
Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder that results in motor impairment due to dopaminergic neuronal loss. The pathology of PD is closely associated with neuroinflammation, which can be characterized by astrocyte activation. Thus, targeting the inflammatory response in astrocytes might provide a novel therapeutic approach. We conducted a luciferase assay on an in-house chemical library to identify compounds with anti-inflammatory effects capable of reducing MPP+-induced NF-κB activity in astrocytes. Among the compounds identified, EI-16004, a novel 3-benzyl-N-phenyl-1H-pyrazole-5-carboxamides, exhibited a significant anti-inflammatory effect by significantly reducing MPP+-induced astrocyte activation. Biochemical analysis and docking simulation indicated that EI-16004 inhibited the MPP+-induced phosphorylation of p65 by attenuating ERK phosphorylation, and EI-16004 reduced pro-inflammatory cytokine and chemokine levels in astrocytes. In vivo studies on the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD model in male C57BL/6 mice showed that EI-16004 ameliorated motor impairment and protected against dopaminergic neuronal loss, and EI-16004 effectively mitigated the MPTP-induced astrocyte activation in striatum (STR) and substantia nigra (SN). These results indicate EI-16004 is a potential neuroprotective agent for the prevention and treatment of astrocyte-mediated neuroinflammatory conditions in PD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) primarily affects the elderly and is characterized by rest tremors, bradykinesia, rigidity, and loss of postural reflexes, which result from the dopaminergic neuronal loss in the nigrostriatal pathway (Jankovic, 2008). Neuroinflammation represents a distinctive pathological feature in PD, marked by the activation of glial cells (Hansson, 2010; Yang & Zhou, 2019). Chronic activation of glial cells in the CNS accelerates neuroinflammation, which can result in the release of pro-inflammatory factors that have toxic effects on surrounding neurons, leading to neurodegeneration (Lee et al., 2019). Astrocytes, a type of glial cells, play a crucial role in brain homeostasis through various coordinated metabolic processes with neurons, such as energy metabolite supply and neurotransmitter recycling functions (Allaman et al., 2011). In response to neuroinflammation or injury, astrocytes can become activated and take on two forms: A1 and A2. A1 astrocytes stimulate production of pro-inflammatory factors, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6, that cause neuronal damage, whereas A2 astrocytes increase neurotrophic factors that is beneficial for neuronal survival and growth and synaptic repair. In the CNS, A2 astrocytes exhibit beneficial effects, whereas A1 astrocytes are associated with detrimental outcomes. Therefore, controlling astrocyte activation and inhibiting the NF-κB signaling pathway, a well-known inflammatory pathway implicated in the pathology of PD, is considered a promising therapeutic strategy for PD (Lee et al., 2019).
Mitogen-activated protein kinases (MAPKs), including extracellular regulated kinase (ERK), p38 and c-Jun N-terminal kinase (JNK), are closely associated with NF-κB, and thus, inhibiting these MAPK subfamily units can result in the inhibition of NF-κB (Chi et al., 2006; Kaminska, 2005). Previous studies have reported that suppressing astrocyte activation and inhibiting MAPK/NF-κB signaling can attenuate dopaminergic neuronal loss (Hong et al., 2022; Kirkley et al., 2019; Lee et al., 2021, 2022), indicating modulators of astrocyte activation with anti-inflammatory effects have therapeutic potential in PD.
EI-16004 is a novel series of 3-benzyl-N-phenyl-1H-pyrazole-5-carboxamides. In previous studies, other series of 3-benzyl-N-phenyl-1H-pyrazole-5-carboxamides were evaluated for their biological activity on glucose-stimulated insulin secretion (GSIS) and found to increase GSIS significantly by regulation PDX-1 activity within β-cells (Jo et al., 2021). Nonetheless, the anti-inflammatory properties of EI-16004 have not yet been explored within the context of neuroinflammation or PD. Effective PD treatment requires drugs to penetrate the blood-brain barrier (BBB) for access to the central nervous system (CNS). We predicted BBB penetration and used logP values to determine whether EI-16004 might effectively penetrate the BBB. BBB penetration was assessed using brain-blood concentration ratios (BB), that is, as [Brain]/[Blood], where [Brain] and [Blood] are the steady-state concentrations of radiolabeled compounds in brain and peripheral blood. A BB ratio of ≥ 2.0 indicates that the substance is readily absorbed into the CNS and is suitable for brain applications. On the other hand, logP values provide a measure of hydrophobicity, and for CNS drugs, logP values range from 0.4 to 5.1 with a median value of 2.8, which indicates optimal hydrophobicity for effective passage through the BBB. EI-16004 has a BB ratio of 3.936 and a logP value of 3.463, indicating that it is likely to cross the BBB effectively (Ghose et al., 2012; Ma et al., 2005; Wager et al., 2010).
Here, we screened an in-house chemical library for substances with anti-inflammatory effects that reduce MPP+-induced NF-κB activity in primary astrocytes. Among the screened substances, EI-16004 emerged as the most effective candidate. Subsequently, we assessed the neuroprotective effects of EI-16004 under astrocyte-mediated neuroinflammatory conditions and explored its impact on PD behavioral characteristics.
Materials and Methods
Materials
The synthesis methods for EI-16004 derivatives can be found in the Supplementary Material. Reagents and chemicals, including Chloroform, Gentamicin, Isopropanol, MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), MPP+ (1-methyl-4-phenylpyridinium), PD98059 (2-(2-Amino-3-methoxyphenyl)-4 H-1-benzopyran-4-one), Paraformaldehyde (PFA), and Poly-L-lysine, were procured from Sigma-Aldrich (St. Louis, MO, USA). Alexa Fluor 488 and Alexa Fluor 568 were obtained from Invitrogen (Waltham, MA, USA), and the Western blot detection reagent (ECL solution) was sourced from Advansta (Menlo Park, CA, USA).
