five patients had smoking histories of at least 30 years, while four others were non-smokers. No general information of the lung donors was collected, because no consent was obtained. The IPF patients' general information and the specimens' usage in each experiment are provided in Supplementary file 1.
RNA-seq library construction and sequencing
Total RNA was extracted from the lung tissues of the patients using the Trizol (invitrogen) according to the manufacturer’s protocol, and ribosomal RNA was removed using the Ribo-Zero™ kit (Epicentre, Madison, WI, USA). Integrity of RNA was examined with the Bioanlyzer 2200 (Agilent). cDNA libraries were prepared using the Illumina TruSeq RNA Sample Preparation kit (Illumina). Fragmented RNA (the average length was approximately 200 bp) were subjected to first-strand and second-strand cDNA synthesis following by adaptor ligation and enrichment with a low cycle according to instructions of NEBNext® Ultra™ RNA Library Prep Kit for Illumina (NEB, USA). The purified library products were evaluated using the Agilent 2200 TapeStation and Qubit®2.0 (Life Technologies, USA). The libraries were paired-end sequenced (PE150, Sequencing reads were 150 bp) at Guangzhou RiboBio Co., Ltd. (Guangzhou, China) using Illumina platform HiSeq3000.
Quality control of RNA sequencing reads
Raw fastq sequences were treated with Trimmomatic tools [18] (v 0.36) using the following options: TRAILING: 20, MINLEN:25, and CROP:25, to remove trailing sequences below a Phred quality score of 20 and to achieve uniform sequence lengths for downstream clustering processes. Sequencing read quality was then inspected using the FastQC software [19]. Adapter removal and read trimming were performed using Trimmomatic. Sequencing reads were trimmed from the end (base quality less than Q20) and filtered by length (less than 25).
Quantification of mRNA expression
Paired-end reads were aligned to the human reference genome hg19 with HISAT2 [20]. HTSeq [21] (v0. 6.0) was used to count the reads numbers mapped to each gene. The whole sample’s expression levels were presented as TPM (Transcripts Per Million), which is the recommended and most common method to estimate the level of gene expression.
Differential expression analysis
The statistically significant DE genes were obtained by an adjusted p value threshold of < 0.05 and |log2(fold change)|> 1 using the DEGseq2 software [22]. Finally, a hierarchical clustering analysis was performed using the R language package ‘gplots’ according to the TPM values of differential genes in different groups. In addition, colors represent different clustering information, such as the similar expression pattern in the same group, including similar functions or participating in the same biological process.
GO terms and KEGG pathway enrichment analysis
All differentially expressed mRNAs were selected for GO and KEGG pathway analyses. GO was performed with KOBAS (version 3.0) software [23]. GO provides label classification of gene function and gene product attributes (http://www.geneontology.org). GO analysis covers three domains: cellular component (CC), molecular function (MF), and biological process (BP). The differentially expressed mRNAs and the enrichment of different pathways were mapped using the KEGG pathways with KOBAS (version 3.0) software.
Target mRNA prediction for DE lncRNAs
In this study, potential target genes for cis- or trans-acting of DE lncRNAs were predicted using different algorithms. Cis-acting target genes were identified by scanning the genome using ORF-finder [24] and BLASTP pipeline [25] (e < 1 × 10–5). Protein-coding genes located within 10 kb upstream or downstream of the lncRNA were obtained as cis-acting targets of the lncRNA. for the prediction of trans-acting target genes, mRNAs that have complementary sequences to lncRNAs were detected by BLASTN (e < 1 × 10–5), and then they were re-screened by the RNAplex tool [26].
Proteomic library construction and data acquisition
For library generation by data-dependent acquisition (DDA), all 11 samples were pooled as a mixture and fractionated by high pH separation with 8 fractions. In addition, all the samples were processed by data-independent acquisition (DIA) individually to assess the proteome differences. First stage mass spectrometry (MS1) and second-stage mass spectrometry (MS2) data were all acquired, and samples acquisition by random order. The iRT kit (Ki3002, Biognosys AG, Switzerland) was added to the samples to calibrate the retention time of extracted peptide peaks. Raw Data of DDA were processed and analyzed by Spectronaut 14 (Biognosys AG, Switzerland) with default settings to generate an initial target list, which contained 94,052 precursors, 87,319 peptides, 9232 proteins, and 9119 protein group. Spectronaut was set up to search the database of uniprot-homo_sapiens.fasta database (version 201,907, 20,414 entries) assuming trypsin as the digestion enzyme. Carbamidomethyl (C) was specified as the fixed modification. Oxidation (M) was specified as the variable modifications. Q value (FDR) cut off on precursor and protein level was applied 1%.
Proteomic analysis
Principal component analysis (PCA) was carried out separately on each data set using the R function ‘prcomp()’ from the package ‘stats’ with default parameters. Hierarchical Cluster Analysis (HCA) was processed with package ‘pheatmap’ (https://CRAN.R-project.org/ package=pheatmap). Volcano plot was drawn using ‘ggplot2’ package [27]. The online tool of Metascape [28] was used to perform GO enrichment analysis. Pathway analysis was processed by KOBAS [23].
Multi-omics analysis
First, we collated the DE RNAs in transcriptomes and subdivided them into mRNA, miRNA, antisense RNA, lincRNA, and lncRNA (which is non-lincRNA and non-antisense). After that, we generated quantitative matrices of these RNAs and the DE proteins, where the RNAs were represented as normalized TPM, and proteins were represented as normalized quantitative signal intensity. Then the R packages ‘mixOmics’ (version 6.14.0) [29] and ‘rgl’ (version 0.105.12) were utilized to conduct the Data Integration Analysis for Biomarker discovery using a Latent cOmponents (DIABLO) analysis [30]. DIABLO is a multivariate integrative classification method that seeks common information and identifies key variables in multiple omics. Based on the analysis method of Partial Least Squares (PLS) and generalized canonical correlation analysis, DIABLO maximizes the common or correlated information between multiple omics datasets by selecting a subset of molecular features and discriminating between multiple phenotypic groups. The ‘block.splsda’ function in the mixOmics’ package was used to integrate the omics and select key genes from each matrix via N-integration with sparse Discriminant Analysis. Then, the ‘plotIndiv’ function was used to provide scatter plots of the PLS–discriminant analysis (PLS–DA) analysis for each block, the ‘plotDiablo’ function was used to visualize the correlation between components from a different matrix, the ‘circosPlot’ function was used to display correlations between selected variable (i.e., RNAs, proteins) in different blocks in a circus, and the ‘cimDiablo’ function was used to generate a heatmap to represent the multi-omics molecular signature expression for each sample.
Classification and GO functional analysis of DE genes in transcription and translation
We categorized the differentially expressed genes at the transcription and protein translation phases and studied the enriched pathways associated with each category of DE genes. We extracted all expression measurements from proteomics and transcriptomics, including log2 fold change (LFC) and FDR adjusted p values, converted the gene IDs of the two matrices into consistent gene names, and merged the two matrices by the gene names. Using R language (version 4.0.3), we classified the genes based on their transcriptional and protein translational differences and plotted them in different colors. The cutoffs used for DE genes were FDR adjusted p < 0.05, fold change > 2 for transcriptome expression and FDR adjusted p < 0.05, fold change > 1.2 for protein levels. For the genes differentially expressed in both stages, we performed GO enrichment analysis using the Metascape tool [28].
