
Abstract
Angiogenesis is a biological process finely tuned by a plethora of pro- and anti-angiogenic molecules, among which vascular endothelial growth factors (VEGFs). Their biological activity is expressed through the interaction with three cognate receptor tyrosine kinases, VEGFR1, 2, and 3. VEGFR2 is the primary regulator of angiogenesis. Ligand-induced VEGFR2 dimerization and activation depend on direct ligand binding to extracellular domains 2 and 3 of receptor and in the establishment of interactions between proximal membrane domains. VEGFR2 domain 7 has been shown to play a crucial role in receptor dimerization and regulation, therefore, representing a convenient target for the allosteric modulation of VEGFR2 activity. The ability to prepare a functional VEGFR2D7 domain represents the starting point to the development of novel VEGFR2 binders acting as allosteric inhibitors of receptor activity. Here, we describe a robust and efficient procedure for the preparation in E. coli of the VEGFR2 domain 7. The protein was obtained with a good yield and was properly folded. It was investigated in a biochemical and structural study, providing information on its conformational arrangement and in solution properties.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Angiogenesis is a biological process consisting in the sprouting of new blood vessels from pre-existing ones [1]. Starting from the pioneering investigations of Judah Folkman [2], molecular mechanisms of angiogenesis continued to be incessantly under analysis due to the recognition of the important role played in several pathologies, from tumors to cardiovascular diseases [3, 4]. As a consequence, the ability to pharmacologically modulate this process is still attracting a considerable biomedical interest. Vascular endothelial growth factors (VEGFs) regulate blood and lymphatic vessel development and homeostasis by binding to and activating the three members of the VEGF-receptor (VEGFR) family of receptor tyrosine kinases (RTKs) [5]. Of the three VEGF receptors, VEGFR2 is the primary regulator of endothelial cell proliferation and migration [6,7,8] and shares with most of all RTKs the same structural organization. This organization consists in an extracellular domain composed of seven immunoglobulin homology domains (Ig), from D1 (located at the N-terminus) to D7 (closest to the cell membrane), a trans-membrane domain, and an intracellular domain endowed of the kinase activity required for receptor downstream signaling. VEGFR2 is expressed in all human vascular endothelial cells and is often overexpressed in highly malignant solid tumors [9, 10]. Aggressive cancerous phenotypes correlate in fact with enhanced VEGFR2 signaling [11]. Ligand binding to VEGFR2 is mediated by Ig domains 2 and 3, while membrane proximal domains are involved, through the establishment of homotypic contacts, in ligand-induced receptor dimerization, ensuring a precise orientation of receptor monomers in the active dimers [12,13,14]. In particular, D4–D4 and D7–D7 homotypic interactions are involved and they are essential for VEGFR2 activation and downstream signaling [15,16,17,18]. In fact, through the analysis of VEGFR2 mutants in which D4 or D7 were substituted with unrelated domains from VEGFR1, the crucial role for these Ig domains in VEGFR2 dimerization was highlighted, furthermore showing that they stabilize VEGFR2 dimers also in the absence of the natural ligand [19]. Finally, it was demonstrated that selected Designed Ankyrin Repeat Proteins (DARPins) interacting with D4 or D7 domains inhibit receptor activity without preventing ligand-mediated receptor dimerization, revealing an allosteric mechanism for receptor inhibition that could represent an attractive option for neovascular diseases treatment [17, 19, 20]. In fact, several molecules, like antibodies [21, 22], peptides [23,24,25,26,27,28], and small organic compounds [29] have been developed so far, as effective tools in strategies aimed to treat diseases that depend on imbalanced angiogenesis, for applications in the diagnostic and therapeutic fields. Most of these molecules have been designed to target the ligand binding domains 2 and 3, but an interesting possibility is also the inhibition of VEGFR2 targeting allosteric sites [17, 22, 30, 31]. As already mentioned, VEGFR2D7 shows a crucial role in receptor dimerization and regulation, therefore, representing a convenient target for the allosteric modulation of VEGFR2 activity. Molecules interfering allosterically with the dimerization and activation of VEGFR2 may overcome the drawbacks associated with the use of conventional anti-angiogenic agents [32, 33]. A starting point to get this aim would be the preparation and characterization of VEGFR2D7 to be used in antibody preparation or in screening assay to select novel VEGFR2 targeting compounds. Thus, here we describe a rapid and reliable methodology for recombinant expression of VEGFR2D7 in E. coli, reporting its biochemical and conformational characterization in solution.
Materials and Methods
Reagents and Methods
Yeast extract and bacto tryptone for preparation of Escherichia coli (E. coli) growth medium, as well as the reagents for preparation of buffer solutions, were supplied by Applichem. Reagents for agarose and polyacrylamide gels electrophoresis were supplied by Sigma Aldrich (TEMED-tetramethylethylenediamine), Euroclone (APS-ammonium persulfate), and Applichem (agarose, acrylamide, SDS, tris, glycine). Molecular weight markers for DNA and proteins were from Sigma-Aldrich and New England Biolabs (NEB), respectively. Also restriction and modification enzymes (calf intestine phosphatase and T4 DNA ligase), as well as Taq DNA polymerase (5 U/μL), were supplied by NEB, while the Pfu Turbo polymerase (2.5 U/μL) was supplied by Agilent Technologies-Stratagene. pETM11 E. coli expression plasmid was supplied by Novagen. DNA purification kits and Ni2+-NTA resin were supplied by Qiagen. Sigma-Genosys took care of the synthesis of the oligonucleotides, while the sequencing service was commissioned at PRIMM srl. The analysis of VEGFR2D7 trypsin digestion was carried out by LC–MS performed on Agilent 1200 Infinity Series (Agilent Technologies) mass spectrometer equipped with an ESI source and a ToF detector and an Aeris widepore 150 × 2.1 mm, 3.6 μm, XB-C8 column (Phenomenex), using a method with a flow rate of 0.2 mL/min and a linear gradient of CH3CN (0.1% TFA) in H2O (0.1% TFA) from 5 to 70%, in 30 min.
VEGFR2D7 Protein Expression and Purification
DNA sequence corresponding to VEGFR2D7 (amino acids 657–765, Uniprot entry: P35968) was amplified by PCR starting from the cDNA of the full VEGFR2 receptor as template (Open Biosystems) and using the oligonucleotide sequences below reported as primers:
Forward: 5′-GCC ATG GCG AGG CAG CTC ACA-3′
Reverse: 5′-CCT CGA GCT ATC AGA TTT CCA AGT TCG TCT TTT C-3′.