Primary Astrocyte Culture
Primary astrocyte cultures were prepared from Sprague Dawley rat pups’ cortices on postnatal day (PND) 1 or 2, following established procedures (Daehan Biolink Co., Ltd., Chungbuk, South Korea). Briefly, the dissected cortices were minced in ice-cold Hanks’ balanced salt solution (HBSS; Welgene, Daegu, South Korea). Subsequently, the tissue was treated with 0.25% trypsin for 15 min at room temperature, followed by three washes with HBSS. The dissociated cells were mechanically separated and then seeded in Dulbecco’s modified Eagle’s medium (DMEM/F12) (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Welgene) and 1% penicillin-streptomycin (Welgene) on poly-L-lysine-coated plastic culture dishes (Sigma-Aldrich, St. Louis, MO, USA). The cultures were maintained under consistent conditions, and experiments were conducted after a 14-day incubation period.
Luciferase Assay
Primary astrocytes were cultured in 96-well plates (1 × 104 cells/well) using DMEM/F12 medium supplemented with 10% FBS and 1% penicillin-streptomycin. For transfection, cells were exposed to 0.1 µg of the 4×NF-κB plasmid (Addgene; Watertown, MA, USA) for 6 h, utilizing the ViaFect™ transfection reagent (Promega; Madison, WI, USA) in Opti-MEM (Gibco). Subsequently, the culture medium was replaced with fresh DMEM/F12 containing 10% FBS and 1% penicillin-streptomycin. After a 3 h pretreatment with 1 µM EI-16004, cells were co-incubated with 250 µM MPP+ for 4 h. Following this, a PBS wash was performed, and cell lysis was achieved using the ONE-Glo™ Luciferase Assay System (Promega). The NF-κB-dependent luciferase reporter activities were quantified using a GENios luminometer (TECAN, Salzburg, Austria).
MTT Assay
Primary astrocytes were seeded in 96-well plates, and treated with different concentrations of EI-16004 for 24 h. Media were then removed, and 200 µL of a 0.5 mg/mL MTT solution in PBS was added to each well. Cells were incubated for 4 h at 37℃, the MTT solution was removed, and cells were lysed using solubilization solution (ethanol: DMSO = 1:1). Amounts of formazan produced were quantified by measuring absorbance using a Multiskan FC microplate reader at 560 nm (Thermo Fisher Scientific; Waltham, MA, USA).
Immunocytochemistry
Primary astrocytes were cultured in poly-L-lysine-coated plastic culture dishes and pre-treated with 1 µM EI-16004 for 6 h. They were then co-incubated with 250 µM MPP+ for 24 h, followed by a PBS wash. Fixed cells were treated with 4% paraformaldehyde (PFA) in pH 7.4 PBS for 15 min at 37 °C. Subsequently, they were subjected to blocking steps first with tris-buffered saline (TBS) and then with TBS-TS (TBS containing 0.1% Triton X-100 and 3% goat serum) for 30 min. Next, the cells were incubated overnight at 4 °C with either GFAP (mouse monoclonal; 1:1000, Cell Signaling Technology, Danvers, MA, USA) or p65 antibody (rabbit monoclonal; 1:500, Cell signaling Technology). On the following day, the cells were washed with TBS and incubated with anti-mouse IgG labeled with Alexa Fluor 488 (3 µL/mL) or anti-rabbit IgG labeled with Alexa Fluor 568 (3 µL/mL) for 3 h at room temperature. Finally, they were stained with DAPI solution (1 µg/mL) for 30 min at 37 °C.
Western Blot Analysis
Cell homogenates were prepared using lysis buffer and centrifuged at 12,000 rpm for 15 min at 4 °C. The resulting supernatants were transferred to fresh tubes, and the protein concentrations were assessed using a BCA protein assay kit (Pierce, Shirley, NY, USA). Equal amounts of protein (15 µg per lane) were separated by electrophoresis on 10% SDS-polyacrylamide gels and subsequently transferred onto Immobilon-PSQ transfer membranes (Millipore, Bedford, MA, USA). After blocking in 5% skim milk for 30 min at room temperature, membranes were incubated overnight at 4 °C with specific primary antibodies: GFAP and p-p38 (mouse monoclonal; 1:1000; Cell Signaling Technology), p38, p-JNK, JNK, p-ERK, ERK, and p-p65 (rabbit monoclonal; 1:1000; Cell Signaling Technology; Danvers, MA, USA), and β-actin (mouse monoclonal; 1:5000; Sigma-Aldrich; Louis, MO, USA), all diluted in TBS-T. Following three 10 min washes, membranes were incubated with secondary antibodies conjugated with horseradish peroxidase (1:10000; Santa Cruz; CA, USA) in TBS-T for 2 h at room temperature. Blots were visualized using enhanced ECL and captured using a cooled CCD camera system (ATTO Ez-Capture; Atto Corp., Tokyo, Japan). Densitometric analysis was performed to calculate fold changes in relative protein levels, with β-actin serving as the loading control.
RNA Isolation and Real Time-PCR
Cell homogenization was carried out using RiboEX™ reagent (GeneAll; Seoul; Korea), followed by vigorous shaking with chloroform and a 15 min incubation. The resulting aqueous phases were carefully transferred to fresh tubes, and isopropanol was added, followed by an overnight incubation at -80℃. After centrifugation at 12,000 rpm for 15 min, the RNA pellets obtained were washed with 75% ethanol and then centrifuged at 8,000 rpm for 5 min. These RNA pellets were air-dried and subsequently dissolved in diethylpyrocarbonate (DEPC) distilled water. The concentration of mRNA was determined, and reverse transcription into cDNA was performed using SuPrimeCript RT Premix (Genetbio Inc.; Daejeon, South Korea). Real-time PCR was carried out utilizing SYBR green master mix (BIOLINE; Taunton, MA, USA) and the CFX Connect System (Bio-Rad Inc.; Hercules, CA, USA). Details of the primer sequences employed in the study can be found in Table 1.