DE analysis of public transcriptome datasets
First, we searched the NCBI’s GEO database [31] for high-quality transcriptomes from lung tissue of IPF patients. As a result, 91 datasets (52 IPF tissues vs. 39 healthy tissues) from four RNA-sequencing projects (GSE52463 [32], GSE83717 [33], GSE92592 [34], and GSE99621 [12]) were identified and downloaded using NCBI’s sratoolkit (http://ncbi.github.io/sra-tools/, version 2.9.6-1). Second, the reads were filtered using the Trimmomatic tool [18] and were mapped to the human reference genome hg38 by STAR [35]. Then the transcript counts were calculated using the featureCounts software [36]. Then, the differential expression analysis was conducted by R package DEseq2 [22] following the standard protocol. The batch biases among different projects were controlled using the design function (design = ~ project + status).
Bleomycin (BLM) IPF mouse model
Twenty-two C57BL/6 male mice were randomly divided into two groups: the IPF group (n = 9), and the control group (n = 13). BLM solution for use was prepared by dissolving 15 mg BLM in 5 mL 0.9% NaCl. Mice were anesthetized via intraperitoneal injection of 1% pentobarbital sodium (50 mg/kg) and fixed on the mouse plate. Either BLM (IPF groups) or saline (control group) 2.1 mg/kg was administered into the glottis using a 100 mL pipette. On day 21 after BLM induction, the establishment of the animal model was confirmed by the presence of progressive pulmonary fibrosis and alveolitis and increased expression of type I collagen (COL I) and Fibronectin in the lung tissue. Thereafter, the mice were sacrificed, their lung tissues were collected for further assays.
Quantitative PCR (qPCR)
RNA in lung tissues collected from patients (six patients from the omics cohort study and three newly recruited IPF patients) and BLM-induced mice was extracted using the Trizol (Invitrogen®). The reverse transcription reaction was conducted following manufacturer's protocol (TaKaRa™). 5uL cDNA was mixed with 01 μL primers and 10 μL 2 × SYBR Green qPCR SuperMix (QiaGen™) in a 20 μL reaction. PCR was performed in LightCycler® 480 II PCR system (Roche™). GAPDH was used as internal control.
Western blot
Tissues from human and BLM-induced mice were lysed with radioimmunoprecipitation (RIPA) lysis buffer (with phenylmethylsulfonyl fluoride (PMSF)) (Beyotime Biotech™); the concentration of the protein solution was measured by the bicinchoninic acid (BCA) protein assay (KeyGene Biotech™). Protein was resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) electrophoresis and then transferred onto methanol pre-wet polyvinylidene difluoride (PVDF) membranes. After incubation with secondary antibodies, the PVDF membranes were mixed with enhanced chemiluminescence (ECL) substrate (Thermo Scientific™), the intensity of light was detected by the Bio-Rad imaging system.
Immunohistochemistry
The tissue sections collected from IPF patients and BLM-induced mice were deparaffinized using xylene and then rehydrated by alcohol solution (85% and 75%) and distilled water. The heat retrieval of antigen was performed by placing the sections in a repair box filled with citric acid (PH6.0) antigen retrieval buffer in a microwave oven. The sections were incubated in 3% hydrogen peroxide for 25 min’ room temperature to block endogenous peroxidases activity. Endogenous antigens were blocked by 3% bovine serum albumin (BSA). The sections were then added with primary antibody (rabbit anti-mouse, Bioss Inc and Sino Biological Inc. for BTNL9 and PLLP, respectively) [dilated by phosphate-buffered saline (PBS)] and incubated in a wet box overnight at 4 ℃. After washing and shaking, the tissue sections were incubated with secondary antibody (anti-rabbit, Wuhan Servicebio Technology Co., Ltd) at room temperature for 50 min. The tissues were stained by 3, 3’-diaminobenzidine (DAB) chromogenic solution and the nucleus was counterstained by hematoxylin stain solution. The stained slides were observed by Nikon® E100 and images were captured by the Nikon DS-U3 camera control unit.
Co-expression network of BTNL9 and PLLP
To probe the possible function pathways of BTNL9 and PLLP, we generated a co-expression network for each based on the public IPF transcriptome datasets prepared in “Target mRNA prediction for DE lncRNAs”. The GSEA software [37] was used to calculate the enrichment score for each gene sets following its official guide. The networks were visualized using the Cytoscape software [38] and the gene set clusters were annotated by the AutoAnnotate application [39].
Results
Quality control of transcriptomes and proteomes
Quality control of RNA-seq reads
The libraries were constructed for the RNA sequencing, and deep sequencing was completed for the ten (six IPF vs. four control) samples that met the quality requirements. The samples had an average of 150,114,026 ± 15,174,766 sequence Reads and 22,517,103,960 ± 2,276,214,831 bases. There was no significant difference between the control group and the IPF group in the measured number of sequences and bases (p > 0.05). The average base error rate was 0.53 ± 0.098%, and there is no significant difference between the control group and the IPF group (p < 0.05). The average GC ratio (GC%) was 47.15 ± 1.80%, the percentage of Q20% bases (error rate < 1%) was 93.87 ± 1.18%, and G30% bases (error rate < 0.1%) was 86.30 ± 2.02%. There was no significant difference in these three indicators between the two groups (p < 0.05). After data filtering, the average clean Q3 ratio is 89.25 ± 1.52%, and the cleaning rate is 90.81 ± 0.83% (Table 1). The bases had a homogeneous distribution along with the sequences, the maximum error rate is < 1%, and the minimum base quality [− 10 × log10(error P)] was above 30 (Fig. S1A–C). The sequence quality was further improved after data filtering (Fig. S1D–F).
Mapping quality of RNA-seq reads
94.7 ± 0.005% reads were successfully mapped to the human reference genome. The detected gene number approached saturation along with the increase of mapped reads, indicating a good sequence depth of this experiment. The averaged mapped genes of all the samples were 27,000 to 33,000 (Fig. S2A). The quality control and comparison results show that the sequencing results met the quality requirements for further analysis. Among the detected RNA sequences, 81.33% derived from exons, 16.1% from intronic, and 2.57% from intergenic reads (Fig. S2B). The detected genes were evenly distributed across chromosomes by comparison with the human gene distribution map (Fig. S2C).
Quality control and quantification of proteomes
Libraries were generated for all 11 samples that met the quality requirements and the proteins were detected and quantified. As a result, 9119 protein groups and 9232 proteins were detected at the QC level of 1% FDR (Spectrum, Peptide, and Protein levels). The levels of 7823 protein groups and 7932 proteins were quantified at the QC level of 1% FDR (precursor and protein levels). The average coefficient of variation (CV) of the precursors was 40.80% and 31.90% for the control and IPF samples, respectively. The median of the precursors’ CV was 40.6% and 32.4% for the control and IPF samples (Fig. S3A). The recovery rate (the ratio of the identified proteins to the indicators in the human protein library) was 68.90% and 77% and the completeness (the ratio of the average number of identified proteins to the number of parent ions quantified in the experiment) was 51.90% and 63.50% for the control and IPF group samples, respectively. The cumulative recovery plot shows that 85% of proteins from the protein spectrum database have been detected in the 11 samples (Fig. S3B). The completeness plot shows that the total completeness of all samples was 83.8%, with 4200 proteins identified in all samples (Fig. S3C). Consistency analysis of the qualitative results showed that 3200 proteins were detected in all samples, and another 3000 proteins were detected in more than half of the samples (Fig. S3D). The heat map of all the detected proteins shows no significant differences in the identification and quantification among all samples (Fig. S3E).