The sequences of restriction enzymes are indicated in bold and the two stop codons are underlined.
The final volume of PCR reaction was of 100 μL containing 50 ng of DNA template, 30 pmol of each primer, dNTPs (0.25 mM each), and 5 units of Pfu Turbo DNA polymerase (2.5 U/μL); the annealing temperature chosen was of 42 °C. The amplified gene, once purified, was digested, for 3 h at 37 °C, in a final volume of 50 μL, with 4 U of NcoI (10 U/μL) and XhoI (20 U/μL) restriction enzymes. Digested gene was successively cloned into the pETM11 expression vector, downstream to the His-tag and TEV recognition site sequences. A 1:3 molar ratio between gene and vector was chosen for ligation reaction performed for 3 h at room temperature (RT), using 100 ng of NcoI–XhoI digested vector, in 10 μL containing 20 U of the T4 DNA Ligase (400 U/μL). The identity of the pETM11-VEGFR2D7 plasmid was confirmed by DNA sequencing. VEGFR2D7 was expressed in Rosetta 2 (DE3) E. coli strain transformed with 100 ng of the recombinant expression vector. Cells were grown over night (O/N) under shaking, at 37 °C, in the presence of kanamycin (50 μg/mL) and chloramphenicol (33 μg/mL) antibiotics and then inoculated in fresh medium containing antibiotics for the preparative culture. Once the exponential bacterial growth phase (0.7/0.8 OD600nm) was reached, cell culture was induced by adding 0.4 mM IPTG and allowed to incubate at 22 °C O/N. The pellet derived from 1 L of culture was suspended in 25 mL of a lysis buffer containing 50 mM Tris–HCl, 150 mM NaCl, 2 mM DTT, pH 8, and Roche protease inhibitors cocktail; the suspension was sonicated for 10 min, by using a Misonix Sonicator 3000 apparatus equipped with a macro tip probe, applying an impulse output of 0.5/1 (= 20/24 Watt). The lysate was centrifuged at 17,000 rpm for 30 min, at 4 °C (Beckman centrifuge equipped with a JA25.50 rotor) and the supernatant and the pellet samples were analyzed by SDS-PAGE on a 15% polyacrylamide gel. After extraction of the recombinant His-tagged VEGFR2D7 from inclusion bodies by means of the lysis buffer containing 10 mM imidazole and 8 M urea, the domain was loaded on Ni2+-NTA resin (40 min, RT) and eluted with high concentration of imidazole (250–300 mM). Fractions eluted from the resin were collected and analyzed by SDS-PAGE and those containing the protein domain were pooled. The concentration of the proteins in solution was determined according to the Bradford’s method, adding the Coomassie Brilliant (Bio-Rad) reagent to the samples and monitoring the absorbance at 595 nm. A solution of 1 μg/μL of bovine serum albumin (BSA) was used as standard. Protein refolding was performed according to the protocol described in Yang et al. [18]. Briefly, VEGFR2D7 was drop wise diluted into a refolding buffer containing 10 mM Tris–HCl, 100 mM NaCl, 3 mM reduced glutathione/0.3 mM oxidized glutathione, Ph 8, with final protein concentration at 0.1 mg/mL. The refolding was carried out at 4 °C for 48 h with stirring. Then, TEV protease was added to refolded protein for His-tag removal. Hydrolysis reaction was performed using a molar ratio of 1:35 (protease:substrate), in the presence of EDTA 0.5 mM, O/N at 4 °C. The cleaved protein was concentrated using the Amicon Ultra system (10,000 MWCO, Millipore) and purified to homogeneity by size-exclusion chromatography using a Superdex 7510/30 column (GE Healthcare) equilibrated in 20 mM Tris–HCl and 120 mM NaCl, pH 7. Finally, it was concentrated again until 0.5 mM. VEGFR2D7 concentration was determined measuring protein absorbance at 280 nm on a Jasco V-550 UV–VIS spectrophotometer, in an 1 cm quartz cell, using a molar extinction coefficient of 7240 cm−1 M−1 [https://web.expasy.org/protparam/]. Double-labeled 15N/13C-VEGFR2D7 was prepared as described in Di Stasi et al. [34], expressing the protein in minimal medium containing heavy nitrogen (15NH4Cl, Sigma, 1 g/L) and carbon (glucose-13C6, Sigma, 1 g/L) sources. Protein expression, refolding, and purification were performed as described for the unlabeled protein.
VEGFR2D7 Trypsin Digestion
Trypsin lyophilized powder (from porcine pancreas-Sigma Aldrich) was solubilized in HCl 1 mM and CaCl2 20 mM, at a concentration of 1 mg/mL. Limited proteolysis reaction was performed on 10 μg of 15N/13C-VEGFR2D7 diluted in 50 μL of Tris–HCl 50 mM, pH 7.5, using a molar ratio protein substrate:trypsin of 100:1. The reaction proceeded at 37 °C for 3 h and then was analyzed by LC–MS.
Circular Dichroism (CD) Analysis
The analysis of VEGFR2D7 secondary structure was performed by CD in the far-UV region, recording protein spectra on a Jasco J-810 spectropolarimeter equipped with a PTC-423S/15 peltier temperature controller. Protein domain was buffer exchanged into 10 mM potassium phosphate buffer pH 7 and experiments were performed at a final protein concentration of 15 μM, using a 0.1-cm path-length quartz cuvette (Hellma). CD spectra were collected at 20 °C, in the wavelength interval comprised between 190 and 260 nm, setting data pitch of 0.2 nm, 10 nm/min scan speed, 2.0 nm bandwidth, and 4 s response as parameters. The data were collected over three averaged scans and expressed as mean residue ellipticity [Ɵ] after subtracting the buffer contribution by using the Spectra Manager software. The CD spectrum of reduced VEGFR2D7, expressed as [Ɵ], was acquired at a final protein concentration of 1.5 μM, in a 1-cm path-length quartz cuvette, using the same experimental conditions and number of scans as VEGFR2D7, but a wavelength interval comprised between 198 and 260 nm, after incubating the protein in 2 mM DTT. CD spectra deconvolution was performed using the CDPro software [http://lamar.colostate.edu/*sreeram/CDPro/] and the protein reference set #4. The secondary structure content values were an average of the three algorithms (SELCON3, CDSSTR, and CONTINLL) used [35,36,37].