Molecular Docking Simulation Analysis
The 3D structure of ERK protein was obtained from the AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/ accessed on January 17, 2022) in Protein Data Bank (PDB) format. Three ligands—EI-16004, PD98059 (CID: 4713), and ATP (CID: 5957)—were sourced from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/ accessed on January 17, 2022, and March 20, 2023) in Structure Data File (SDF) format. These ligands were converted to PDB format using the AutoDockFRprepare_ligand software [PMID: 26629955]. The 3D structure of EI-16004 was generated in SDF format using MarvinSketch version 23.4 software (ChemAxon, Budapest, Hungary) and then converted to PDB format employing the AutoDockFRprepare_ligand software [PMID: 26629955]. Computational docking simulations were performed between ERK protein and EI-16004, PD98059, or ATP using AutoDock Vina 1.2 in AMDock version 1.5.2, employing the ‘Simple Docking’ mode [PMID: 32,938,494]. The grid box dimensions were set to 70 Å for x, y, and z, respectively. The grid was centered on specific xyz-coordinates to explore the entire predicted binding site: EI-16004 (3.60, 1.70, and − 0.70 Å), PD98059 (0.30, 0.30, and 0 Å), and ATP (0.20, − 6.20, and − 13.70 Å). Docking conformations and receptor-ligand interactions were visualized using Discovery Studio Visualizer v21.1.0.20298 (BIOVIA, San Diego, CA, USA).
Animals and Treatment
Male C57BL/6 mice at the age of 6 weeks were procured from Daehan Biolink (Chungbuk, Korea). These mice were randomly assigned to one of three groups, each containing five animals per cage: an untreated control group, an MPTP group, or an MPTP + 5 mg/kg EI-16004 group. The mice were housed in controlled conditions, maintaining a temperature range of 20–23 °C under a 12 h light-dark cycle, and had access to food and water ad libitum throughout the study period. The animals in the MPTP group and MPTP + 5 mg/kg EI-16004 group received intraperitoneal (i.p.) injections of MPTP at a dose of 30 mg/kg daily for 5 consecutive days. The untreated control group received an equivalent volume of 0.1 M PBS via i.p. injection. One hour after the MPTP injection, the MPTP + 5 mg/kg EI-16004 group received EI-16004 (dissolved in corn oil containing 5% DMSO) at a dose of 5 mg/kg daily on the day of the MPTP injection and for 2 additional consecutive days after the last MPTP administration. The untreated control group and the MPTP group received the same volume of corn oil containing 5% DMSO via i.p. injection as the MPTP + 5 mg/kg EI-16004 group.
Rota-Rod Test
Mice were pre-trained for 3 days to acclimate to the rota-rod (5-lane accelerating rota-rod; JD-A-07, JEUNG DO BIO & PLANT CO., LTD., Korea) for 180 s. Training was performed using four consecutive runs per session, and the rod speed was increased stepwise over the training period from 20 to 30 rpm. Following the pre-training phase, mice were subjected to testing at various time points, specifically at 2, 6, 24, 48, and 72 h after the final MPTP injections. The testing sessions consisted of four trials at a fixed rod speed of 30 rpm.
Tissue Preparation
For histological analysis, mice were anesthetized 72 h after the final MPTP administration (on postnatal day 59) using ethyl ether. Subsequently, they were intracardially perfused with 0.1 M PBS (pH 7.4) containing 0.9% NaCl and fixed with 0.1 M PBS containing 4% paraformaldehyde (PFA). The brains were carefully extracted, post-fixed in the same PFA solution, and then transferred to 30% sucrose at 4 °C overnight. After cryoprotection, the brains were sectioned serially into 40 μm-thick slices in the coronal plane using a freezing microtome (MICROM, Walldorf, Germany). These sections were preserved in Dulbecco’s phosphate-buffered saline (DPBS) solution containing 0.1% sodium azide at 4 °C.
Diaminobenzidine (DAB) Immunohistochemistry
Brain Sect. (40 μm) underwent a 30 min incubation with 0.6% H2O2 in TBS, followed by three 10 min TBS washes. Subsequently, the sections were blocked with TBS-TS for 30 min and then exposed to the primary antibody, anti-tyrosine hydroxylase (TH) (mouse monoclonal; 1:5000; Chemicon, Rolling Meadows, IL, USA), in TBS-TS overnight at 4 °C. Afterward, the sections were treated with suitable biotinylated secondary antibodies, either goat anti-mouse or anti-rabbit IgG, in TBS for 3 h at room temperature. This was followed by incubation in an avidin-biotin complex (ABC) solution (Vectastain ABC reagent Elite Kit, Vector Laboratories, Burlingame, CA, USA) in TBS for 1 h at room temperature. Upon development in DAB solution, images were captured using a Nikon ECLIPSE TE 2000-U microscope (Nikon, Tokyo, Japan). Quantitative assessment of dopaminergic neurons was performed as follows: five to seven sections per mouse containing the substantia nigra (SN) were selected. The number of TH-positive dopaminergic neurons in these sections was counted and divided by the respective section areas. The average counts for TH-positive neurons in five to seven sections per mouse were then calculated. For the quantitative analysis of TH immunostaining in the striatum (STR), TH expression intensities (assessed in five to seven sections per mouse) were measured utilizing FluorChem SP software (Alpha Innotech, San Leandro, CA, USA).
