
Abstract
The mammalian expression system plays a key central role in the production of therapeutic recombinant proteins. Conspicuously, any improvements in the expression system which lead to a higher expression level would have an impact especially in bio-pharmaceutical industries. In the current study, to take steps toward the improvement of expression of recombinant protein, first, we established a stable HEK293 cell line to overexpress a well-known cytoprotective and antioxidant gene, Nrf2. Next, we transiently expressed human recombinant coagulation factor VII, as an example of human recombinant protein, in the engineered-HEK293 cell line. Our results revealed that the established cells had a higher growth rate and were able to endure to UV-induced oxidative stress. Furthermore, within our expectation, our results revealed that the expression level of recombinant FVII in Nrf2-engineered HEK293 cells (315 ng/ml) was higher than the HEK293 (198 ng/ml) cells and it was functional in a coagulation test assay. Moreover, our new cell line could be a suitable cell to express other recombinant proteins especially for large-scale production of recombinant protein under other culture condition such as lower serum and suspension culture that imposed advantages especially in terms of cost benefits in bio-pharmaceutical industries.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Development of recombinant proteins as safe and efficient therapeutic modalities is of great interest in the view point of both cell and molecular scientists and bio-pharmaceutical industries. In this way, recombinant proteins are usually produced in living cells such as microbial, yeast and animals. The major recombinant proteins with therapeutic application in the clinic need posttranslational processing including aggregation, folding, solubility, proteolytic processing and glycosylation [4, 8, 32], indicating a few suitable expression hosts are available to produce them.
For example, glycol-engineering and upstream process optimization has been employed to improve the glycan composition of the bio-therapeutics mainly by choosing a proper host for the protein expression. These attempts among others have addressed some challenges which mentioned above [16]. It is noteworthy to say that more than half of therapeutic proteins are produced by mammalian expression systems [27]. The host cells for mammalian expression system include Chinese hamster ovary (CHO), baby hamster kidney (BHK), human embryo kidney (HEK293), human retinal cells and mouse myeloma cell lines (Sp2/0 and NS0) [3, 6, 9, 25, 36].
However, due to many reasons such as low expression, high production costs and the low survival of the host cells under unfavorable culture condition, the production of recombinant therapeutic proteins has always been challenging [4, 26]. Therefore, developing a strategy to improve, optimize and address mammalian expression systems is the focus of many investigations. Using genetically modified mammalian hosts with increased proliferation and survival could be one of the efficient strategies to address the challenges [6, 9, 20, 35].
To develop stable and higher host cell-producing recombinant proteins, gene-editing tools such as ZFNs, TALENs and CRISPR/Cas9 have been used to knock-in or knock-out of a particular gene in certain loci of the host cells recently [11].
Supporting this notion, strategies such as genetic engineering of mammalian cells by cell cycle control genes (cyclins), proto-oncogenes, growth factor and anti-apoptotic genes have been applied to improve mammalian hosts in terms of expression of recombinant protein [6, 7, 17, 29]. However, possibly due to commercialization or potential conflicts, the details are unavailable in the literature and remain unpublished.
Among the mammalian expression systems, human expression systems have a great advantage; being that subsequent products have less immunogenic potential. Therefore, currently, the production of bio-therapeutic proteins in human cells is increasingly important [1, 3].
HEK293 and fibrosarcoma HT-1080 are examples of approved human cell lines used for production of therapeutic recombinant proteins. HEK293 cells have been and continue to be used to produce research-grade proteins [22, 34]. More recently, five therapeutic agents produced in HEK293 cells have been approved by the FDA or the European Medicines Agency (EMA) [3]. In this study, we genetically manipulated HEK293 cells by nuclear factor erythroid-derived like 2 (Nrf2) to improve expression of human coagulation factor VII recombinant protein.