Transcriptome analysis of IPF
Differential gene expression
We identified the DE genes between samples through two cutoffs: log fold change (|log2(fold change)|> 1) and significance level (FDR adjusted p < 0.05). In comparison with normal lung tissue, a total of 2531 genes were significantly differentially expressed in the lungs of patients with end-stage IPF, including 1772 up-regulated and 759 down-regulated genes (Fig. 1A). Clustered heatmap (Fig. 1B) shows that the control and IPF groups could be well separated by the genes, while the samples of IPF number 1, 4, and 5 had a clearer contrast with controls.

DE mRNAs and GO enrichment analysis. A Volcano plot of DE genes in transcription phase. The cutoff was set as |log2(fold change)|> 1 and adjusted p value < 0.05. The fold changes and p values were calculated by DESeq2. B Heatmap of the expression (TPM) of DE genes. mRNA expression (TPM) of each gene was log10 transformed and are displayed as colors ranging from red to blue as shown in the key. Both rows and columns are clustered using correlation distance and average linkage. C Bar plot of significantly enriched gene sets, classified into biological processes, cellular components, and molecular functions. The enriched gene sets are classified into biological processes, cellular components, and molecular functions. For each term, the bar in the left is the log10 transformed the enrichment score, and the bar in the right is the number of genes that fall into the term
GO and KEGG enrichment analysis was performed to probe the biological processes and signaling pathways associated with the DE genes (Fig. 1C). IPF lung tissues had a significant enrichment of biological processes and functional pathways that dominate the mechanism of IPF when compared to control tissues. These enriched pathways influence the progression of IPF at the biological process, cellular structure, and molecular function levels. Eight of the top ten pathways are related to the immune system activities and inflammatory response. Furthermore, there are also the pathways of the construction of ECM, which replaces normal Alveolar tissue and deposits abnormally in IPF [16, 40].
Differential lncRNA expression
To investigate the regulatory impact of lncRNA in the end-stage IFP patients’ gene expression, we first quantified their expression and identified DE lncRNAs. The results showed that a total of 604 lncRNAs were significantly differentially expressed in IPF lung tissue, including 410 up-regulated genes and 194 down-regulated genes (Fig. 2A). Clustered heatmap (Fig. 2B) showed that the expression of these DE lncRNAs could separate the IPF samples from the control samples. In addition, IPF samples 1, 4, and 5 showed a clearer contrast to the control samples than the other three IPF samples.

DE lncRNAs and GO enrichment analysis. A Volcano plot of DE lncRNAs in transcription phase. The cutoff was set as |log2(fold change)|> 1 and adjusted p value < 0.05. The fold changes and p values were calculated by DESeq2. B Heatmap of the expression of the DE lncRNAs. Con1–Con5 represent the four control lung tissues, while Exp1–Exp6 represent the six IPF lung tissues. lncRNA expression levels (TPM) was log10 transformed and are displayed as colors ranging from red to blue as shown in the key. Both rows and columns are clustered using correlation distance and average linkage. C Dot plot of significantly enriched gene sets. Each circle represents a term, the color is the log10 transformed the enrichment score, and the circle size is the number of genes that fall into the term
As the lncRNAs mainly function by regulating the protein-coding target genes, we predicted the potential target genes of cis-regulation and trans-regulation for the lncRNAs. We then performed GO enrichment analysis on the target genes and analyzed the results with the significance threshold of FDR adjusted p < 0.05 (Fig. 2C). Most of the enriched pathways were associated with the structure and function of lung epithelial apical junction, such as apical junction assembly and tight junction assembly. This implies that the DE lncRNAs in IPF may mainly promote the process of epithelial–mesenchymal transition (EMT), cell migration, accelerated fibrosis progression, innate immunity, as well as cellular differentiation and proliferation [41, 42]. Besides, there are also two pathways related to apoptosis, such as the cysteine-type endopeptidase activity involved in apoptotic process.
Proteomics analysis of IPF
Principal component analysis was performed on the protein expression data using the PLS–DA method, and the top 2 components were plotted in Fig. 3A. The results showed that the end-stage IPF tissues were more concentrated on the graph compared with normal tissues, indicating a higher homogeneity of protein expression and a more consistent within-group expression profile. We performed a Welch’s ANOVA test on the protein quantifications and defined the DE proteins by a threshold of adjusted p < 0.05 and fold change > 1.5. As a result, we got 1532 DE proteins in IPF tissues, including 1231 up-regulated proteins and 301 down-regulated proteins (Fig. 3B).

DE proteins and GO enrichment analysis. A PLS–DA plot. Displays the first two components of all samples. The components were calculated using the Projection to Latent Structures–Discriminant Analysis (PLS–DA) method. B Volcano plot of the DE proteins. C Heatmap of the enriched GO sets. The color represents the enrichment score of each gene set. C1–C5 represent the five control samples, D1–D5 represent the six IPF samples. D Bar plot of the enriched KEGG pathways. The bars represent the number of genes fall in the pathway and the FDR adjusted p value of the enrichment analysis. The horizontal coordinate is the percentage of enriched proteins to all the differential proteins
Figure 3C shows the top 10 enrichment results under the three categories of Biological Process, Molecular Function, and Cellular Component. We note that these enriched gene clusters were mainly focused on the negative regulation of TOR and TORC1 signaling, which are associated with the decreased metabolism and protein production, autophagy, and extracellular matrix (ECM) production in end-stage IPF [43,44,45].
The DE proteins were significantly enriched in 13 KEGG pathways (FDR adjusted p < 0.05) (Fig. 3D). According to previous studies, five of them are associated with the pathology of end-stage IFP. The RAS signaling pathway is associated with cell apoptosis and regeneration [46, 47], the tight junction and gap junction are associated with cell regeneration and junction construction [41, 42], the mTOR signaling pathway regulates cell growth and metabolism [43,44,45], and nucleotide excision repair is associated with wound repair [48, 49].
Multi-omics analysis
By interactively analyzing the expression matrices of RNAs of different types and proteins, we identified the key genes of each type that drive the discrimination between IPF and control tissues and investigated the correlations between the ncRNAs and the expression of mRNAs and proteins.
Using the DIABLO method, we identified the genes contributing most to the discrimination between IPF and control tissues. These top-contributing genes include 20 mRNAs, 20 proteins, ten lncRNAs, ten lincRNAs, ten antisense RNAs, and ten miRNAs. These top-contributing genes include 20 mRNAs, 20 proteins, 10 lncRNAs, 10 lincRNAs, 10 antisense RNAs and 10 miRNAs. Only the top-10 genes were kept from three types of ncRNAs, this is because they each have relatively small gene numbers (from 59 to 279).
First, we display the discrimination of the IPF samples and control samples by the PLS–DA plot (Fig. 4A). In the PCA plots, the control samples and IPF samples 1, 2, 4, and 5 were clustered closely in all blocks, while the IPF samples 6 and 3 were at longer distances from the other IPF samples. Among the six blocks of the expression matrix, the IPF samples are more discrete in the mRNA and protein blocks, while they are more homogeneous in the ncRNA blocks. Figure 4B shows the correlation structure between components from each expression matrix. There are very strong associations between ncRNAs, mRNA, and proteins, and the correlation coefficients between any two datasets ≥ 0.98. The results indicate a good matrices design that favors the separation of the two groups.