Double Fluorescence Immunohistochemisty
Brain Sect. (40 μm in thickness) underwent a 30 min blocking step with TBS-TS at room temperature. Subsequently, they were exposed to primary antibodies: anti-GFAP antibody (mouse polyclonal; 1:500, Cell Signaling, MA, USA) or anti-ionized calcium-binding adaptor molecule 1 (Iba-1) antibody (rabbit polyclonal; 1:500, Wako, Tokyo, Japan) in TBS-TS overnight at 4 °C. After thorough washing with TBS, the sections were subjected to a 3 h incubation with anti-mouse IgG labeled with Alexa Fluor 488 (3 µL/mL) and anti-rabbit IgG labeled with Alexa Flour 568 (3 µL/mL) at room temperature. Images of the substantia nigra (SN) and striatum (STR) were captured using a LS30 fluorescence microscope (Leam Solution, Seoul, Korea), with two images obtained per mouse for both SN and STR regions. Subsequently, fluorescence intensities were quantified across the entire images of the left and right hemispheres using Image J software and then averaged.
Statistical Analysis
The statistical analysis was performed using Prism Ver. 9.5.1 (GraphPad Software Inc., San Diego, CA, USA). Descriptive and comparative statistics were conducted to analyze the data. The mean ± SEM of independent determinations from a minimum of three experimental replicates were used to express the data. For data represented in vitro studies and immunohistochemistry analysis, a one-way repeated measures analysis of variance (ANOVA) was employed, followed by Tukey’s post-hoc test. For data represented behavioral test, two-way ANOVA was employed, followed by Tukey’s post-hoc test. The level of statistical significance was set at p < 0.05.
Results
Screening of the In-House Chemical Library for Anti-Inflammatory Candidates
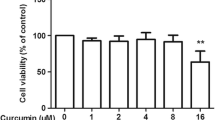
The in-house chemical library was provided by Prof. Hwayoung Yun (College of Pharmacy of Pusan National University, Busan, Korea), and anti-inflammatory effects were evaluated by luciferase assay using primary astrocytes. Primary astrocytes were pretreated with 19 compounds from the in-house chemical library at 1 µM for 3 h and then co-treated with 250 µM MPP+ for 3 h. Luciferase assay results confirmed that MPP+ increased NF-κB activity and showed that EI-16004 most effectively attenuated NF-κB activity (Fig. 1A; F22,207 = 14.38, p < 0.0001). The structures of EI-16004 and its derivatives are shown in Fig. 1B and Supplemental Material. In consideration of the potential impact on cell viability and to assess whether the observed decrease in NF-κB levels was attributable to the anti-inflammatory properties of EI-16004 rather than cytotoxic effects, we conducted a comprehensive evaluation of the compound’s cytotoxicity. Cell viability assays were performed on primary astrocytes, utilizing concentrations ranging from 0.01 to 10 µM. MTT assays showed that EI-16004 did not affect the cell viability of primary astrocytes (Fig. 1C; F4,35 < 5.0, p > 0.05). Based on these results, EI-16004 at 1 µM was chosen for further study.

(A) Relative luminescence units (RLU) were used to assess NF-κB promotor activity in primary astrocytes. EI-16004 effectively attenuated MPP+-induced NF-κB activity (n = 10). (B) The structure of EI-16004. (C) Cell viability of primary astrocytes was evaluated using an MTT assay after treatment with varying concentrations of EI-16004 (0.01, 0.1, 1, or 10 µM) for 24 h (n = 8). Results are presented as means ± SEMs. *** p < 0.001 vs. naïve control and ### p < 0.001, ## p < 0.01, # p < 0.05 vs. MPP+ control. The statistical significance was evaluated through the utilization of one-way ANOVA and subsequently verified using Tukey’s multiple comparison test
EI-16004 Downregulated Inflammatory Response in Primary Astrocytes
To confirm the anti-inflammatory effect of EI-16004, immunocytochemistry and western blot analysis were performed on primary astrocytes. Primary astrocytes were pretreated with 1 µM EI-16004 for 6 h, and then co-treated with 250 µM MPP+ for 30 min. Interestingly, p65 was translocated to nuclei when astrocytes were treated with MPP+, and this translocation was inhibited by EI-16004 (Fig. 2A). Western blot showed that MPP+ upregulated the levels of phosphorylated p65 and that EI-16004 significantly inhibited MPP+-induced p65 phosphorylation (Fig. 2B, C; F2, 6 = 12.68, p = 0.007). In addition, real-time PCR data indicated that MPP+ increased the expressions of pro-inflammatory cytokines (IL-1β, TNF-α, and IL-6) and of a chemokine (CCL2) in primary cultured astrocytes, and these increases were also significantly suppressed by EI-16004 (Fig. 2D; IL-1β, F2,12 = 161.7, p < 0.0001; TNF-α, F2,12 = 27.61, p < 0.0001; IL-6, F2,12 = 48.93, p < 0.0001; CCL2, F2,12 = 40.38, p < 0.0001). These results show that the observed anti-inflammatory effect of EI-16004 in primary astrocytes was due to NF- κB signaling pathway inhibition.

Anti-inflammatory effects of EI-16004 in primary astrocytes. (A) Representative images showed that EI-16004 inhibited the MPP+-induced nuclear translocation of p65. Scale bar = 50 μm. (B) Western blot showed that EI-16004 suppressed MPP+-induced p65 phosphorylation. (C) Densitometric western blot results. Three independent experiments were performed (n = 3). β-actin was used as a loading control. (D) Real-time PCR showed that EI-16004 significantly reduced MPP+-induced increases in pro-inflammatory cytokine and chemokine levels. Five independent experiments were performed (n = 5). Results are presented as means ± SEMs. *** p < 0.001, ** p < 0.01 vs. naïve control and ### p < 0.001, ## p < 0.01, # p < 0.05 vs. MPP+ control. The statistical significance was evaluated through the utilization of one-way ANOVA and subsequently verified using Tukey’s multiple comparison test
EI-16004 Inhibited MPP+-induced Astrocyte Activation
It was previously demonstrated that MPP+ induces astrocyte activation and neuroinflammation by upregulating GFAP and pro-inflammatory cytokines and chemokine expressions in primary astrocytes (Lee et al., 2022). After pre-treating astrocytes with 1 µM EI-16004 for 6 h and subsequently co-treating them with 250 µM MPP+ for 24 h, both immunocytochemistry and western blot analysis revealed that EI-16004 effectively mitigated the MPP+-induced upregulation of GFAP (Fig. 3A-C; F2,6 = 17.93, p = 0.0029). These results suggest that EI-16004 suppresses MPP+-induced astrocyte activation.