Nuclear Factor E2-Related Factor-2 (Nrf-2), also known as NFE2L2 is a transcription factor encoded by the NFE2L2 gene. Nrf2 plays a crucial role in the antioxidant defense system and protect the cells against stressful condition. Additionally, it is involved in cell proliferation, differentiation and chemoprevention through the Nrf2-ARE pathway activation [24] and cytoprotective enzyme induction [21, 24, 33]. It has also been shown that Nrf2 protects different cell types and organs from toxic agents including radical oxygen species (ROSs), chemotherapeutic agents and pathogens [33, 38].
Therefore, to explore the unique advantage of Nrf2 in this project, we genetically engineered HEK293 cells. Next, as an example of recombinant protein, we expressed recombinant human coagulation FVII in the Nrf2- engineered cells. Our initial results revealed that Nrf2-engineered cells had improved FVII expression, and were also more robust in their proliferation and more refractory to the unfavorable stress condition.
Materials and Methods
Plasmids and Bacteria
In this account, pcDNA4B and pcDNA3.1 (Thermo, USA) plasmids were used in this study. The E. coli Top10 bacterial strain (Thermo, USA) was used as a host for cloning of the constructs. The pcDNA3.1-Nrf2 recombinant vector and pcDNA3.1-FVII were available from previous studies [13, 24].
Sub-cloning of FVII
Second, pcDNA3.1-FVII was sub-cloned into pcDNA4B vector. Briefly, first the E.coli bacteria of Top10 strain containing pcDNA3.1-FVII construct was cultured and plasmid extraction was performed according to the manufacturer’s protocol (Roche, Germany). Human FVII was amplified from the recombinant construct by polymerase chain reaction (PCR). Primer pairs for amplification of full length of human FVII containing Kozak sequence, EcoRI (forward) and Not I (reverse) restriction enzyme sites consisted of forward: 5′-ACG AAT TCA CCA TGG TGG TCT CCC AGG CCC TCA GGC TC-3′ and reverse: 5′-TAG CGG CCG CGG GAA ATG GGG CTC GCA GG-3′. PCR was performed using Platinum Taq DNA polymerase (Thermo, USA). Then, the PCR product was purified using the high pure PCR product purification kit (Roche, Germany). The purified FVII fragment, as well as pcDNA4B vector, was cut with EcoRI and Not I restriction enzymes and was ligated using T4 DNA ligase (Roche, Germany) followed by transformation into the competent E.coli.
Cell Culture
HEK293 was obtained from the National Cell Bank of Iran and cultivated in T25 flask. The cell line was grown in RPMI-1640 medium (Gibco-BRL, Germany) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/mL streptomycin (Gibco-BRL, Germany).
Establishment of Stable Nrf-2-Engineered HEK293 Cells
HEK293 cells (5 × 105) were seeded and upon reaching 70% confluence were transfected with 2 µg of pcDNA3.1-Nrf2 DNA using the FuGENE HD transfection reagent (Roche, Germany) according to the manufacturer’s protocol. The empty vector, pcDNA3.1, was also transfected and used as a control (HEK-V). HEK293 cells containing pcDNA3.1-Nrf2 construct (HEK-Nrf2) were selected in a medium containing 300 µg/mL geneticin (Roche, Germany) for at least 20 days. Stable clones were generated by dilution of cells and their culture in 96-well culture plates and cultivated for at least 20 generations. Several stable clones were established. The experiments were performed on the two clones (HEK-Nrf2 (C1) and HEK-Nrf2 (C2)). Expression of Nrf2 at RNA level was evaluated in the first, 10th, and 20th passages of HEK-Nrf2 (C1) and HEK-Nrf2 (C2) stable clones.
Cell Proliferation Assay
Different cell groups including HEK-V, HEK-Nrf2 (C1) and HEK-Nrf2 (C2) were seeded in 96-well plate at the density of 1 × 104 cells/well. Then, the cells were cultured in CO2 incubator at 37 °C for two days followed by cell proliferation assay using water-soluble tetrazolium salts-1 (WST-1) kit (Roche, Germany) according to manufacturer’s protocol. Next, the absorbance was measured using ELx800 absorbance microplate reader at 450 nm. The cells were also exposed to UV irradiation for 10 min (the exposure time was optimized) followed by cell proliferation assay.