Multi-omics analysis using DIABLO method. A PLS–DA plot of expression matrices of proteins and RNAs. The distances between samples represent the discrimination between two conditions, which were calculated based on the top-contributing genes in each gene type. The samples from different groups are represented with different colors. B Correlation structure between the expression of different gene types. Colors indicate the class of each sample. The number in the bottom left are the correlation coefficients between two expression matrices. C Clustered heatmap of the mix-omics signature variables. Con1–Con5 represent the four control lung tissues, while IPF1–IPF5 represent the six IPF lung tissues. Both rows and columns are clustered using correlation distance and average linkage. This map displays the scaled expression of above the top-contributing genes from six matrices
Second, we created a clustered heat map representing the multi-omics profiles of all the samples (Fig. 4C). The image shows that these top-contributing genes from six matrices well represent the separation of gene expression features of the IPF and control group. The only exception is the IPF sample 6, the expression characteristics of which are similar to neither the control nor the other IPF tissues. This result is consistent with the PLS–DA plot, in which IPF is also clearly discriminated from other IPF samples on the first component (x-axis). As the most important contributors to the expression characteristics, the top-contributing proteins include ROM01, T22D3, MIS12, ZN384, LHPL2, TANC2, DESI1, MEA1, ARID2, NFRKB, PKP2, MTG1, RIPR2, ARHGP, DPOA2, GNB1L, YETS2, IKZF1, MBOA2, and CEP57.
Third, we produced a Circos plot displaying the relationship between and within the top-contributing genes from the six matrices, the cutoff for the correlation coefficient was set as > 0.9 (Fig. 5). The strong correlations between mRNAs, proteins, and the ncRNAs indicate a universal regulatory effect of these ncRNAs on mRNA transcription and protein translation. Compared to the proteins, mRNAs had more strong links with the regulatory ncRNAs. Among the ncRNAs, the lincRNA has the most links with protein and mRNA, suggesting a significant regulatory role in IPF. In this co-expression network, the most-contribute variables are the lincRNAs ENST00000437698.1 and ENST00000442197.1, the antisense RNAs ENST00000519197.1 and ENST00000566738.1, the lncRNAs NR_110255.1 and NR_024344.1, and the miRNAs NR_030340.1 and NR_030408.1.

Circos plot of the correlations between genes of different types. Red links stand for positive correlation and blue links stands for negative correlations. The orange line stands for the expression level in IPF tissues, while the blue line stands for the expression in control tissues
DE genes classification and functional enrichment analysis
Genes significantly differentially expressed at both transcriptional and translational phases were extracted and classified into four categories based on the trend they were regulated. The threshold of FDR adjusted p value was set as < 0.05, and threshold of fold change was set as > 2 for transcriptome and > 1.2 for proteome. The results are displayed in the quadrant diagram (Fig. 6A).

DE genes in both omics and GO enrichment analysis. A Quadrant plot of gene expression in transcription and translation phase. B Bar plot of enriched pathways of genes upregulated in transcription and translation phases. C Bar plot of enriched pathways of genes downregulated in transcription and translation phases. D Clustered network of the enriched terms for the up-regulated genes. E Clustered network of the enriched terms for the down-regulated genes
Classification of DE genes
A total of 78 genes were differentially expressed in both transcriptome and proteome, and they were divided into four categories according to their regulation. 24 genes were significantly up-regulated in both omics, such as TUBB3, IGLV1-47, and CAPS. A total of 46 genes were significantly downregulated in both omics, including AGER, BNTL9, and RETN. Eight genes had opposite regulation trends, three genes were significantly down-regulated in the transcriptome but up-regulated in the proteome: CSK, RAC2, and SEMA5B. Five other genes were significantly up-regulated during transcription but down-regulated during translation: EPS8L1, GON7, HOMER2, IGLV8-61, and PROC.
GO enrichment analysis
The 24 genes were most frequently located on chromosomes 4 and 11, which had 3 and 4 genes, respectively. Twenty-one genes had four or more isoforms, suggesting that isoforms may be more active in the lung tissue of patients with severe IPF. These genes are significantly enriched in seven biological pathways and high-level GO terms (Fig. 6B). The enrichment network shows that the enriched functions were clustered in the biological process of regeneration and cell morphogenesis involved in differentiation (Fig. 6D). These over-activated pathways participated in the cell regeneration, differentiation, and intercellular sequential generation, which probably due to the deteriorated tissue damage and regeneration processes in the end-stages IPF patients [1, 50].
These genes downregulated in both omics mainly locate on chromosomes 1, 9, 17, and 19. The genes were significantly enriched in 12 biological pathways and high-level GO terms (Fig. 6C). The enrichment network shows that the most enriched terms were the biological process of myeloid leukocyte activation, regulation of IL-1 β production, cell–cell communication, cellular extravasation, and lipid localization (Fig. 6E). These significant compromised functions and biological processes in the end-stage IPF lung tissues might be associated with reduced immune activities and the damage and obliteration of the alveolar tissue [51].
Identification of potential biomarkers
To further validate the expression of these 78 DE genes we obtained in the previous step, we further checked for their expression in the 91 IPF transcriptomes from public databases. The results showed that in comparing the IPF lung tissues and healthy lung tissues, 66 genes had significant DE with adjusted p values < 0.05 (Supplementary file 2). Among these 66 genes, we further identified 13 genes that had the most significant fold changes and adjusted p values in all three experiments (Table 2).
Literature review shows that approximately half of these 13 candidate genes have been reported involving in IPF mechanism or differentially expressed, which proves the efficiency of our research approach. Four genes have been reported to have significant impacts on the pathology of IPF {S100A4 [52], STX11 [53], THY1 [54], and TUBB3[55]}, another three have been reported DE expression in IPF yet not validated {BTNL9 [56], SELENBP1 [57], and PLLP[58]}, while no study had been reported in IPF for six genes (ADGRL2, CA4, IGLV1-47, LIMCH1, MID1IP1, and QDPR).
Focusing on the three DE genes that have been reported in previous studies, we selected two, BTNL9 and PLLP, for further validation after investigating their known impacts on human biology and pathology. BTNL9 is a biomarker and prognosis indicator for several types of lung cancers [59,60,61], and it is involved in the extracellular matrix–receptor (ECM-receptor) pathway [62]. PLLP encodes the membrane protein Plasmolipin, which functions in the epithelial development [63] and migration [64]. Although SELENBP1 is a cancer-preventing gene which inhibits lung adenocarcinoma growth [65], it had no reported involvement in fibrosis-associated processes, such as epithelial development and ECM generation. Besides, BTNL9 and PLLP have more significant fold change and adjusted p value than SELENBP1. Thus, we determined to focus validation on BTNL9 and PLLP.
BTNL9 and PLLP expression in lung tissues of IPF patients and BLM-induced mice
Both genes’ mRNA transcript expression was quantified using qPCR, their protein expression in lung tissues was detected by Western blotting, and their subcellular expression was investigated by IHC staining. qPCR assay showed that PLLP’s mRNA expression was significantly reduced in IPF patients (Wilcoxon test, p < 0.01) (Fig. 7B), while BTNL9’s mRNA expression had a non-significant reduction (Fig. 7A). In BLM-induced mouse model, mRNA transcription of both BTNL9 and PLLP were significantly decreased (Wilcoxon test, p < 0.05, Fig. 7D and E). Western blotting assay showed that both genes had decreased protein expression in both IPF patients and in BLM-induced mice (Fig. 7C and F).