EI-16004 effectively suppressed astrocyte activation. (A) Representative images showing that EI-16004 attenuated GFAP fluorescence intensity (an astrocyte marker). Cell nuclei were counterstained with DAPI; scale bar = 50 μm. (B) Western blot confirmed that EI-16004 reduced MPP+-induced astrocyte activation. (C) Densitometric western blot results. Three independent experiments were performed (n = 3). β-actin was used as a loading control. Results are presented as means ± SEMs. ** p < 0.01 vs. naïve control and # p < 0.05 vs. MPP+ control. The statistical significance was evaluated through the utilization of one-way ANOVA and subsequently verified using Tukey’s multiple comparison test
EI-16004 Demonstrated Inhibition of ERK-NF-κB Pathway During Astrocyte Activation
Previous studies have demonstrated that MAPKs (mitogen-activated protein kinases) are involved in astrocyte activation (Lee et al., 2021, 2022). We investigated the underlying mechanism responsible for the anti-inflammatory effect of EI-16004 by pretreating astrocytes at 1 µM for 6 h, followed by co-treating them with 250 µM MPP+ for 30 min. Western blot revealed a significant increase in the levels of phosphorylated ERK, JNK, and p38 in astrocytes following exposure to MPP+. However, EI-16004 demonstrated its efficacy in reducing the phosphorylation of ERK while not affecting the phosphorylation of JNK and p38 (Fig. 4A, B; F2,9 = 7.739, p = 0.0111). To further investigate the relationship between ERK and astrocyte activation, primary astrocytes were pretreated with 20 µM PD98059 (an ERK inhibitor) for 2 h and then co-treated with 250 µM MPP+ for 24 h. Western blot then showed that PD98059 suppressed MPP+-induced GFAP increases in primary astrocytes (Fig. 4 C, D; F2,6 = 22.86, p = 0.0016). These results indicate that EI-16004 reduces astrocyte activation via the ERK-p65 pathway.

EI-16004 inhibited astrocyte activation via the ERK pathway. (A) Western blot analysis revealed that EI-16004 inhibited the phosphorylation of ERK induced by MPP+ in primary astrocytes. (B) Densitometric western blot results. Four independent experiments were performed (n = 4). ERK, JNK, p38 were used as a loading control. (C) Western blot analysis showed that PD98059 effectively suppressed MPP+-induced astrocyte activation. (D) Densitometric western blot results. Three independent experiments were performed (n = 3). β-actin was used as a loading control. Results are presented as means ± SEMs. ** p < 0.01 vs. naïve control and ## p < 0.01, # p < 0.05 vs. MPP+ control. The statistical significance was evaluated through the utilization of one-way ANOVA and subsequently verified using Tukey’s multiple comparison test
Docking Simulations and Binding Affinity Analysis of the Interaction Between EI-16004 and ERK
Docking simulations were conducted to assess the interaction between EI-16004 and ERK, with PD98059, a well-established ERK inhibitor, serving as a positive control. The results demonstrated that both EI-16004 and PD98059 bound to the ATP-binding site of ERK. Specifically, the methyl group of EI-16004 formed hydrogen bonds with Gln, Ala, Ile, and Leu within the active site, while the hydroxyl phenyl ring of EI-16004 engaged in hydrophobic interactions with Val, Ala, and Ile (see Fig. 5A and B). The binding affinities for ERK with EI-16004, PD98059, and ATP were determined as -7.3, -7.4, and − 6.5 kcal/mol, respectively (refer to Table 2). These docking simulations indicated that EI-16004 and PD98059 exhibited comparable binding affinities to ERK, both surpassing the affinity observed with ATP.

Molecular docking analysis of EI-16004, PD98059, and ATP bound to ERK. (A) 2D diagrams illustrating the various contacts established between ERK and the respective ligands. The colors of the dashed lines denote the types of interactions occurring between ERK binding site residues and the ligands. (B) 3D interactions visualizing the structure of the binding pocket of ERK and the corresponding ligands
EI-16004 Effectively Improved MPTP-Induced Motor Dysfunction in the PD Model
The effect of EI-16004 on motility was also evaluated in an MPTP-induced mouse model of PD. Mice were pre-trained on the rota-rod for 3 days. The following day, mice were intraperitoneally (i.p.) co-treated with MPTP (30 mg/kg) and EI-16004 (5 mg/kg) daily for 5 consecutive days and post-treated EI-16004 (5 mg/kg) for 2 consecutive days (Fig. 6A). All MPTP-treated mice immediately fell off the rotating rod 2 h after the last MPTP injection, indicating weakened motor function. Interestingly, motor function recovered significantly faster in the 5 mg/kg EI-16004-treated MPTP group than in the MPTP group at 6 h after last MPTP injections. At 24 h after last MPTP injections, mice in the both MPTP treated groups completely recovered (Fig. 6B; F(treatment)2,60 = 11.34, p < 0.0001; F(time)4,60 = 27.21, p < 0.0001; F(interaction)8,60 = 7.045, p < 0.0001).