Transient Expression of rFVII in HEK293-Nrf2 Stable Cell Line
HEK-Nrf2 (C1) and HEK-Nrf2 (C2) stable cells (5 × 105) were seeded and upon reaching 70% confluence were transfected with 2 µg of pcDNA4B–FVII or pcDNA4B vector using the FuGENE HD transfection reagent (Roche, Germany) according to the manufacturer’s protocol. HEK293 cells were also transfected by PcDNA4B-FVII to express FVII transiently, as another control group, (HEK-FVII). The expression of FVII was demonstrated by RT-PCR (using internal primers), western blot and enzyme-linked immunosorbent assay (ELISA) methods.
Enzyme Linked Immune Sorbent Assay
ELISA was performed to evaluate the expression of FVII protein in the culture medium of HEK293, HEK-FVII, HEK-Nrf2 (C1)-FVII and HEK-Nrf2 (C2)-FVII cells. Relatively, 72 h after transfection, the medium was harvested and the expression of FVII protein was evaluated by FVII immunoassay kit (Abcam, UK) according to the manufacturer’s protocol and the OD values were measured at 450 nm using a microplate reader.
Western Blot
The culture medium of the FVII-overexpressing HEK293 cells, as well as HEK-V was concentrated using Vivaspin6 Sartourious 3 kDa filter (Sartorius, Germany). Protein concentration was performed using Bradford assay kit (BIO Rad, USA). Western blot analysis for FVII and β-actin was performed as described previously [13]. For the detection of the protein bands, the PVDF membrane was washed four times with PBS containing 0.1% Tween20, and developed by ECL reagents (Amersham, UK). Image lab software was applied to semi-quantify the western blot bands.
Coagulation Activity of the Recombinant FVII
The coagulation function of the expressed rFVII in the medium of cells was measured using prothrombin time test as described previously [13, 23]. Thromboplastin preparations utilized in the assay were from human (Hoechst Canada Inc., Behring Diagnostics, Montreal, Quebec, Canada).
Statistical Analysis
At each data point, the mean and standard deviation (SD) were calculated and statistically analyzed using student’s t test. p < 0.05 which was considered significant.
Results
Sub-cloning of FVII from pcDNA 3.1 to pcDNA4B Vector
FVII cDNA was isolated from a previously available vector, pcDNA3.1-FVII, by PCR. The isolated cDNA was cloned into pcDNA4B vector. The existence of the insert was confirmed by PCR and restriction enzyme digestion. There was a single band of expected size of PCR product in the recombinant vector and no band was observed after PCR amplification of FVII from empty vector (Fig. 1a). Correspondingly, restriction enzyme double digestion revealed the existence of FVII in the recombinant construct (Fig. 1b).

Sub-cloning of FVII from pcDNA 3.1 to pcDNA4B vector. FVII cDNA was isolated from previously available vector, pcDNA3.1-FVII, by PCR. The isolated cDNA was cloned into pcDNA4B vector. Existence of the insert in the new vector was checked by PCR and restriction enzymes digestion. a Electrophoresis of the PCR product on two present agarose gel indicated the expected size of PCR band can be observed in the recombinant construct (lane 2) while no bond was observed from the empty vector(lane 1). b Existence of the insert was also confirmed restriction enzymes digestion. The existence of expected size of FVII insert (lane 1) and absence of this band in the empty vector following double digestion, 1300 bp, were indicating the successful of sub-cloning. M; 100 bp ladder marker
Establishment of Stable HEK293 Cell Expressing Nrf2
Post-transfection of the HEK293 with the recombinant pcDNA3.1-Nrf2 or empty plasmid, the stable cells were selected in the presence of geneticin. Several single cell clones were established by serial dilution of cells in 96-well plates. The over expression of Nrf2 gene was analysed by RT-PCR in two stable clones, HEK-Nrf2 (C1) and HEK-Nrf2 (C2), in the first, 10th, and 20th passages (Fig. 2a–c). No PCR product was detected in HEK-V cells. However, in the stable HEK293-Nrf2(C1) and HEK293-Nrf2 (C2) cells a PCR band was detected in the first, 10th ,and 20th generations (Fig. 2a–c) indicating the stable expression of Nrf2.