Fluorescence Spectroscopic Analysis
Spectroscopic measurements were taken on a Cary Eclipse spectrofluorimeter, registering spectra at 20 °C in a 1.0 cm quartz cell (Hellma), using samples prepared diluting the protein more than 100 fold, from 20 mM Tris–HCl and 120 mM NaCl, pH 7 in phosphate buffer 10 mM pH 7 containing increasing concentration (0.5 M; 1 M; 1.5 M; 2 M; 2.5 M; 3 M; 4 M; 5 M; 6 M) of guanidine hydrochloride (GuHCl, Sigma). GuHCl was diluted starting from an 8 M stock solution. After acquisition of VEGFR2D7 spectrum in native conditions (potassium phosphate buffer 10 mM, pH 7), the samples of protein in the presence of the above-mentioned GuHCl concentrations were left to equilibrate for 90 min at RT. All the experiments were carried out at a protein concentration of 4.5 μM, setting a scan speed of 120 nm/min, exciting the samples at 295 nm and recording the emission spectra in the range 300–500 nm. The excitation and the emission slit widths were set at 5 nm. VEGFR2D7 was also analyzed in reducing conditions, incubating the protein (4.5 μM) in phosphate buffer 10 mM pH 7 with 2 mM DTT, following the tryptophan emission in the range 300–500 nm. Denaturation curve was obtained reporting the values of tryptophan fluorescence emission registered at 350 nm in the function of GuHCl molar concentration. Data were analyzed using the software GraphPad Prism (version 6), setting a non-linear regression fit and a sigmoidal behavior for the curve showing the fluorescence intensity at 350 nm as a function of the guanidine hydrochloride concentration.
Static Light Scattering Analysis
The oligomeric state of VEGFR2D7 was evaluated using a SEC-LS method consisting of a semi-preparative size-exclusion chromatography analysis performed on a Superdex7510/30 (GE Healthcare) coupled to a light scattering spectrometer, equipped with a laser with λ = 658 nm, (mini DAWN TREOS-Wyatt Technology) and a differential refractive index detector (Shodex RI-101). 1 mg of the protein sample was loaded on Superdex 7510/30 equilibrated with 20 mM Tris–HCl, 120 mM NaCl, pH 7. The scattering data were processed by using the Astra (5.3.4.14 version, Wyatt Technology Corporation) software supplied with the instrument, providing the value of VEGFR2D7 average molecular weight.
NMR Spectroscopy Studies
All NMR experiments were carried out at 298 K using an Inova 600 MHz spectrometer (Varian Inc.), equipped with a cryogenic probe optimized for 1H detection. NMR sample consisted of 100 μM 15N/13C-VEGFR2D7 in 90% H2O/10% 2H2O (v/v), containing 20 mM Tris–HCl and 120 mM NaCl pH 7. One-dimensional (1D) 1H spectra were acquired with a spectral width of 6712.0 Hz, relaxation delay 1.0 s, 7 k data points for acquisition, and 16 k for transformation. Nuclear Overhauser effect spectroscopy (NOESY) was acquired with 32 or 64 scans per t1 increment with a spectral width of 6712.0 Hz along both t1 and t2, 2048 × 256 data points in t2 and t1, respectively, and recycle delay 1.0 s. Water suppression was achieved by means of Double Pulsed Field Gradient Spin-Echo (DPFGSE) sequence [38, 39]. 2D [15N, 1H] heteronuclear single quantum coherence (HSQC) spectra were acquired with 1024 (HN) × 128 (N) data points and eight scans. Spectral widths of 6712.0 and 2066.3 Hz were used in the HN and N dimensions, respectively. The data were apodized with a square cosine window function and zero filled to a matrix of size 4096 × 1024 before Fourier transformation and baseline correction. 1D 1H spectrum was analyzed by using ChemAxon software (http://www.chemaxon.com) while bi-dimensional spectra were processed with SPARKY [40] and analyzed with CARA (Computer Aided Resonance Assignment) software [41] (http://cara.nmr.ch). Proton chemical shift were calibrated using DSS as internal reference, while 15N was calibrated indirectly, using an external reference. Diffusion-ordered NMR spectroscopy was performed with the pulsed gradient spin-echo (PGSE) NMR technique [42]. The translational self-diffusion coefficient D can be calculated with the equation I = I0 exp[−Dγ2δ2G2(Δ−δ/3)], where I0 is the measured signal intensity of a set of resonances at the smaller gradient strength, I is the corresponding observed peak intensity, D is the diffusion constant, γ is the proton gyromagnetic ratio, δ is the diffusion gradient length, G is the gradient strength, and Δ is the diffusion delay [43]. The hydrodynamic radius (rH) of the protein was evaluated with the Stokes–Einstein equation: Dt = kBT/f, where f (= 6πηrH) is the translational friction coefficient, η is the viscosity of the solution, kB is Boltzmann’s constant, and T is the temperature in kelvin. The hydrodynamic properties of the structural model of VEGFR2D7 (3KVQ.pdb) were evaluated by using the software HYDROPRO [44].
Results
Cloning, Expression and Refolding of VEGFR2 Domain 7
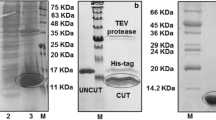
Human VEGFR2D7 protein domain spans residues from 657 to 765 of VEGFR2 including conserved disulfide bonds between Cys688 and Cys737 and Cys740 and Cys745 typical of the immunoglobulin super family fold [18] (Fig. 1). Its gene was amplified from the cDNA of the entire receptor and successively cloned in pETM11 expression plasmid, in order to obtain a recombinant protein fused with an His-tag sequence to its N-terminal. His-tagged VEGFR2D7 was insoluble expressed in Rosetta 2 (DE3) E. coli strain (Fig. 2a), extracted in 50 mM Tris–HCl, 150 mM NaCl, 2 mM DTT, 8 M Urea, pH 8 from inclusion bodies and purified by affinity chromatography on Ni+2-NTA resin. VEGFR2D7 eluted from resin at imidazole concentration from 250 to 300 mM, with a final wash at 500 mM imidazole. Fractions containing VEGFR2D7 were pooled and refolded according to the protocol described in Yang et al. [18], slightly modified to speed up the procedure. In particular, the protein domain was dropwise diluted in a buffer free of urea, containing 10 mM Tris–HCl, 100 mM NaCl, pH 8, and the mix of 3 mM reduced glutathione/0.3 mM oxidized glutathione to facilitate disulfide bridges formation. Protein was diluted in renaturation buffer at a final concentration of 0.1 mg/mL and left under stirring at 4 °C for about 48 h. At the end, we obtained a clear protein solution, not requiring an additional filtration step. TEV protease was added to the sample to remove the poly-histidine tag. The cleavage reaction proceeded with a cutting efficiency of over 90% (Fig. 2b). Digested VEGFR2D7 was purified to homogeneity with only a single chromatography step, by using size-exclusion chromatography in Tris–HCl 20 mM, NaCl 120 mM, pH 7 (Fig. 3), instead of the three chromatography steps previously reported [18]. Fractions corresponding to purified domain were pooled and concentrated until 0.5 mM (Fig. 2c). The final yield of purified VEGFR2D7 was of ∼ 7 mg of homogeneous product per liter of cultured bacteria.