EI-16004 ameliorated MPTP-induced motor dysfunction in mice. (A) Schematic of the in vivo experiment. (B) Motor function was evaluated using a rota-rod test. Mice were pre-trained for three days to adapt to the rod for 180 s. Tests were performed 2, 6, 24, 48 and 72 h after last MPTP injections (n = 5 mice/group). Results are presented as means ± SEMs. *** p < 0.001 vs. treatment-naïve control and ### p < 0.001 vs. the MPTP treated control. The statistical significance was evaluated through the utilization of two-way ANOVA and subsequently verified using Tukey’s multiple comparison test
EI-16004 Attenuated the Loss of Dopaminergic Neurons Induced by MPTP in STR and SN
TH immunostaining, encompassing the nigrostriatal pathway, which includes the STR and SN, was performed to evaluate whether EI-16004 exhibited a neuroprotective effect on dopaminergic neurons in a mouse model of MPTP-induced PD. The results of TH immunostaining demonstrated a significant reduction in dopaminergic neuron levels in the MPTP-treated group when compared to the untreated control group. Importantly, EI-16004 effectively mitigated this MPTP-induced loss of dopaminergic neurons (Fig. 7A). A densitometric quantitative analysis of STR and TH-positive cell counts analysis in SN showed that EI-16004 exerted neuroprotective effects in the PD mouse model (Fig. 7B, C; F2,12 = 44.49, p < 0.0001; F2,12 = 11.68, p = 0.0015).

EI-16004 prevented the loss of dopaminergic neurons in the MPTP-induced mouse model of PD. (A) The neuroprotective effects of EI-16004 on the nigrostriatal pathway were assessed through immunohistochemical analysis. Scale bar = 100 μm. (B) Densitometry was used to assess STR TH levels (n = 5 mice/group). (C) TH-positive dopaminergic neurons in the SN were counted (n = 5 mice/group). Results are presented as means ± SEMs. *** p < 0.001, ** p < 0.01 vs. treatment-naïve control and ### p < 0.001, # p < 0.05 vs. the MPTP treated control. The statistical significance was evaluated through the utilization of one-way ANOVA and subsequently verified using Tukey’s multiple comparison test
EI-16004 Suppressed MPTP-Induced Astrocyte Activation in the PD Model
Chronic neuroinflammation is characterized by the activation of glial cells, such as astrocytes and microglia. Double immunohistochemistry was carried out utilizing both GFAP and Iba-1 antibodies to assess the impact of EI-16004 on glial activation within the mouse model of MPTP-induced PD. Animals in the MPTP-treated group showed greater astrocyte activation in the STR and SN than the untreated control group, and EI-16004 treatment significantly suppressed astrocyte activation in the STR (Fig. 8A, B; F2.12 = 105.7, p < 0.0001; Iba-1, F2,12 < 5.0, p > 0.05) and SN (Fig. 8C, D; GFAP, F2,12 = 44.35, p < 0.0001; Iba-1, F2,12 < 5.0, p > 0.05). These results demonstrated that EI-16004 has neuroprotective effect by suppressing astrocyte activation.

EI-16004 reduced MPTP-induced astrocyte activation in STR and SN. (A-D) Striatum (A, B) and substantia nigra (C, D), STR and SN sections were double immunostained using GFAP (astrocyte marker) and Iba-1 (microglial marker) antibodies. Scale bar = 100 μm. Quantitative analysis of GFAP (green) and Iba-1 (red) fluorescence intensities (n = 5 mice/group). Results are presented as means ± SEMs. *** p < 0.001 vs. treatment-naïve control and ### p < 0.001 vs. the MPTP treated control. The statistical significance was evaluated through the utilization of one-way ANOVA and subsequently verified using Tukey’s multiple comparison test
Discussion
PD is a prevalent neurodegenerative condition associated with aging. It is primarily characterized by motor impairments resulting from the degeneration of dopaminergic neurons in the nigrostriatal pathway, leading to reduced dopamine synthesis (Latif et al., 2021). Current treatment options for PD, such as levodopa, dopamine agonists, monoamine oxidase B (MAO-B) inhibitors, and catechol-O-methyltransferase (COMT) inhibitors, primarily aim to alleviate symptoms by either directly or indirectly supporting dopamine levels. However, these medications are limited by their side effects, including dyskinesia (abnormal involuntary movements), motor fluctuations, hallucinations, nausea, sleep disturbances, and edema (Marsili et al., 2017). Additionally, they do not halt the progression of dopaminergic neuronal loss, which is a hallmark of PD, nor do they provide a cure. Given the increasing number of individuals affected by PD due to the growing elderly population (Tysnes & Storstein, 2017), there is an urgent demand for therapeutic agents that can comprehensively address this condition effectively.
Numerous studies have demonstrated that PD is characterized by the progressive loss of nigrostriatal neurons, a process primarily attributed to chronic neuroinflammation triggered by the activation of glial cells, specifically microglia and astrocytes (Kempuraj et al., 2016; Kwon & Koh, 2020; Lee et al., 2019; Vila et al., 2001). Microglia and astrocytes can adopt distinct phenotypic states: M1 and M2 for microglia, and A1 and A2 for astrocytes. M1 microglia produce pro-inflammatory cytokines and iNOS, whereas M2 microglia exhibit anti-inflammatory properties. Similarly, A1 astrocytes upregulate pro-inflammatory factors like TNF-α, IL-1β, and IL-6, while A2 astrocytes increase the expression of neurotrophic factors that support neuronal survival and growth. In PD, activated microglia release pro-inflammatory cytokines and iNOS, triggering astrocyte activation and disrupting brain homeostasis, ultimately leading to the degeneration of dopaminergic neurons (Lee et al., 2019). Given this underlying mechanism, targeting the activation of glial cells along the M1-A1 pathway, which is closely associated with neuroinflammation, emerges as a pivotal therapeutic strategy to slow down the accelerated neurodegeneration in PD.