Establishment of stable HEK293 cells overexpressing Nrf2. HEK293 cells transfected either with pcDNA3.1-Nrf2 or pcDNA3.1 vector. The stable cells were selected in the presence of geneticin. RT-PCR was performed to detect overexpression of Nrf2 in two selected stable clones (C1 and C2) at first, 10th and 20th passages. a RT-PCR analysis of Nrf2 overexpressing in HEK293-Nrf2 (C1), HEK293-Nrf2 (C2) and control group (HEK-V) at first passage. b RT-PCR analysis of those mentioned cells at 10th passage. c RT-PCR analysis of different cell groups at 20th passage. d Expression of β-actin. HEK293-Nrf2 (C1) and HEK293-Nrf2 (C2) expressed Nrf2 even at 20th passage
Nrf2 Increased Proliferation and Protected the Cells to UV Irradiation
To determine whether Nrf2 overexpression influenced cell proliferation or not, HEK-Nrf2 (C1), HEK-Nrf2 (C2) and HEK-V cells were cultivated in 96-well plate for 3 days followed by cell proliferation assay. Interestingly, the number of cells in the HEK-Nrf2 groups was higher than HEK-V group indicating Nrf2 conferred an advantage to the cells proliferation.
Notably, one of the well-known functions of Nrf2 is antioxidant activity. Therefore, next to evaluate the functional activity of Nrf2 in the stable cells, they exposed to UV irradiation followed by cell proliferation assay. Within our expectation, Nrf2 protected the cells from UV-induced cell death owing to cytoprotective function of Nrf2 (Fig. 3). These results were further confirmed by colony assay in which the number of colonies in HEK-Nrf2 cells was higher than the number of HEK-V cells following exposure to UV irradiation (data not shown).

Cell viability assay in different cell groups. Stable clones, HEK-Nrf2 (C1), HEK-Nrf2 (C2) and HEK-V, were cultivated in 96-well plate followed by WST-1 assay. The cells were also exposed to UV irradiation followed by cell proliferation assay. The viability of UV-exposed HEK-Nrf2 (C1) and UV-exposed HEK-Nrf2 (C2) was more than UV-exposed HEK-V, as control group. Nrf-2 protected the cells against UV-induced cell death (Mean ± SD, **p < 0.01)
Stable HEK293-Nrf2 Cells Expressed rFVII Higher Than HEK293 Cells
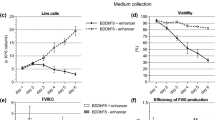
Next to investigate whether establishment of Nrf2-overexpressing HEK293 cells influence the expression level of any recombinant protein, the stable HEK-Nrf2 (C1) and HEK-Nrf2 (C2) cells and the stable HEK-V cells were transfected with pcDNA4B-FVII construct transiently (Fig. 4a, b). The detected bands in western blot analysis were semi-quantified to measure the expression level of FVII protein using Image lab software. As it shown in Fig. 4c, HEK-Nrf2 (C1)-FVII and HEK-Nrf2 (C2)-FVII expressed FVII about 1.7-fold more than HEK-FVII (Fig. 4c). In addition, the expression of rhFVII in the cells was also evaluated by ELISA. The expression level of FVII in the HEK-Nrf2 (C1)-FVII and HEK-Nrf2 (C2)-FVII cells was higher than HEK-FVII (Fig. 4d), 315 ng/ml, 300nglml and 198 ng/ml, respectively. Finally, the functional activity of FVII was evaluated by prothrombin time test. Due to the presence of recombinant FVII in the medium of HEK-Nrf2 (C1)-FVII, HEK-Nrf2 (C2)-FVII and HEK-FVII cells clotting time decreased in comparison to HEK-V group (Fig. 5).