VEGFR2D7 nucleotide and amino acid sequences. a VEGFR2D7 gene sequence cloned in pETM11 expression vector between NcoI (red) and XhoI (green) endonucleases restriction sites. In blue, nucleotides corresponding to His-tag, underlined the 2 stop codons. b Amino acid sequence encompassing VEGFR2 residues 657–765. Highlighted in yellow, the four VEGFR2D7 cysteine residues (Color figure online)

15% SDS-PAGE analysis of VEGFR2D7. a VEGFR2D7 expression in E. coli Rosetta2 (DE3) strain, at 22 °C, overnight in lanes 1 and 2 are showed soluble fractions of not induced and induced cells (0.4 mM IPTG), respectively. In lanes 3 and 4 are reported the insoluble fractions of not induced and induced samples, respectively. In red circle is highlighted His-tagged VEGFR2D7 (15.3 kDa). In the last lane is reported the BSA standard (3 μg). b VEGFR2D7 before (lane 1) and after (lane 2) TEV protease hydrolysis. c VEGFR2D7 after last purification step by size-exclusion chromatography. M, Molecular weight marker

VEGFR2D7 chromatographic profile recorded at 214 and 280 nm. TEV digestion mix of VEGFR2D7 was purified by size-exclusion chromatography on Superdex 7510/30. The peak collected at the retention volume of 13.38 mL corresponds to domain 7, as shown on polyacrylamide gel in the inset. TEV protease and contaminants present in the reaction mix were eluted in peaks collected upstream of that of our interest, while the peak eluted downstream contains the His-tag as, in native conditions, it aggregates, migrating at a higher molecular weight respect to its theoretical one (3000 Da)
NMR studies required the expression of the 15N/13C-labeled protein. Therefore, VEGFR2D7 was expressed in minimal medium in the presence of heavy nitrogen and carbon sources consisting of 15NH4Cl and d-glucose-13C6, respectively . The double-labeled 15N/13C-VEGFR2D7 was obtained with the same yield achieved for the unlabeled protein domain; expression, refolding, and purification were carried out as described for the unlabeled protein.
A biochemical and conformational characterization of VEGFR2D7 was performed in order to verify if the protein was correctly folded. In order to estimate the oxidation state of cysteine residues, we performed a trypsin digestion on VEGFR2D7. The protease was added to 15N/13C-VEGFR2D7 in a molar ratio enzyme:protein domain of 1:100. LC–MS analysis of digested sample was performed after 3 h of incubation at 37 °C. We identified six fragments covering 88% of the protein sequence (Fig. 4a), among which a fragment eluting at the retention time of 18.67 min, showing a mass peak of 6217.42 Da. This mass value is in agreement with those calculated (6218.75 Da) for the fragments E30–K747 and V665–K702 with all Cys residues oxidized (Fig. 4b) and joined by a disulfide bond. We are not able to determine which Cys residues formed the disulfide bonds. In fact, after trypsin digestion, 3 out of 4 cysteine residues are included in the fragment E30–K747 preventing the assignment of cysteine residues involved in disulfide bonds. We can only confirm that the Cys residues are oxidized in the folded protein.

VEGFR2D7 trypsin proteolytic reaction mixture analyzed by LC–MS after 3 h of incubation at 37°C. a VEGFR2D7 protein sequence and theoretical pattern of peptide fragments obtained after trypsin digestion. The trypsin cleavage sites are underlined and the corresponding residue is numbered. Numbering refers to the human VEGFR2 protein sequence. Cysteine residues are in bold. balist of peptide fragments obtained from VEGFR2D7 trypsin digestion and identified by LC–MS; bexperimental (exp) and theoretical (th) molecular weight (MW) of the identified peptide fragments; cretention time (TR) of the identified peptide fragments; dpeptide putatively corresponding to the two fragments harboring the cysteine residues (E730–K747 and V665–K702) in the oxidized state and joined by the disulfide bridge
Circular Dichroism Characterization
The secondary structures of VEGFR2D7 were estimated in solution by far-UV CD spectroscopy. The 2.7-Å resolution crystal structure of VEGFR2 domain 7 reported by Yang et al., shows that it is an all-β protein as expected for an Ig-like domain [18]. The CD spectrum recorded for VEGFR2D7 at 20 °C, in phosphate buffer 10 mM, pH 7 on protein at a concentration of 15 μM, displays a distinctive minimum at 217 nm (Fig. 5a) that is indicative of the presence of β-sheet structures [45]. The minimum under 200 nm could, instead, take into account the loop interconnecting β-sheets or unordered regions [46]. CD spectrum deconvolution revealed an average secondary structure content of ∼ 33% β-sheet region, which is in agreement with the secondary structure analysis derived from X-ray structure (2% helix, 38% β-sheet) [18]. Overall, the CD analysis in the far-UV region suggests that recombinant protein domain assumes in solution the fold expected for VEGFRD7 and similar immunoglobulin-like domains [18, 31, 34]. A thermal denaturation curve was obtained heating protein sample in the temperature range comprised between 5 and 75 °C, monitoring the signal at 217 nm (data not shown). Unfortunately, the protein cooled after thermal denaturation did not recover its initial CD spectrum (data not shown) suggesting that the denaturation process is not reversible in our experimental conditions. The reason of this irreversibility maybe resides in the onset of aggregation phenomena at high temperature. We also performed CD analysis after reducing the protein with 2 mM DTT. The far-UV CD spectrum (Fig. 5b) showed the disappearance of the minimum at 217 nm hidden by a large increase of the band below 200 nm, indicating that the disulfide bonds reduction partially modify the protein structure.