Validation experiment for BTNL9 and PLLP. A, B: qPCR of BTNL9 and PLLP’s expression in human IPF tissues. C Western blot of BTNL9 and PLLP’s expression in human IPF tissues. D, E: qPCR of BTNL9 and PLLP’s expression in mouse model. F Western blot of BTNL9 and PLLP’s expression in mouse model
Using IHC technology, we stained and imaged the BTNL9 and PLLP proteins in the lung tissues from IPF patients (Supplementary Fig. 4) and BLM-induced mice (Supplementary Fig. 5). In healthy human lung tissues, BTNL9 protein was expressed in the nuclear membrane of type 1 AEC cells, which is consistent with previous studies [66]. Besides, it was also expressed in the nuclear membrane of a number of type 2 AEC cells. While in lung tissues from IPF patients or BLM-induced mice, the expression significantly decreased, no staining areas could be found in the cells within fibrotic foci. Moreover, it also had decreased expression in other cell types, including type 1 and 2 AEC cells, the cytoplasm of lung bronchiolar epithelial cells. PLLP has a very high expression on the cell membrane of type 1 AEC cells of healthy lung tissues. In contrast, in In lung tissues from IPF patients or BLM-induced mice, the expression of PLLP decreased, especially type 1 AEC cells in the fibrotic foci.
Co-expression networks of BTNL9 and PLLP
The co-expression network demonstrates the promoted and inhibited gene sets associated with the expression of specific genes. The co-expression network of BTNL9 shows that its expression is associated with the promotion of endothelium establishment, vessel endothelium migration, and construction of cell–cell junction. BTNL9’s expression is associated with the inhibited pathways, such as immune system activity, production of extracellular matrix, and cilium production (Fig. 8). The co-expression network of PLLP shows that its expression is associated with the promotion of endothelium development, cell membrane, and cell junction development. It is associated with the inhibited pathways, such as abnormal respiratory function, immune system activity, and cilium production (Fig. 9).

Co-expression network of BTNL9. Red nods are enriched gene sets, and blue nodes are inhibited gene sets. Node size represents the gene number of the gene set, and edge width stands for the similarity between gene sets

Co-expression network of PLLP. Red nods are enriched gene sets, and blue nodes are inhibited gene sets. Node size represents the gene number of the gene set, and edge width stands for the similarity between gene sets
Discussion
IPF is a progressive interstitial lung disease. IPF patients suffer deteriorating pulmonary fibrosis and their average survival time after diagnosis is 3–5 years. IPF is now widely recognized as the consequence of excessive myofibroblast proliferation and extracellular matrix deposition initiated by malfunctioned wound repair process in aged lung epithelial cells. However, current research is still some way from fully understanding the pathogenesis of IPF, and to date, only two antifibrotic drugs have shown valid therapeutic effects on IPF in clinical trials [67, 68].
Over the last two decades, studies on IPF’s whole-genomics, including gene mutation, transcriptomics, and proteomics, have provided new perspectives for understanding the pathogenesis and pathological process of IPF, identifying biomarkers for diagnosis and prognosis, and searching for new therapeutic targets [1]. In addition to traditional genomic analysis, recent gene expression studies have confirmed the roles of long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) in the pathogenesis and progression of and IFP [69,70,71,72,73,74,75]. Recently, the multi-omics analysis started being applied in revealing the IPF mechanism. Konigsberg et al. described the molecular landscape of IPF by integratively analyzing DNA methylome, transcriptome, and proteome using the “mixOmics” tool [15].
To further the multi-omics study of IPF, we designed this study to profile the integrative feature of IPF and identify new biomarkers. In this experiment, we depicted the expression characteristics and gene expression correlation network of IPF patients using an integrative analysis of the transcriptomes and proteomes of end-stage IPF patients’ lung tissues. During transcription, DE genes in IPF patients were mainly enriched in immune-related pathways and some ECM-related pathways. DE genes in protein translation, on the other hand, were mainly enriched in biological functions and pathways associated with extracellular matrix production and deposition, such as negative regulation of TOR and TORC1 signaling, intracellular organelle part, and gap junction. This suggested that the upregulated transcription of immune-related genes might lead to the enhanced production of proteins associated with ECM production. The differences in gene expression characteristics during the transcriptional and protein translation phases indicate the significance of ncRNA’s regulative impact in IPF. The multi-omics analysis of the proteomics and expression matrix of five RNA subtypes revealed that non-coding RNAs are highly involved in the progress of IPF. Among them, lincRNAs have more correlation links to the mRNA and proteins. Antisense RNAs, lncRNAs, and miRNAs also had strong correlations. Due to the limitations of this study, we were unable to conclude specific causal relationships between these interacting variables, which need to be investigated in the future using a new experimental design.
We further investigated the DE genes in both phases and identified novel biomarkers for IPF. Twenty-two genes were significantly up-regulated during both transcriptional and translation, while 46 were significantly down-regulated. GO enrichment analysis revealed that, the most prominent processes in end-stage IPF patients include the enhanced activities of AEC injury and repair, increased ECM production, and compromised immune activities.
Among the DE genes in both the transcription and translation phase, we examined the expression of BTNL9 and PLLP probed their possible roles in IPF. BTNL9 encodes the protein Butyrophilin-Like-9 which is involved in cell-mediated immunity via the pathway of Class I MHC mediated antigen processing and presentation [76]. RNA-seq studies showed that it is down-regulated in IPF [77, 78] and chronic hypersensitivity pneumonitis [79]. Using qPCR and Western blot, we validate its reduced mRNA and protein expression in both IPF patients and BLM-induced mice. Co-expression analysis indicates that BTNL9 is associated with reduced immune response and might slow down IPF progression by inhibiting ECM production. It also might promote the wound healing of injured AEC by enhancing endothelium regeneration and cell–cell adhesion. PLLP encodes the plasmolipin which is involved in the development and differentiation of epithelial cells [80, 81]. Our study validated that PLLP is down-regulated in the lung tissues from both IPF patients and BLM-induced mice. PLLP might protect the tissue by enhancing the development of endothelium, cell membrane, and cell–cell junction. Its downregulation is associated with impairment of respiratory function, which is consistent with previous observation in COPD patients [82]. These results indicate that both genes might play protective roles in IPF and their downregulation in IPF is associated with IPF progressions, such as increased immune responses, ECM production, and impaired wound healing. BTNL9 inhibits excessive proliferation in lung tissue, prohibiting tumor development and fibroblast proliferation. It can also work as a biomarker for IPF. Besides, it is also noteworthy that both genes significantly impact the development and function of cilia (Figs. 8, 9), which is also the main function target of TMEM231, the DE gene with the greatest increase in Konigsberg et al.’s study [15]. This commonality in the findings of both studies indicates that cilia-associated pathways might be a promising direction of IPF mechanism investigation and treatment development.
The authors acknowledge that this experiment has certain limitations. First, due to the rarity of the IPF incidence and the decreasing clinical application of biopsy in the diagnosis of IPF, the sample size of our study was relatively small, which might affect the credibility of our results. Second, although the end-stage IPF patients were recruited following strict criteria, a certain degree of heterogeneity was still observed in sample six, which might have been caused by different phenotypic subgroups [83]. Nevertheless, the high homogeneity of the other samples might guarantee the credibility of our results. Third, although the expression and possible roles of BTNL9 and PLLP have been preliminarily probed, their functional pathways need to be further validated in future research.
In summary, in this study we sequenced and analyzed the transcriptomes and proteomes of end-stage IPF patients, portraying the landscape of end-stage IPF patient's whole-genome expression composed of DE genes, enriched biological processes, and the regulating networks. Based on this, we identified two IPF potential biomarker genes downregulated in both IPF patients and BLM-induced mice, BTNL9 and PLLP, which might protect against ECM production and promoting wound repair in alveolar epithelial cells. Our results reveal the most prominent pathological processes of IPF in the transcription and translation phase and provide an efficient strategy for future research on IPF mechanisms and biomarker identification.