Expression of FVII in the stable clones. HEK-Nrf2 (C1), HEK-Nrf2 (C2) and HEK cells were transfected with pcDNA4B-FVII. HEK cells were also transfected with empty pcDNA4B as control. a Evaluation of FVII mRNA expression by RT-PCR using internal primers. HEK-Nrf2 (C1)-FVII, HEK-Nrf2 (C2)-FVII and HEK-FVII that transfected with FVII-containing construct expressed FVII mRNA, 240 bp PCR product, while in the cells transfected with empty vector no expression was observed. b I Western blot analysis was performed to detect FVII protein. The culture medium of the cells transfected with FVII constructs as well as the cells transfected with empty vector was subjected to western blot. HEK-Nrf2 (C1)-FVII, HEK-Nrf2 (C2)-FVII and HEK-FVII cells expressed FVII protein. b II Semi quantification of western blot bands using Image lab software. c ELISA for FVII. Cell culture medium of the cells was harvested in different interval time. Maximum expression was detected 72 h post transfection. The highest expression level of FVII protein was detected in the HEK293-Nrf2 (C1)-FVII and the HEK293-Nrf2 (C2)-FVII groups. No expression of FVII was detected in the medium of the cells transfected with empty vector (pcDNA 4B). (Mean ± SD, *p < 0.05)

Prothrombin time assay. Similar to PNP (Pooled normal plasma) recombinant FVII in the culture medium of the cells transfected with FVII construct decreased clotting time in comparison to the culture medium of cells transfected with the empty vector (pcDNA4B) (Mean ± SD,**p < 0.01, ***p < 0.001)
Discussion
Higher efficiency and production of recombinant proteins in mammalian cell lines are essential nowadays. However, the expression of recombinant protein in the mammalian expression system is challenging. Supporting this notion, currently addressing the challenges is a priority for biotechnological industries. Manipulation of biosynthetic and metabolic pathways by synthetic approaches has already been employed to improve the production of recombinant protein and alternative expression systems [4, 36].
Gupta and et al. have been revealed that increased expression of PYC2 gene in CHO cells have resulted in enhancing of metabolic activity of the cells thereby had positive effects on the overall cell performance. Furthermore, PYC2 render CHO cells as an expression platform for cell line development to produce a potential therapeutic protein in large scale and also well-controlled upstream process in a bioreactor [10, 12].
In this study, we tried to establish a versatile and refractory cell line under the potential harsh microenvironment of cell the culture by a genetically engineered method.
In our study, we genetically engineered HEK293 cells by a well-known cytoprotective gene, Nrf2.
Protein expression can be lost due to posttranslational modification [2, 7, 19]. Furthermore, cell growth rate can be influenced by cell culture conditions. For example, Yang and Schmelzer in their studies showed that pH can affect the activity of extracellular glycosidases [28, 37]. Conspicuously, several stresses such as nutrient depletion, heat shock and oxidative stress are among the commonplace and famous causes of incorrect protein folding in the mammalian-based expression system [18]. Therefore, in this study, the numerous advantages of Nrf2 have been employed to improve the mammalian expression system. As an example of a human recombinant protein and also due to our previous experiences dealing with the expression of recombinant human coagulation FVII, we expressed rFVII in Nrf2-engineered HEK293 cells.
Our results revealed the improvement of the expression of FVII in HEK-Nrf2 cells in comparison with the expression of FVII in HEK293 cells. This might be owing to antioxidant, anti-apoptotic, and cytoprotective properties of Nrf2 [24]. It has been reported that oxidative stress, serum deprivation, and hypoxia are the most important stresses known in vitro and in vivo, which adversely affect cellular function. Under this stress condition, the cytoprotective functions of Nrf2 activate and lead to up-regulation of a variety of genes/proteins including antioxidant factors [24]. Supporting this notion, our results revealed that Nrf2-engineered-HEK293 cells had a higher survival under UV-induced cell death. Moreover, we have already shown that Nrf2 overexpression in the mesenchymal stem cells (MSCs) increased their resistance to different stresses including hypoxia, serum deprivation and oxidative stresses [24].