While most PD research related to neuroinflammation has concentrated on microglia, it is important to recognize that these essential, nervous system-specific immune cells are involved in a multifaceted array of functions encompassing brain development, neural environment maintenance, injury response, and the repair process (Novak & Koh, 2013; Shechter et al., 2013). Microglia can adopt various phenotypes, adapting their functions to preserve tissue homeostasis (Li & Barres, 2018; Olah et al., 2011). Notably, during the process of accelerated neurodegeneration due to neuroinflammation, microglial activation plays a pivotal role (Lee et al., 2019; Orihuela et al., 2016; Tang & Le, 2016). However, in the present study, it was found EI-16004 failed to mitigate microglial activation nor exert anti-inflammatory effects in microglia (data not shown). Interestingly, EI-16004 did attenuate astrocyte activation and demonstrated anti-inflammatory effects on astrocytes. These findings suggest that EI-16004 specifically exerts an anti-inflammatory effect in astrocytes, thereby reducing astrocyte activation. Furthermore, EI-16004 was found to protect against the loss of dopaminergic neurons and improve motor dysfunction by effectively mitigating astrocyte activation in the MPTP-induced PD mouse model. In the present study, it is evident that MPTP treatment did not lead to statistically significant microglial activation in either the SN or the STR in our PD mouse model (Fig. 8A-D). This observed lack of microglial activation in these regions may be attributed to the specific experimental timeline we adopted. Microglia are known to undergo rapid activation in response to MPTP, releasing pro-inflammatory factors and facilitating the recruitment and activation of astrocytes. This process typically unfolds within a span of several hours to a day. However, our animals were sacrificed 3 days post-MPTP administration, a time point at which microglia may have transitioned to an inactivated state, while astrocytes were actively responding, as previously reported (Lee et al., 2019). The focus of our study on astrocyte activation during this particular temporal window may explain the absence of significant microglial activation in both the substantia nigra and striatum. This temporal consideration underscores the dynamic nature of glial cell responses and offers insights into the observed outcomes. A previous study has reported that preventing MPTP-induced dopaminergic neuronal loss can be achieved by selectively knocking out the NF-κB gene in astrocytes (Kirkley et al., 2019). Therefore, we propose that by modulating the activation of A1 astrocytes, which represent the final stage in the M1-A1 pathway associated with neuroinflammation, we can alleviate the detrimental effects of neuroinflammation and preserve the integrity of dopaminergic neurons in PD patients.
Astrocyte-mediated neuroinflammation can be triggered through various signaling pathways, including AKT, NLRP3, and NF-κB pathways. Previous studies have indicated that modulating these pathways holds promise for alleviating neurodegenerative conditions (Cai et al., 2022; Liu et al., 2021; Rai et al., 2017). The NF-κB pathway, renowned for its involvement in inflammation, becomes activated in response to different stimuli, such as inflammation and cytokine signaling. Activation of NF-κB leads to the upregulation of inflammatory factors, including pro-inflammatory cytokines and chemokines like IL-1β, IL-6, TNF-α, and CCL2 (Madhi et al., 2021). Additionally, phosphorylation of p65, a critical component of NF-κB signaling, is closely associated with MAPKs. Consequently, regulating MAPKs effectively suppresses the expression of pro-inflammatory NF-κB products (Oeckinghaus et al., 2011). Several studies have suggested that manipulating these mechanisms could potentially modulate astrocyte activation (Choi et al., 2018; Lee et al., 2022; Liu et al., 2017). In particular, ERK, a prominent member of the MAPK family, which was the central focus of this study, plays a pivotal role in a multitude of cellular processes, including cell growth, development, and proliferation (Mebratu & Tesfaigzi, 2009). However, excessive activation of the ERK/MAPK pathway has been associated with tumorigenesis (Fang & Richardson, 2005; Mishima et al., 2002). Consequently, there is ongoing research to explore the potential anti-cancer effects of regulating ERK to suppress tumor cell proliferation (Sugiura et al., 2021). Moreover, the ERK/MAPK signaling pathway is also involved in other extracellular signaling pathways related to angiogenesis and platelet production (Guo et al., 2020) and modulation of the this signaling pathway has been investigated for improving conditions like rheumatoid arthritis and neuroinflammation (Li et al., 2020; Ohori, 2008). Given the diverse effects associated with ERK, precise regulation of its activity is crucial. Previous studies have demonstrated that controlling ERK activity can effectively suppress the differentiation and proliferation of cancer cells, leading to potential anti-cancer effects. Several drugs, including ASN007, cobimetinib, binimetinib, and ulixertinib, have been developed to specifically target ERK for cancer treatment (Germann et al., 2017; Koelblinger et al., 2017; Tran et al., 2015). The involvement of the ERK signaling pathway in PD has been proposed (Colucci-D’Amato et al., 2003; Hong et al., 2022; Rai et al., 2019). However, the development of specific ERK inhibitors for PD treatment is still an active area of research. In the present study, we investigated a chemical compound called EI-16004 that we screened from an in-house chemical library. The results showed that EI-16004 demonstrated significant anti-inflammatory effects in primary astrocytes. Additionally, we found that EI-16004 modulates astrocyte activation through the ERK-p65 pathway, as demonstrated by the use of PD98059, a well-known ERK inhibitor. To further evaluate the potential of EI-16004 and compare it with PD98059, we utilized the pre-ADMET tool (https://preadmet.webservice.bmdrc.org/) to predict the physiochemical properties of these compounds (Supplementary Table 1). The analysis revealed that EI-16004 has a logP value of 3.463, indicating its hydrophobic properities, and a BBB value of 3.936, suggesting good penetration through the BBB. In contrast, PD98059 has a logP value of 2.19, also indicating hydrophobicity, but a low BBB value of 0.026, suggesting limited penetration through the BBB. Despite the similarities in their binding sites (Fig. 5), one advantageous property of EI-16004 is its ability to effectively cross the BBB, unlike PD98059. Based on these findings, the study suggests that EI-16004, with its capacity to penetrate the BBB and target ERK, could serve as a potential therapeutic agent for PD, a condition closely associated with neuroinflammation.