VEGFR2D7 conformational studies by CD spectroscopy. a CD spectrum, of VEGFR2D7 (15 μM) recorded in 10 mM potassium phosphate buffer pH 7 at 20 °C. b CD spectrum VEGFR2D7 (1.5 μM) recorded in 10 mM potassium phosphate buffer pH 7, at 20 °C, after DTT reduction. [Ɵ] is mean residue ellipticity. Spectra reported in A and B were acquired in a quartz cuvette, respectively, of 0.1-cm and 1-cm path length
Steady-State Fluorescence Spectroscopy Studies
VEGFR2D7 amino acid sequence presents one single tryptophan residue (Trp700) that is positioned against the disulfide bond between cysteine residues 688 and 737 and it is part of the hydrophobic core [18]. The presence of this single Trp residue allowed us to study the folding properties of our protein domain by steady-state fluorescence spectroscopy. The excitation wavelength was set at 295 nm to follow the emission of Trp excluding the contribution of the other aromatic residues. The fluorescence emission spectrum of VEGFR2D7 in phosphate buffer 10 mM pH 7, at room temperature, shows a typical curve centered around 350 nm (Fig. 6a). The wavelength of the maximum suggests that the Trp residue is positioned in a polar environment and is probably partially exposed to the solvent. To verify the folding stability of our domain, we performed a chemical denaturation titrating VEGFR2D7 with guanidine hydrochloride (GuHCl). We observed an increase of fluorescence intensity at 350 nm and the absence of any wavelength shift (Fig. 6a). The lack of any wavelength shift suggests that the Trp residue experiences a similar environment in the fully folded form as well as in the unfolded one. Reporting the fluorescence intensity observed at 350 nm in function of GuHCl, we obtained a sigmoidal denaturation curve with a value of GuHCl concentration at which half of the protein molecules are unfolded ([GuHCl]50%) of 1.77 ± 0.07 M, as expected for a globular domain unfolding through a two-state mechanism and indicating a good chemical stability (Fig. 6b). The increase of the fluorescence intensity could be related to the increase of distance in the unfolded state of the Trp from the disulfide bond which in the folded state acts as quencher [47]. Reducing VEGFR2D7 with 2 mM DTT, we observed a Trp fluorescence emission spectrum comparable to that observed in the presence of high GuHCl concentration (Fig. 6c).

VEGFR2D7 analysis by steady-state fluorescence spectroscopy. a Emission spectra of protein domain recorded in the presence of increasing concentration of GuHCl. b Dependence of the fluorescence intensity at 350 nm as function of the guanidine hydrochloride concentration. The curve represents the result of the non-linear regression analysis (R2 = 0.99). c Emission spectra of VEGFR2D7 recorded in the presence (blue curve) or absence (red curve) of 2 mM DTT. a.u is arbitrary unit
Static Light Scattering Analysis
Information on the molecular weight and oligomeric state of the VEGFR2 receptor domain 7 was obtained by combining size-exclusion chromatography with the light scattering analysis performed on the peak eluted, by using as mobile phase a buffer composed of Tris–HCl 20 mM, NaCl 120 mM, pH 7, at the retention volume of 13.38 mL (Fig. 7). This analysis provided for VEGFR2D7 a molecular weight value of 12,230 ± 122 Da, as indicated by the light scattering signals (black dots in Fig. 7), in good agreement with the theoretical one calculated for our protein domain (12,341.01 Da) and consistent with a monomeric state in solution.

VEGFR2D7 light scattering analysis in 20 mM Tris–HCl, 120 mM NaCl, pH 7. Analytical size-exclusion chromatography profile monitored at 280 nm is indicated by the solid lane, while the light scattering signals are indicated by black dots
NMR Studies
To assess VEGFR2D7 aggregation state in solution, DOSY (diffusion-ordered spectroscopy) experiments [42] were conducted in 90% H2O/10% 2H2O (v/v), containing 20 mM Tris–HCl and 120 mM NaCl pH 7, at a protein concentration of 100 μM. DOSY measurements indicate, for the protein, a diffusion coefficient (Dt) equal to (0.70 ± 0.08) × 10−10 m2/s that corresponds, by using the Stokes–Einstein equation, to a hydrodynamic radius (rH) equal to 1.93 nm. The rH calculated from VEGFR2D7 X-ray structure (3KVQ.pdb) by using HYDROPRO software [44] afforded a value of 1.76 nm in agreement with the experimental NMR value (1.93 nm) indicating that the protein assumes a compact monomeric conformation in solution. The 1H-spectrum of VEGFR2D7 (Fig. 8) showed a good chemical shift dispersion on both amide and aliphatic regions, suggesting the protein folds into a unique well-defined structure. Furthermore, the observation of a large amount of NOE cross peaks (Fig. 9a) confirms that unlabeled VEGFR2D7 adopts a globular conformation. The 2D [1H, 15N] HSQC spectrum of the uniformly 15N-labeled sample (Fig. 9b) exhibits a large number of resonances, which are well dispersed over a chemical shift range of 4.0 ppm in the proton dimension and 30 ppm in the nitrogen dimension further confirming that the VEGFR1D7 adopts a unique well-defined structure. Indeed, the VEGFR2D7 chemical shift dispersions shown in Fig. 9b largely reflect a β-sheet structure.