The HEK293 parental cell line has been adapted to serum-free suspension growth and is preferentially utilized for transient gene expression due to the higher transfection efficiencies [30, 31]. We think our HEK-Nrf2 cells might compare favorably to the original cell line under serum-free suspension cultivation. In this project, we expressed rFVII in HEK293 cells. Of note, due to several drawbacks of obtaining of coagulation FVII in human plasma including the possibility of transmission of blood-derived infections, development and improvement of mammalian expression system to produce rFVII are under investigation.
Currently, the therapeutic rFVII is expressed in BHK by Novo Nordisk (https://www.rxlist.com/novoseven-drug). We also have expressed rFVII in CHO cell line [13, 23]. However, expression of a human recombinant protein in human origin cells could be advantageous. The high similarity of post-translation modification and the absence of antigenic residues are main advantages for the production of human recombinant protein in human cell-based expression systems [4]. A number of studies have been indicated that HEK293 cell line has a promising potential to produce human recombinant proteins produced in large-scale using serum-free suspension technology [31]. Furthermore, this cell line has a relatively rapid growth and high rate of transfection [5, 34]. Furthermore, commercially available HEK293 cell lines which are adapted for suspension culture conditions (e.g. FreeStyle 293-F cells) allow for the production of large-scale recombinant proteins [13].
Overall, in an attempt to establish and improve a cell line for the production of human recombinant proteins this study was conducted. We successfully established an Nrf2-engineered cell line that has a higher growth rate and is refractory to the potential stresses-induced cell death. As an example of human recombinant protein, we expressed coagulation FVII in the established cell line. Our initial results indicated that the established cell line increased the expression of the recombinant protein. Our new cell line might have advantages for the large-scale production of recombinant protein under lower serum and suspension culture condition that conferred superiority especially in terms of cost benefits in biotechnological industries. However, further studies are required in this regards including an increased expression for other model recombinant proteins and additional mechanistic evidence of Nrf2 relieving the burden of recombinant target protein expression.
Conclusion
It is concluded that this new cell line can be used as a suitable cell line to express other recombinant proteins particularly for large-scale production of recombinant protein under other culture situation including lower serum and suspension culture that imposed advantages strongly in terms of cost benefits in bio-pharmaceutical industries.
References
Berlec, A., & Štrukelj, B. (2013). Current state and recent advances in biopharmaceutical production in Escherichia coli, yeasts and mammalian cells. Journal of Industrial Microbiology & Biotechnology, 40(3–4), 257–274.
Dalton, A. C., & Barton, W. A. (2014). Over-expression of secreted proteins from mammalian cell lines. Protein Science, 23(5), 517–525.
Dumont, J., Euwart, D., Mei, B., Estes, S., & Kshirsagar, R. (2016). Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Critical Reviews in Biotechnology, 36(6), 1110–1122.
Durocher, Y., & Butler, M. (2009). Expression systems for therapeutic glycoprotein production. Current Opinion in Biotechnology, 20(6), 700–707.
Durocher, Y., Perret, S., & Kamen, A. (2002). High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Research, 30(2), e9–e9.
Estes, S., & Melville, M. (2013). Mammalian cell line developments in speed and efficiency. In W. Zhou & A. Kantardjieff (Eds.), Mammalian cell cultures for biologics manufacturing (pp. 11–33). Berlin: Springer.
Ghaderi, D., Taylor, R. E., Padler-Karavani, V., Diaz, S., & Varki, A. (2010). Implications of the presence of N-glycolylneuraminic acid in recombinant therapeutic glycoproteins. Nature Biotechnology, 28, 863.