Data availability
The authors declare that all data supporting the findings of this study are available from the corresponding authors on reasonable request.
Code availability
The authors declare that code for data analysis in this study are available from the corresponding authors on reasonable request.
References
Meyer KC, Nathan SD (2019) Idiopathic pulmonary fibrosis. Humana Press, Cham
Lawson WE, Grant SW, Ambrosini V, Womble KE, Dawson EP, Lane KB et al (2004) Genetic mutations in surfactant protein C are a rare cause of sporadic cases of IPF. Thorax 59:977–980. https://doi.org/10.1136/thx.2004.026336
Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL et al (2009) Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet 84:52–59. https://doi.org/10.1016/j.ajhg.2008.11.010
Kropski JA, Blackwell TS, Loyd JE (2015) The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J 45:1717–1727. https://doi.org/10.1183/09031936.00163814
Kaur A, Mathai SK, Schwartz DA (2017) Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front Med (Lausanne) 4:154. https://doi.org/10.3389/fmed.2017.00154
Korfei M, Schmitt S, Ruppert C, Henneke I, Markart P, Loeh B et al (2011) Comparative proteomic analysis of lung tissue from patients with idiopathic pulmonary fibrosis (IPF) and lung transplant donor lungs. J Proteome Res 10:2185–2205. https://doi.org/10.1021/pr1009355
Foster MW, Morrison LD, Todd JL, Snyder LD, Thompson JW, Soderblom EJ et al (2015) Quantitative proteomics of bronchoalveolar lavage fluid in idiopathic pulmonary fibrosis. J Proteome Res 14:1238–1249. https://doi.org/10.1021/pr501149m
O’Dwyer DN, Norman KC, Xia M, Huang Y, Gurczynski SJ, Ashley SL et al (2017) The peripheral blood proteome signature of idiopathic pulmonary fibrosis is distinct from normal and is associated with novel immunological processes. Sci Rep 7:46560. https://doi.org/10.1038/srep46560
Niu R, Liu Y, Zhang Y, Zhang Y, Wang H, Wang Y et al (2017) iTRAQ-based proteomics reveals novel biomarkers for idiopathic pulmonary fibrosis. PLoS ONE 12:e0170741. https://doi.org/10.1371/journal.pone.0170741
Tian Y, Li H, Gao Y, Liu C, Qiu T, Wu H et al (2019) Quantitative proteomic characterization of lung tissue in idiopathic pulmonary fibrosis. Clin Proteomics 16:6. https://doi.org/10.1186/s12014-019-9226-4
Landi C, Bergantini L, Cameli P, d’Alessandro M, Carleo A, Shaba E et al (2020) Idiopathic pulmonary fibrosis serum proteomic analysis before and after nintedanib therapy. Sci Rep 10:9378. https://doi.org/10.1038/s41598-020-66296-z
Luzina IG, Salcedo MV, Rojas-Pena ML, Wyman AE, Galvin JR, Sachdeva A et al (2018) Transcriptomic evidence of immune activation in macroscopically normal-appearing and scarred lung tissues in idiopathic pulmonary fibrosis. Cell Immunol 325:1–13. https://doi.org/10.1016/j.cellimm.2018.01.002
Sheu CC, Chang WA, Tsai MJ, Liao SH, Chong IW, Kuo PL (2019) Gene expression changes associated with nintedanib treatment in idiopathic pulmonary fibrosis fibroblasts: a next-generation sequencing and bioinformatics study. J Clin Med 8:308. https://doi.org/10.3390/jcm8030308
Sheu CC, Chang WA, Tsai MJ, Liao SH, Chong IW, Kuo PL (2019) Bioinformatic analysis of next-generation sequencing data to identify dysregulated genes in fibroblasts of idiopathic pulmonary fibrosis. Int J Mol Med 43:1643–1656. https://doi.org/10.3892/ijmm.2019.4086
Konigsberg IR, Borie R, Walts AD, Cardwell J, Rojas M, Metzger F et al (2021) Molecular signatures of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. https://doi.org/10.1165/rcmb.2020-0546OC
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ et al (2018) Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 198:e44–e68. https://doi.org/10.1164/rccm.201807-1255ST
Group of Interstitial Lung Diseases RDB, Association CM (2016) Consensus of Chinese experts on diagnosis and treatment of idiopathic pulmonary fibrosis. Chin J Tuberc Respir Dis 39:427–432. https://doi.org/10.3760/cma.j.issn.1001-0939.2016.06.005
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Andrews S (2010) FASTQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 26 Apr 2010
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12:357–360. https://doi.org/10.1038/nmeth.3317
Anders S, Pyl PT, Huber W (2014) HTSeq—a python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. https://doi.org/10.1093/bioinformatics/btu638
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S et al (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316-322. https://doi.org/10.1093/nar/gkr483
Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU et al (2003) Database resources of the National Center for Biotechnology. Nucleic Acids Res 31:28–33. https://doi.org/10.1093/nar/gkg033
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410. https://doi.org/10.1016/s0022-2836(05)80360-2
Tafer H, Hofacker IL (2008) RNAplex: a fast tool for RNA-RNA interaction search. Bioinformatics 24:2657–2663. https://doi.org/10.1093/bioinformatics/btn193
Wickham H (2016) ggplot2. Springer International Publishing, Cham
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O et al (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10:1523. https://doi.org/10.1038/s41467-019-09234-6
Rohart F, Gautier B, Singh A, Le Cao KA (2017) mixOmics: an R package for ‘omics feature selection and multiple data integration. PLoS Comput Biol 13:e1005752. https://doi.org/10.1371/journal.pcbi.1005752
Singh A, Shannon CP, Gautier B, Rohart F, Vacher M, Tebbutt SJ et al (2019) DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 35:3055–3062. https://doi.org/10.1093/bioinformatics/bty1054
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M et al (2013) NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41:D991-995. https://doi.org/10.1093/nar/gks1193
Nance T, Smith KS, Anaya V, Richardson R, Ho L, Pala M et al (2014) Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS ONE 9:e92111. https://doi.org/10.1371/journal.pone.0092111
Vukmirovic M, Herazo-Maya JD, Blackmon J, Skodric-Trifunovic V, Jovanovic D, Pavlovic S et al (2017) Identification and validation of differentially expressed transcripts by RNA-sequencing of formalin-fixed, paraffin-embedded (FFPE) lung tissue from patients with idiopathic pulmonary fibrosis. BMC Pulm Med 17:15. https://doi.org/10.1186/s12890-016-0356-4
Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ et al (2017) Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8:14532. https://doi.org/10.1038/ncomms14532
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. https://doi.org/10.1093/bioinformatics/bts635
Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30:923–930. https://doi.org/10.1093/bioinformatics/btt656
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102:15545–15550. https://doi.org/10.1073/pnas.0506580102
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. https://doi.org/10.1101/gr.1239303
Kucera M, Isserlin R, Arkhangorodsky A, Bader GD (2016) AutoAnnotate: a cytoscape app for summarizing networks with semantic annotations. F1000Res 5:1717. https://doi.org/10.12688/f1000research.9090.1
Richeldi L, Collard HR, Jones MG (2017) Idiopathic pulmonary fibrosis. Lancet 389:1941–1952. https://doi.org/10.1016/s0140-6736(17)30866-8