The 600-MHz 1H NMR spectrum of unlabeled VEGFR2D7 protein in a 90/10 (v/v) H2O/2H2O mixture at 298 K. NMR sample consisted of 0.10 mM protein domain in 10 mM Tris–HCl and 120 mM NaCl, pH 7

VEGFR2D7 in solution NMR characterization. a Section of the 2D [1H, 1H] NOESY (100 ms) spectrum of the unlabeled VEGFR2D7 in a 90/10 (v/v) H2O/2H2O mixture at 298 K. b 2D [1H, 15N] HSQC spectrum of 15N-labeled VEGFR2D7 acquired in a 90/10 (v/v) H2O/2H2O mixture at 298 K. Both NMR samples consisted of 0.10 mM protein domain in 10 mM Tris–HCl and 120 mM NaCl, pH 7
Discussion and Conclusions
In the last decades, the study of the mechanisms of angiogenesis and the research of new molecules able to modulate this process have been largely explored, resulting in the development of several anti-angiogenic agents as therapeutic tools aimed at inhibition of tumor growth and metastasis and to threat ocular pathologies. The most common anti-angiogenic therapies rely on an approach based on sequestration of VEGF through a specific antibody, like bevacizumab [21], thus blocking VEGFR2 activation. But this strategy is not without drawbacks, because a systemic destruction of the normal vasculature is also observed in this case [48]. Another possible approach consists in the use of specific antibodies against domains D2–3 [22] or receptor binder peptides [49,50,51,52,53,54] inhibiting VEGF binding to VEGFR2, even if they must be administered in large amounts to successfully compete with VEGF for blocking receptor activity. In addition, kinase inhibitors are administered to block VEGFR2 kinase domain and activation of intracellular signaling cascade. Unfortunately, their lack of receptor specificity correlates to a non-safety profile [29]. The presence of D4–D4 and D7–D7 homotypic contacts in VEGFR2 extracellular portion underlines the therapeutic potential of an approach aimed to target these allosteric sites as a strategy to bind the receptor, inactivating its downstream signal cascade and offering a promising alternative to the classical anti-angiogenic approaches [17, 20]. In fact, compounds able to inhibit VEGFR2 without competing with VEGF are desirable as VEGFR antagonists to overcome drawbacks related to the use of classical anti-VEGF compounds and so for diagnostic purposes. The starting point to get this aim was the preparation of a properly folded VEGFR2D7 domain subsequently characterized in a biochemical and structural study. The obtainment of VEGFR2D7 will give the possibility to use the protein domain, to perform structural studies in solution and to set up screening assays, using for example the phage display technology, in order to find new molecules, antibodies, or peptides, able to disrupt VEGFR2D7 homotypic contacts, preventing VEGFR2 activation. In this study, we report a procedure to prepare a functional VEGFR2 domain 7. Furthermore, we analyzed its molecular properties in solution combining different spectroscopic techniques. VEGFR2D7 has already been expressed in bacterial host for crystallographic studies. In this work, we report an improved and faster procedure but, at the same time, as well as effective compared to that described in literature [18]. To this end, the nucleotide sequence corresponding to domain 7 was successfully cloned into the expression vector pETM11 that allowed to express the recombinant protein with an N-terminal His-tag sequence, then easily removed with TEV, a more specific and suitable protease than Thrombin previously employed [18]. The protein was expressed with a yield of ∼ 200 mg/mL, but in an insoluble form, which required the application of a refolding procedure. At first, we operated as described in Yang et al, diluting the protein, solubilized in 8 M urea, in a buffer without denaturant agent. Once performed the refolding procedure, after tag removal, we directly passed to the step of size-exclusion chromatography to obtain the homogeneously purified protein, skipping both the filtration step after refolding and the two purification steps on anionic exchanger carried out before and after the gel filtration one performed by Yang et al. In this way, we were able to develop a strategy that streamlines and speeds up the procedure for the obtainment of the final protein product. Furthermore, we obtained, with the same yield reported for the unlabeled protein, the double-labeled 15N/13C-VEGFR2D7, following the experimental procedure described above. Purity and identity of recombinant VEGFR2D7 were confirmed by SDS-PAGE analyses. Unfortunately, we were unable to determine the exact mass of the full-length VEGFR2D7 by mass spectrometry mainly because ionization problem both in ESI and MALDI source. Then, we verified the oxidation state of the four cysteines performing a trypsin digestion of VEGFR2D7. The identification of a mass peak corresponding to two reduced tryptic fragments joined by a disulfide bond revealed that all four cysteines are reduced. Unfortunately, the distribution of tryptic cleavage sites does not allow us to identify the Cys involved in the single disulfide bridges because a tryptic fragment possesses three out of four cys residues. The refolded protein was characterized in solution using different spectroscopic techniques to verify if the recombinant VEGFR2D7 is properly folded. We showed by light scattering and DOSY experiments that VEGFR2D7 is a monomer up to 1 × 10−4 M. These data are in agreement with analytical centrifugation experiments performed by Yang et al [18]. Besides, the rH measured by DOSY experiments for the protein (i.e., ∼ 19 Å) is in agreement with the rH values calculated from VEGFR2D7 X-ray structure and for other compact proteins of a similar size [55]. The chemical denaturation shows a sigmoidal behavior as expected for a two-state unfolding transition of a globular protein and the half transition value is in agreement with what is reported for similar Ig-like domains [34, 56]. Steady-state fluorescence analysis indicates that the single Trp residue is quenched in the folded state as expected because of its close distance with disulfide bond [47] between cysteine residues 688 and 737 as revealed by X-ray crystallography. To confirm this hypothesis, we found an increase of Trp fluorescence emission after incubating VEGFR2D7 in the presence of DTT which, by reducing domain disulfide bridges, dampens sulfur atoms' quenching effect observed in the folded state of the protein. However, disulfide reduction causes also a partial protein structure modification, as revealed by CD analysis. So, the increase of Trp fluorescence intensity could be related to both the increase of the distance from sulfur atoms due to the bond breakage and the change of conformation.
In the X-ray structure, Trp side chain points towards protein interior. However, fluorescence emission spectrum shows a maximum around 350 nm which is typical of a Trp exposed to the solvent or, in general, in a polar environment. To explain this behavior, we can consider that the presence of two polarizable atoms like sulfur may influence the microenvironment of Trp. Furthermore, inspection of X-ray structure shows that Trp is partially accessible to the solvent and two water molecules are located at a closer distance (less than 5 Å) [18]. However, we cannot exclude that in solution, the protein may show a less compact structure as reported from X-ray structure.
Finally, CD and NMR analyses show that the recombinant protein prevalently folds in a β-sheet structure, as expected for an immunoglobulin-like domain and in agreement with VEGFR2D7 crystal structure (3KVQ.pdb) [18].
The ability to prepare a functional and folded VEGFR2D7 is important to allow structural studies in solution or to develop novel molecules, as antibodies or peptides, interacting with VEGFR2. In fact, the recombinant VEGFR2D7 could be used to set up screening assays to find binders able to disrupt VEGFR2D7 homotypic contacts needful for VEGFR2 activation. These molecules could act as modulators of angiogenesis, enabing them suitable for new therapeutic exploitation.
In conclusion, we reported a fast and efficient protocol to prepare recombinant VEGFR2D7 domain. The protein was obtained with a good yield and was properly folded as assessed in solution combining different spectroscopic techniques. The ability to prepare a functional VEGFR2D7 domain is crucial to develop and investigate novel VEGFR2 binders acting as allosteric inhibitors targeting receptor membrane proximal domain 7.