Ghaderi, D., Zhang, M., Hurtado-Ziola, N., & Varki, A. (2012). Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnology and Genetic Engineering Reviews, 28(1), 147–176.
Gupta, S. K., Dangi, A. K., Smita, M., Dwivedi, S., & Shukla, P. (2019). Effectual bioprocess development for protein production. In P. Shukla (Ed.), Applied Microbiology and bioengineering (pp. 203–227). Cambridge: Academic Press.
Gupta, S. K., Sharma, A., Kushwaha, H., & Shukla, P. (2017). Over-expression of a codon optimized yeast cytosolic pyruvate carboxylase (PYC2) in CHO cells for an augmented lactate metabolism. Frontiers in Pharmacology, 8, 463.
Gupta, S. K., & Shukla, P. (2017). Sophisticated cloning, fermentation, and purification technologies for an enhanced therapeutic protein production: A review. Frontiers in Pharmacology, 8, 419.
Gupta, S. K., Srivastava, S. K., Sharma, A., Nalage, V. H., Salvi, D., Kushwaha, H., Chitnis, N. B., & Shukla, P. (2017). Metabolic engineering of CHO cells for the development of a robust protein production platform. PloS ONE, 12(8), e0181455.
Halabian, R., Roudkenar, M. H., Esmaeili, N. S., Masroori, N., Roushandeh, A., & Najafabadi, A. (2009). Establishment of a cell line expressing recombinant factor VII and its subsequent conversion to active form FVIIa through hepsin. Genetic Engineering Method.Vox Sanguinis, 96(4), 309–315.
https://www.rxlist.com/novoseven-drug., N. c. f. v. r.
Invitrogen Corporation, C. CA, http://www.invitrogen.com.
Gupta, K., S. and Shukla, P. (2018). Glycosylation control technologies for recombinant therapeutic proteins. Applied Microbiology and Biotechnology, 102(24), 10457–10468.
Kayser, K., Lin, N., Allison, D., Donahue, L., & Caple, M. (2006). Cell line engineering methods for improving productivity. BioProcess International, 4(5), 6–13.
Kopito, R. R. (2000). Aggresomes, inclusion bodies and protein aggregation. Trends in Cell Biology, 10(12), 524–530.
Kuriakose, A., Chirmule, N., & Nair, P. (2016). Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. Journal of Immunology Research. https://doi.org/10.1155/2016/1298473
Lai, T., Yang, Y., & Ng, S. K. (2013). Advances in mammalian cell line development technologies for recombinant protein production. Pharmaceuticals, 6(5), 579–603.
Lee, J.-M., & Johnson, J. A. (2004). An important role of Nrf2-ARE pathway in the cellular defense mechanism. BMB Reports, 37(2), 139–143.
Liste-Calleja, L., Lecina, M., Schucht, R., Wirth, D., Hauser, H., & Cairó, J. J. (2015). “Hek293 as a recombinant protein factory: three different approaches for protein production. BMC Proceedings, 9(9): P74.
Masroori, N., Halabian, R., Mohammadipour, M., Roushandeh, A. M., Rouhbakhsh, M., Najafabadi, A. J., Fathabad, M. E., Salimi, M., Shokrgozar, M. A., & Roudkenar, M. H. (2010). High-level expression of functional recombinant human coagulation factor VII in insect cells. Biotechnology Letters, 32(6), 803–809.
Mohammadzadeh, M., Halabian, R., Gharehbaghian, A., Amirizadeh, N., Jahanian-Najafabadi, A., Roushandeh, A. M., & Roudkenar, M. H. (2012). “Nrf-2 overexpression in mesenchymal stem cells reduces oxidative stress-induced apoptosis and cytotoxicity. Cell Stress and Chaperones, 17(5), 553–565.
Movahed, M., Roudkenar, M. H., Bahadori, M., Mohammadipour, M., Jalili, M. A., & Amiri, F. (2018). Establishment of Stable CHO Cell Line Expressing recombinant human haptoglobin: Toward new haptoglobin-based therapeutics. Iranian Journal of Science and Technology Transactions A: Science, 42(3), 1097–1103.