Kulkarni T, de Andrade J, Zhou Y, Luckhardt T, Thannickal VJ (2016) Alveolar epithelial disintegrity in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 311:L185-191. https://doi.org/10.1152/ajplung.00115.2016
Rout-Pitt N, Farrow N, Parsons D, Donnelley M (2018) Epithelial mesenchymal transition (EMT): a universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir Res 19:136. https://doi.org/10.1186/s12931-018-0834-8
Demetriades C, Doumpas N, Teleman Aurelio A (2014) Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell 156:786–799. https://doi.org/10.1016/j.cell.2014.01.024
Plate M, Guillotin D, Chambers RC (2020) The promise of mTOR as a therapeutic target pathway in idiopathic pulmonary fibrosis. Eur Respir Rev 29:200269. https://doi.org/10.1183/16000617.0269-2020
Romero Y, Bueno M, Ramirez R, Alvarez D, Sembrat JC, Goncharova EA et al (2016) mTORC1 activation decreases autophagy in aging and idiopathic pulmonary fibrosis and contributes to apoptosis resistance in IPF fibroblasts. Aging Cell 15:1103–1112. https://doi.org/10.1111/acel.12514
Hosseinzadeh A, Javad-Moosavi SA, Reiter RJ, Hemati K, Ghaznavi H, Mehrzadi S (2018) Idiopathic pulmonary fibrosis (IPF) signaling pathways and protective roles of melatonin. Life Sci 201:17–29. https://doi.org/10.1016/j.lfs.2018.03.032
Nataraj D, Ernst A, Kalluri R (2010) Idiopathic pulmonary fibrosis is associated with endothelial to mesenchymal transition. Am J Respir Cell Mol Biol 43:129–130. https://doi.org/10.1165/rcmb.2010-0044ED
Hunninghake GW, Gadek JE, Lawley TJ, Crystal RG (1981) Mechanisms of neutrophil accumulation in the lungs of patients with idiopathic pulmonary fibrosis. J Clin Invest 68:259–269. https://doi.org/10.1172/jci110242
Bucala R, Spiegel LA, Chesney J, Hogan M, Cerami A (1994) Circulating fibrocytes define a new leukocyte subpopulation that mediates tissue repair. Mol Med 1:71–81
Beers MF, Morrisey EE (2011) The three R’s of lung health and disease: repair, remodeling, and regeneration. J Clin Invest 121:2065–2073. https://doi.org/10.1172/JCI45961
Chambers RC, Mercer PF (2015) Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann Am Thorac Soc 12(Suppl 1):S16-20. https://doi.org/10.1513/AnnalsATS.201410-448MG
Li Y, Bao J, Bian Y, Erben U, Wang P, Song K et al (2018) S100A4(+) macrophages are necessary for pulmonary fibrosis by activating lung fibroblasts. Front Immunol 9:1776. https://doi.org/10.3389/fimmu.2018.01776
Cui Y, Ji J, Hou J, Tan Y, Han X (2021) Identification of key candidate genes involved in the progression of idiopathic pulmonary fibrosis. Molecules. https://doi.org/10.3390/molecules26041123
Chen L, Tang R-Z, Ruan J, Zhu X-B, Yang Y (2019) Up-regulation of THY1 attenuates interstitial pulmonary fibrosis and promotes lung fibroblast apoptosis during acute interstitial pneumonia by blockade of the WNT signaling pathway. Cell Cycle 18:670–681
Chilosi M, Calio A, Rossi A, Gilioli E, Pedica F, Montagna L et al (2017) Epithelial to mesenchymal transition-related proteins ZEB1, beta-catenin, and beta-tubulin-III in idiopathic pulmonary fibrosis. Mod Pathol 30:26–38. https://doi.org/10.1038/modpathol.2016.147
DePianto DJ, Chandriani S, Abbas AR, Jia G, N’Diaye EN, Caplazi P et al (2015) Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax 70:48–56. https://doi.org/10.1136/thoraxjnl-2013-204596
Bauer Y, Tedrow J, de Bernard S, Birker-Robaczewska M, Gibson KF, Guardela BJ et al (2015) A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 52:217–231. https://doi.org/10.1165/rcmb.2013-0310OC
Wang F, Li P, Li FS (2020) Integrated analysis of a gene correlation network identifies critical regulation of fibrosis by lncRNAs and TFs in idiopathic pulmonary fibrosis. Biomed Res Int 2020:6537462. https://doi.org/10.1155/2020/6537462
Hsu Y-L, Hung J-Y, Lee Y-L, Chen F-W, Chang K-F, Chang W-A et al (2017) Identification of novel gene expression signature in lung adenocarcinoma by using next-generation sequencing data and bioinformatics analysis. Oncotarget. https://doi.org/10.18632/oncotarget.21022
Ma W, Liang J, Zhang S, Ningdong H, Chen Z, Tian D (2020) Butyrophilin-like 9 regulates immune infiltration and serves as a prognostic marker in lung adenocarcinoma. BMC Cancer. https://doi.org/10.21203/rs.3.rs-113263/v1
Wang Y, Zhao N, Zhang X, Li Z, Liang Z, Yang J et al (2020) Bibliometrics analysis of butyrophilins as immune regulators [1992–2019] and implications for cancer prognosis. Front Immunol. https://doi.org/10.3389/fimmu.2020.01187
Bao Y, Wang L, Shi L, Yun F, Liu X, Chen Y et al (2019) Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell Mol Biol Lett 24:38. https://doi.org/10.1186/s11658-019-0162-0
Le Guelte A, Macara IG (2015) Plasmolipin—a new player in endocytosis and epithelial development. EMBO J 34:1147–1148. https://doi.org/10.15252/embj.201591448
You J, Corley SM, Wen L, Hodge C, Hollhumer R, Madigan MC et al (2018) RNA-Seq analysis and comparison of corneal epithelium in keratoconus and myopia patients. Sci Rep 8:389. https://doi.org/10.1038/s41598-017-18480-x
Caswell DR, Chuang CH, Ma RK, Winters IP, Snyder EL, Winslow MM (2018) Tumor suppressor activity of Selenbp1, a direct Nkx2-1 target, in lung adenocarcinoma. Mol Cancer Res 16:1737–1749. https://doi.org/10.1158/1541-7786.Mcr-18-0392
Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H et al (2017) A subcellular map of the human proteome. Science 356:eaal3321. https://doi.org/10.1126/science.aal3321
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U et al (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370:2071–2082. https://doi.org/10.1056/NEJMoa1402584
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK et al (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 370:2083–2092. https://doi.org/10.1056/NEJMoa1402582
Tzouvelekis A, Kaminski N (2015) Epigenetics in idiopathic pulmonary fibrosis. Biochem Cell Biol 93:159–170. https://doi.org/10.1139/bcb-2014-0126
Huang C, Yang Y, Liu L (2015) Interaction of long noncoding RNAs and microRNAs in the pathogenesis of idiopathic pulmonary fibrosis. Physiol Genom 47:463–469. https://doi.org/10.1152/physiolgenomics.00064.2015
Molyneaux PL, Willis-Owen SAG, Cox MJ, James P, Cowman S, Loebinger M et al (2017) Host-microbial interactions in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 195:1640–1650. https://doi.org/10.1164/rccm.201607-1408OC
Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN et al (2014) Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med 2:548–556. https://doi.org/10.1016/S2213-2600(14)70069-4
Pandit KV, Milosevic J (2015) MicroRNA regulatory networks in idiopathic pulmonary fibrosis. Biochem Cell Biol 93:129–137. https://doi.org/10.1139/bcb-2014-0101
Mizuno K, Mataki H, Seki N, Kumamoto T, Kamikawaji K, Inoue H (2017) MicroRNAs in non-small cell lung cancer and idiopathic pulmonary fibrosis. J Hum Genet 62:57–65. https://doi.org/10.1038/jhg.2016.98
Yang IV, Schwartz DA (2015) Epigenetics of idiopathic pulmonary fibrosis. Transl Res 165:48–60. https://doi.org/10.1016/j.trsl.2014.03.011