Abbreviations
- VEGFs:
-
Vascular endothelial growth factors
- VEGFRs:
-
Vascular endothelial growth factor receptors
- RTKs:
-
Receptor tyrosine kinases
- TEMED:
-
Tetramethylethylenediamine
- APS:
-
Ammonium persulfate
- dNTPs:
-
Deoxynucleotide triphosphates
- IPTG:
-
Isopropyl β-d-1-thiogalactopyranoside
- O.D.600 :
-
Optical density at 600 nm
- Ni-NTA resin:
-
Nickel-charged nitrilotriacetic resin
- TEV protease:
-
Tobacco etch virus protease
- EDTA:
-
Ethylenediaminetetraacetic acid
- DTT:
-
Dithiothreitol
- RP-HPLC:
-
Reversed-phase high-performance liquid chromatography
- TFA:
-
Trifluoroacetic acid
- LC–MS:
-
Liquid chromatography–mass spectrometry
- Trp:
-
Tryptophan
- DSS:
-
4,4-Dimethyl-4-silapentane-1-sulfonic acid
References
Patel-Hett, S., & D’Amore, P. A. (2011). Signal transduction in vasculogenesis and developmental angiogenesis. The International Journal of Developmental Biology, 55, 353–363.
Ribatti, D. (2008). Judah Folkman, a pioneer in the study of angiogenesis. Angiogenesis, 11, 3–10.
Carmeliet, P. (2003). Angiogenesis in health and disease. Nature Medicine, 9, 653–660.
Shibuya, M. (2014). VEGF-VEGFR signals in health and disease. Biomolecules & Therapeutics, 22, 1–9.
Olsson, A. K., Dimberg, A., Kreuger, J., & Claesson-Welsh, L. (2006). VEGF receptor signalling-in control of vascular function. Nature Reviews Molecular Cell Biology, 7, 359–371.
Gerhardt, H., Golding, M., Fruttiger, M., Ruhrberg, C., Lundkvist, A., Abramsson, A., et al. (2003). VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. The Journal of Cell Biology, 161, 1163–1177.
Takahashi, H., & Shibuya, M. (2005). The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clinical Science, 109, 227–241.
Lania, G., Ferrentino, R., & Baldini, A. (2015). TBX1 represses Vegfr2 gene expression and enhances the cardiac fate of VEGFR2+ cells. PLoS ONE, 10, e0138525.
Smith, N. R., Baker, D., James, N. H., Ratcliffe, K., Jenkins, M., Ashton, S. E., et al. (2010). Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 16, 3548–3561.
Yamagishi, N., Teshima-Kondo, S., Masuda, K., Nishida, K., Kuwano, Y., Dang, D. T., et al. (2013). Chronic inhibition of tumor cell-derived VEGF enhances the malignant phenotype of colorectal cancer cells. BMC Cancer, 13, 229.
Chatterjee, S., Heukamp, L. C., Siobal, M., Schottle, J., Wieczorek, C., Peifer, M., et al. (2013). Tumor VEGF:VEGFR2 autocrine feed-forward loop triggers angiogenesis in lung cancer. The Journal of Clinical Investigation, 123, 1732–1740.
Ruch, C., Skiniotis, G., Steinmetz, M. O., Walz, T., & Ballmer-Hofer, K. (2007). Structure of a VEGF-VEGF receptor complex determined by electron microscopy. Nature Structural & Molecular Biology, 14, 249–250.
Kisko, K., Brozzo, M. S., Missimer, J., Schleier, T., Menzel, A., Leppanen, V. M., et al. (2011). Structural analysis of vascular endothelial growth factor receptor-2/ligand complexes by small-angle X-ray solution scattering. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 25, 2980–2986.
Shinkai, A., Ito, M., Anazawa, H., Yamaguchi, S., Shitara, K., & Shibuya, M. (1998). Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. The Journal of Biological Chemistry, 273, 31283–31288.
Dosch, D. D., & Ballmer-Hofer, K. (2010). Transmembrane domain-mediated orientation of receptor monomers in active VEGFR-2 dimers. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 24, 32–38.
Brozzo, M. S., Bjelic, S., Kisko, K., Schleier, T., Leppanen, V. M., Alitalo, K., et al. (2012). Thermodynamic and structural description of allosterically regulated VEGFR-2 dimerization. Blood, 119, 1781–1788.
Hyde, C. A., Giese, A., Stuttfeld, E., Abram, Saliba J., Villemagne, D., Schleier, T., et al. (2012). Targeting extracellular domains D4 and D7 of vascular endothelial growth factor receptor 2 reveals allosteric receptor regulatory sites. Molecular and Cellular Biology, 32, 3802–3813.
Yang, Y., Xie, P., Opatowsky, Y., & Schlessinger, J. (2010). Direct contacts between extracellular membrane-proximal domains are required for VEGF receptor activation and cell signaling. Proceedings of the National academy of Sciences of the United States of America, 107, 1906–1911.
Sarabipour, S., Ballmer-Hofer, K., & Hristova, K. (2016). VEGFR-2 conformational switch in response to ligand binding. eLife, 5, e13876.
Thieltges, K. M., Avramovic, D., Piscitelli, C. L., Markovic-Mueller, S., Binz, H. K., & Ballmer-Hofer, K. (2018). Characterization of a drug-targetable allosteric site regulating vascular endothelial growth factor signaling. Angiogenesis, 21, 533–543.
Ellis L. M. (2005). Bevacizumab. Nature Reviews Drug Discovery, Suppl S8–9.
Krupitskaya, Y., & Wakelee, H. A. (2009). Ramucirumab a fully human mAb to the transmembrane signaling tyrosine kinase VEGFR-2 for the potential treatment of cancer. Current Opinion in Investigational Drugs, 10, 597–605.
D’Andrea, L. D., Del Gatto, A., De Rosa, L., Romanelli, A., & Pedone, C. (2009). Peptides targeting angiogenesis related growth factor receptors. Current Pharmaceutical Design, 15, 2414–2429.
Feng, S., Zou, L., Ni, Q., Zhang, X., Li, Q., Zheng, L., et al. (2014). Modulation, bioinformatic screening, and assessment of small molecular peptides targeting the vascular endothelial growth factor receptor. Cell Biochemistry and Biophysics, 70, 1913–1921.
Di Stasi, R., De Rosa, L., Romanelli, A., & D’Andrea, L. D. (2016). Peptides interacting with growth factor receptors regulating angiogenesis. Frontiers in Medicinal Chemistry, 9, 103–160.
De Rosa, L., Di Stasi, R., & D’Andrea, L. D. (2018). Pro-angiogenic peptides in biomedicine. Archives of Biochemistry and Biophysics, 660, 72–86.
D’Andrea, L. D., Romanelli, A., Di Stasi, R., & Pedone, C. (2010). Bioinorganic aspects of angiogenesis. Dalton Transactions, 39, 7625–7636.