Palomares, L. A., Estrada-Mondaca, S., & Ramirez, O. T. (2004). Production of recombinant proteins: challenges and solutions. Methods in Molecular Biology, 267, 15–52.
Picanço-Castro, V., Tage Biaggio, R., Tadeu Cova, D., & Swiech, K. (2013). Production of recombinant therapeutic proteins in human cells: Current achievements and future perspectives. Protein and Peptide Letters, 20(12), 1373–1381.
Schmelzer, A. E., & Miller, W. M. (2002). “Effects of osmoprotectant compounds on NCAM polysialylation under hyperosmotic stress and elevated pCO2. Biotechnology and Bioengineering, 77(4), 359–368.
Shukla, A. A., & Thömmes, J. (2010). Recent advances in large-scale production of monoclonal antibodies and related proteins. Trends in Biotechnology, 28(5), 253–261.
Sun, X., Hia, H. C., Goh, P. E., & Yap, M. G. (2008). High-density transient gene expression in suspension-adapted 293 EBNA1 cells. Biotechnology and Bioengineering, 99(1), 108–116.
Swiech, K., Kamen, A., Ansorge, S., Durocher, Y., Picanço-Castro, V., Russo-Carbolante, E. M. S., Neto, M. S. A., & Covas, D. T. (2011). Transient transfection of serum-free suspension HEK 293 cell culture for efficient production of human rFVIII. BMC Biotechnology, 11, 114–114.
Swiech, K., Picanço-Castro, V., & Covas, D. T. (2012). Human cells: new platform for recombinant therapeutic protein production. Protein Expression and Purification, 84(1), 147–153.
Thimmulappa, R. K., Lee, H., Rangasamy, T., Reddy, S. P., Yamamoto, M., Kensler, T. W., & Biswal, S. (2016). Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. The Journal of Clinical Investigation, 116(4), 984–995.
Thomas, P., & Smart, T. G. (2005). HEK293 cell line: a vehicle for the expression of recombinant proteins. Journal of Pharmacological and Toxicological Methods, 51(3), 187–200.
Walsh, G. (2014). Biopharmaceutical benchmarks 2014. Nature Biotechnology, 32, 992.
Wurm, F. M. (2004). Production of recombinant protein therapeutics in cultivated mammalian cells. Nature Biotechnology, 22(11), 1393.
Yang, M., & Butler, M. (2000). Effect of ammonia on the glycosylation of human recombinant erythropoietin in culture. Biotechnology Progress, 16(5), 751–759.
Zhaleh, F., Amiri, F., Mohammadzadeh-Vardin, M., Bahadori, M., Harati, M. D., Roudkenar, M. H., & Saki, S. (2016). Nuclear factor erythroid-2 related factor 2 overexpressed mesenchymal stem cells transplantation, improves renal function, decreases injuries markers and increases repair markers in glycerol-induced Acute kidney injury rats. Iranian Journal of Basic medical Sciences, 19(3), 323.
Acknowledgements
This study was supported by the Blood Transfusion Research Center, High Institute for Research and Education in Transfusion Medicine, Iran, Grant No. 1395-01-33-1933.
Author information
Authors and Affiliations
Contributions
ZAb, FA and MM collected all data, samples and also accomplished all cellular and molecular tests. MHR controlled and managed the project, wrote the manuscript and finalized it. All authors revised the article carefully and confirmed the edited version of the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical Approval
There are no ethical problems for this project.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abbasi-Malati, Z., Amiri, F., Mohammadipour, M. et al. HEK293 Cells Overexpressing Nuclear Factor E2-Related Factor-2 Improve Expression of Recombinant Coagulation Factor VII. Mol Biotechnol 61, 317–324 (2019). https://doi.org/10.1007/s12033-019-00160-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-019-00160-y