Stelzer G, Rosen N, Plaschkes I, Zimmerman S, Twik M, Fishilevich S et al (2016) The genecards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinform. https://doi.org/10.1002/cpbi.5
Li D, Liu Y, Wang B (2020) Identification of transcriptomic markers for developing idiopathic pulmonary fibrosis: an integrative analysis of gene expression profiles. Int J Clin Exp Pathol 13:1698–1706. https://www.ncbi.nlm.nih.gov/pubmed/32782692. Accessed 15 July 2020
Wang Z, Zhu J, Chen F, Ma L (2019) Weighted gene coexpression network analysis identifies key genes and pathways associated with idiopathic pulmonary fibrosis. Med Sci Monit 25:4285–4304. https://doi.org/10.12659/MSM.916828
Furusawa H, Cardwell JH, Okamoto T, Walts AD, Konigsberg IR, Kurche JS et al (2020) Chronic hypersensitivity pneumonitis, an interstitial lung disease with distinct molecular signatures. Am J Respir Crit Care Med 202:1430–1444. https://doi.org/10.1164/rccm.202001-0134OC
Fischer I, Durrie R, Sapirstein VS (1994) Plasmolipin: the other myelin proteolipid. A review of studies on its structure, expression, and function. Neurochem Res 19:959–966. https://doi.org/10.1007/bf00968705
Rodríguez-Fraticelli AE, Bagwell J, Bosch-Fortea M, Boncompain G, Reglero-Real N, García-León MJ et al (2015) Developmental regulation of apical endocytosis controls epithelial patterning in vertebrate tubular organs. Nat Cell Biol 17:241–250. https://doi.org/10.1038/ncb3106
Zhang Y-H, Hoopmann MR, Castaldi PJ, Simonsen KA, Midha MK, Cho MH et al (2021) Lung proteomic biomarkers associated with chronic obstructive pulmonary disease. medRxiv. https://doi.org/10.1101/2021.04.07.21255030
Mohning MP, Swigris JJ, Olson AL (2019) Idiopathic pulmonary fibrosis: the epidemiology and natural history of disease. In: Meyer KC, Nathan SD (eds) Idiopathic pulmonary fibrosis. Springer International Publishing, Cham, pp 11–35 (Respiratory Medicine)
Funding
This work was supported by the National Natural Science Foundation of China (Project No. 81871736), Bureau of traditional Chinese Medicine Scientific Research Project of Guangdong (Project No. 20192048), The First Affiliated Hospital Of Guangzhou Medical University (ZH201915) (funds from GMU), Guangzhou Institute of Respiratory Health Open Project (Funds provided by China Evergrande Group, Project No. 2020GIRHHMS04), the Zhongnanshan Medical Foundation of Guangdong Province (Project No: ZNSA-2021005 and Project No: ZNSA-2020001), the University of Macau (grant numbers: FHS-CRDA-029-002-2017, and MYRG2018-00071-FHS), the Science and Technology Development Fund, Macau SAR (File No. 0004/2019/AFJ and 0011/2019/AKP), and Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (Project No. 2018PT31048). Funding for open access charge: National Natural Science Foundation of China.
Author information
Authors and Affiliations
Contributions
All authors participated in the study design, the interpretation of the results, and the drafting and revision of the manuscript. PZ and SS conceived the idea and conducted the experiments, JW collected the specimens and conducted the experiments, SS analyzed the data and drafted the first version of the manuscript, XDZ and BS supervised the research. All authors reviewed and commented on the manuscript and approved the final draft. The authors would like to express their sincere thanks to Prof. Yingying Gu and Dr. Zhucheng Chen from The First Affiliated Hospital of Guangzhou Medical University for her support in determining the HRCT and histological patterns of the patients and confirming the IPF diagnosis. Besides, we would like to thank all members of Professor Sun’s lab and Professor Zhang’s lab for their kind help in conducting the experiments and preparing this manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
All procedures in this study involving human participants and animals were performed with the approval and according to the guidelines of the ethics committee of The First Affiliated Hospital of Guangzhou Medical University. All procedures were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all individuals for being included in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
18_2021_4094_MOESM1_ESM.pdf
Supplementary file1 Quality control results of RNA sequencing. A, B, and C are QC plots for raw sequences, and D, E, and F are QC plots for filtered sequences. A and D: Percentage of base content along position in reads. The fluctuations around 150th of the read position are caused by the read length limitation. B and E: error rates of bases along the position in the reads. C and F: the quality score of bases along the position in the reads. The quality score was calculated as -10×log10 (error P) (PDF 468 KB)
18_2021_4094_MOESM2_ESM.pdf
Supplementary file2 Distribution of mapped reads. A. Saturation map of all samples. It displays the increase of mapped genes along with the percentage of mapped reads. “Con” stands for control samples, and “Exp” stands for IPF samples. B. Mapped reads distribution. C. Reads density of a sample in the chromosomes. Red stands for forward strands, blue stands for reverse strands (PDF 842 KB)
18_2021_4094_MOESM3_ESM.pdf
Supplementary file3 Quality control of proteome. A. Density distribution of precursors. CV: coefficient of variation. “C” stands for control group and “D” stands for IPF group. B. Cumulative recovery plot for identified proteins. C. Completeness plot of the identified proteins. D. Identified proteins in each sample. C.1–C.5 represent the five control samples, D.1–D.6 represent the six IPF samples. E. Heatmap of the intensity of all quantified proteins (PDF 560 KB)
18_2021_4094_MOESM4_ESM.png
Supplementary file4 IHC results in human lung tissues. This figure displays the IHC staining of BTNL9 and PLLP in healthy and IPF lung tissues. The hematoxylin-stained nucleus is blue, and the DAB-positive area is brownish yellow. “×100” represents images captured through a 100 times lens, while “×400” represents images captured through a 400 times lens. BTNL9 stained nuclei were observed in AEC I cells of healthy lung tissue, while not observed in those of IPF lung tissues. PLLP staining was observed in AEC I cell membrane of healthy lung tissue, while not observed in those of IPF lung tissues (PNG 7897 KB)
18_2021_4094_MOESM5_ESM.png
Supplementary file5 IHC results in mouse lung tissues. This figure displays the IHC staining of BTNL9 and PLLP in lung tissues from BLM-induced mice and healthy ones. The hematoxylin-stained nucleus is blue, and the DAB-positive area is brownish yellow. “×100” represents images captured through a 100 times lens, while “×400” represents images captured through a 400 times lens. The comparison displays the destruction of the lung tissues in BLM-induced mice; both BTNL9 and PLLP showed decreased expression in the cytosol of lung bronchioles epithelial cells, and they increased expressed in alveoli cells (PNG 8832 KB)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Zheng, P., Sun, S., Wang, J. et al. Integrative omics analysis identifies biomarkers of idiopathic pulmonary fibrosis. Cell. Mol. Life Sci. 79, 66 (2022). https://doi.org/10.1007/s00018-021-04094-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-021-04094-0