D’Andrea, L. D., De Rosa, L., Vigliotti, C., & Cataldi, M. (2017). VEGF mimic peptides: Potential applications in central nervous system therapeutics. New Horizons in Translational Medicine, 3, 233–251.
Mendel, D. B., Laird, A. D., Xin, X., Louie, S. G., Christensen, J. G., Li, G., et al. (2003). In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research, 9, 327–337.
Kendrew, J., Eberlein, C., Hedberg, B., McDaid, K., Smith, N. R., Weir, H. M., et al. (2011). An antibody targeted to VEGFR-2 Ig domains 4–7 inhibits VEGFR-2 activation and VEGFR-2-dependent angiogenesis without affecting ligand binding. Molecular Cancer Therapeutics, 10, 770–783.
Di Stasi, R., De Rosa, L., Diana, D., Fattorusso, R., & D’Andrea, L. D. (2019). Human recombinant VEGFR2D4 biochemical characterization to investigate novel anti-VEGFR2D4 antibodies for allosteric targeting of VEGFR2. Molecular Biotechnology, 61, 513–520.
Keunen, O., Johansson, M., Oudin, A., Sanzey, M., Rahim, S. A., Fack, F., et al. (2011). Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proceedings of the National academy of Sciences of the United States of America, 108, 3749–3754.
Chung, A. S., Kowanetz, M., Wu, X., Zhuang, G., Ngu, H., Finkle, D., et al. (2012). Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. The Journal of Pathology, 227, 404–416.
Di Stasi, R., Diana, D., Capasso, D., Palumbo, R., Romanelli, A., Pedone, C., et al. (2010). VEGFR1(D2) in drug discovery: Expression and molecular characterization. Biopolymers, 94, 800–809.
Provencher, S. W., & Glockner, J. (1981). Estimation of globular protein secondary structure from circular dichroism. Biochemistry, 20, 33–37.
Manavalan, P., & Johnson, W. C., Jr. (1987). Variable selection method improves the prediction of protein secondary structure from circular dichroism spectra. Analytical Biochemistry, 167, 76–85.
Sreerama, N., & Woody, R. W. (2000). Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Analytical Biochemistry, 287, 252–260.
Hwang, T. L., & Shaka, A. J. (1995). Water suppression that works. Excitation sculpting using arbitrary wave-forms and pulsed-field gradients. Journal of Magnetic Resonance Series A, 112, 275–279.
Dalvit, C. (1998). Efficient multiple-solvent suppression for the study of the interactions of organic solvents with biomolecules. Journal of Biomolecular NMR, 11, 437–444.
Goddard, T. D., & Kneller, D. G. (2004). SPARKY 3. California: University of San Francisco.
Keller, R. L. J. (2004). The computer aided resonance assignement tutorial. Newport Beach: CANTINA.
Morris, K. F., & Jr, Johnson C. S. (1992). Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. Journal of the American Chemical Society, 114, 3139–3141.
Price, W. S., Nara, M., & Arata, Y. (1997). A pulsed field gradient NMR study of the aggregation and hydration of parvalbumin. Biophysical Chemistry, 65, 179–187.
Garcia de la Torre, J., Huertas, M. L., & Carrasco, B. (2000). HYDRONMR: Prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. Journal of Magnetic Resonance, 147, 138–146.
Chemes, L. B., Alonso, L. G., Noval, M. G., & de Prat-Gay, G. (2012). Circular dichroism techniques for the analysis of intrinsically disordered proteins and domains. Methods in Molecular Biology, 895, 387–404.
Greenfield, N. J. (2006). Using circular dichroism spectra to estimate protein secondary structure. Nature Protocols, 1, 2876–2890.
Chen, Y., & Barkley, M. D. (1998). Toward understanding tryptophan fluorescence in proteins. Biochemistry, 37, 9976–9982.
Yang, Y., Zhang, Y., Cao, Z., Ji, H., Yang, X., Iwamoto, H., et al. (2013). Anti-VEGF- and anti-VEGF receptor-induced vascular alteration in mouse healthy tissues. Proceedings of the National academy of Sciences of the United States of America, 110, 12018–12023.
Gautier, B., Goncalves, V., Diana, D., Di Stasi, R., Teillet, F., Lenoir, C., et al. (2010). Biochemical and structural analysis of the binding determinants of a vascular endothelial growth factor receptor peptidic antagonist. Journal of Medicinal Chemistry, 53, 4428–4440.
Basile, A., Del Gatto, A., Diana, D., Di Stasi, R., Falco, A., Festa, M., et al. (2011). Characterization of a designed vascular endothelial growth factor receptor antagonist helical peptide with antiangiogenic activity in vivo. Journal of Medicinal Chemistry, 54, 1391–1400.
De Rosa, L., Diana, D., Basile, A., Russomanno, A., Isernia, C., Turco, M. C., et al. (2014). Design, structural and biological characterization of a VEGF inhibitor beta-hairpin-constrained peptide. European Journal of Medicinal Chemistry, 73, 210–216.
Wang, L., Coric, P., Broussy, S., Di Stasi, R., Zhou, L., D’Andrea, L. D., et al. (2019). Structural studies of the binding of an antagonistic cyclic peptide to the VEGFR1 domain 2. European Journal of Medicinal Chemistry, 169, 65–75.
Ruegg, C., Hasmim, M., Lejeune, F. J., & Alghisi, G. C. (2006). Antiangiogenic peptides and proteins: From experimental tools to clinical drugs. Biochimica et Biophysica Acta, 1765, 155–177.
Sulochana, K. N., & Ge, R. (2007). Developing antiangiogenic peptide drugs for angiogenesis-related diseases. Current Pharmaceutical Design, 13, 2074–2086.
Wilkins, D. K., Grimshaw, S. B., Receveur, V., Dobson, C. M., Jones, J. A., & Smith, L. J. (1999). Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry, 38, 16424–16431.
Clarke, J., Cota, E., Fowler, S. B., & Hamill, S. J. (1999). Folding studies of immunoglobulin-like beta-sandwich proteins suggest that they share a common folding pathway. Structure, 7, 1145–1153.
Acknowledgements
LDR is supported by Fondazione Umberto Veronesi-Post-Doctoral Fellowship 2019. We would like to thank Mr Leopoldo Zona for technical assistance and Dr Luigi Russo for skillful help with HYDROPRO.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Di Stasi, R., Diana, D., De Rosa, L. et al. Biochemical and Conformational Characterization of Recombinant VEGFR2 Domain 7. Mol Biotechnol 61, 860–872 (2019). https://doi.org/10.1007/s12033-019-00211-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-019-00211-4