In this study, we assessed the potential of EI-16004 in protecting dopaminergic neurons in a mouse model of PD by targeting astrocyte activation and neuroinflammation. The results indicate that EI-16004 effectively reduced astrocyte activation by inhibiting the phosphorylation of p65 and ERK, leading to decreased expression of pro-inflammatory cytokines and chemokines, as well as an improvement in PD-like symptoms. Additionally, we demonstrated that EI-16004 significantly protects against dopaminergic neuronal loss in a PD mouse model. These findings suggest the therapeutic potential of EI-16004 in the treatment of PD by targeting astrocyte activation and neuroinflammation through modulation of the ERK-p65 pathway. The present study contributes to the growing understanding of the role of neuroinflammation in PD and present a promising avenue for the development of effective treatments for PD and related disorders.
Data Availability
The data supporting the findings of this study can be made available by contacting the corresponding author upon a reasonable request.
References
Allaman, I., Bélanger, M., & Magistretti, P. J. (2011). Astrocyte-neuron metabolic relationships: For better and for worse. Trends in Neurosciences, 34(2), 76–87. https://doi.org/10.1016/j.tins.2010.12.001.
Cai, L., Gong, Q., Qi, L., Xu, T., Suo, Q., Li, X., et al. (2022). ACT001 attenuates microglia-mediated neuroinflammation after traumatic brain injury via inhibiting AKT/NFκB/NLRP3 pathway. Cell Communication and Signaling, 20(1), 1–23. https://doi.org/10.1186/s12964-022-00862-y.
Chi, H., Barry, S. P., Roth, R. J., Wu, J. J., Jones, E. A., Bennett, A. M., & Flavell, R. A. (2006). Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci U S A, 103(7), 2274–2279. https://doi.org/10.1073/pnas.0510965103.
Choi, J. H., Jang, M., Lee, J., Il, Chung, W. S., & Cho, I. H. (2018). Neuroprotective effects of a traditional multi-herbal medicine kyung-ok-ko in an animal model of parkinson’s disease: Inhibition of mapks and nf-kb pathways and activation of keap1-nrf2 pathway. Frontiers in Pharmacology, 9(December), 1–15. https://doi.org/10.3389/fphar.2018.01444.
Colucci-D’Amato, L., Perrone-Capano, C., & Di Porzio, U. (2003). Chronic activation of ERK and neurodegenerative diseases. Bioessays, 25(11), 1085–1095. https://doi.org/10.1002/bies.10355.
Fang, J. Y., & Richardson, B. C. (2005). The MAPK signalling pathways and colorectal cancer. Lancet Oncology, 6(5), 322–327. https://doi.org/10.1016/S1470-2045(05)70168-6.
Germann, U. A., Furey, B. F., Markland, W., Hoover, R. R., Aronov, A. M., Roix, J. J., et al. (2017). Targeting the MAPK signaling pathway in cancer: Promising preclinical activity with the novel selective ERK1/2 inhibitor BVD-523 (ulixertinib). Molecular Cancer Therapeutics, 16(11), 2351–2363. https://doi.org/10.1158/1535-7163.MCT-17-0456.
Ghose, A. K., Herbertz, T., Hudkins, R. L., Dorsey, B. D., & Mallamo, J. P. (2012). Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. Acs Chemical Neuroscience, 3(1), 50–68. https://doi.org/10.1021/cn200100h.
Guo, Y., Pan, W., Liu, S., Shen, Z., Xu, Y., & Hu, L. (2020). ERK/MAPK signalling pathway and tumorigenesis (review). Exp Ther Med, 1997–2007. https://doi.org/10.3892/etm.2020.8454.
Hansson, E. (2010). Long-term pain, neuroinflammation and glial activation. Scand J Pain, 1(2), 67–72. https://doi.org/10.1016/j.sjpain.2010.01.002.
Hong, D. G., Lee, S., Kim, J., Yang, S., Lee, M., Ahn, J., et al. (2022). Anti-inflammatory and neuroprotective effects of Morin in an MPTP-Induced Parkinson’s Disease Model. International Journal of Molecular Sciences, 23(18). https://doi.org/10.3390/ijms231810578.
Jankovic, J. (2008). Parkinson’s disease: Clinical features and diagnosis. Journal of Neurology, Neurosurgery and Psychiatry, 79(4), 368–376. https://doi.org/10.1136/jnnp.2007.131045.
Jo, J., Lee, D., Park, Y. H., Choi, H., Han, J., Park, D. H., et al. (2021). Discovery and optimization of novel 3-benzyl-N-phenyl-1H-pyrazole-5-carboxamides as bifunctional antidiabetic agents stimulating both insulin secretion and glucose uptake. European Journal of Medicinal Chemistry, 217, 113325. https://doi.org/10.1016/j.ejmech.2021.113325.
Kaminska, B. (2005). MAPK signalling pathways as molecular targets for anti-inflammatory therapy - from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta Proteins Proteom, 1754(1–2), 253–262. https://doi.org/10.1016/j.bbapap.2005.08.017.
Kempuraj, D., Thangavel, R., Natteru, P. A., Selvakumar, G. P., Saeed, D., Zahoor, H., et al. (2016). Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine, 1(1), 1003.
Kirkley, K. S., Popichak, K. A., Hammond, S. L., Davies, C., Hunt, L., & Tjalkens, R. B. (2019). Genetic suppression of IKK2/NF-κB in astrocytes inhibits neuroinflammation and reduces neuronal loss in the MPTP-Probenecid model of Parkinson’s disease. Neurobiology of Diseases, 127(November 2018), 193–209. https://doi.org/10.1016/j.nbd.2019.02.020.
Koelblinger, P., Dornbierer, J., & Dummer, R. (2017). A review of binimetinib for the treatment of mutant cutaneous melanoma. Future Oncology, 13(20), 1–12. https://doi.org/10.2217/fon-2017-0170.
Kwon, H. S., & Koh, S. H. (2020). Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl Neurodegener, 9(1), 1–12. https://doi.org/10.1186/s40035-020-00221-2.
Latif, S., Jahangeer, M., Maknoon Razia, D., Ashiq, M., Ghaffar, A., Akram, M., et al. (2021). Dopamine in Parkinson’s disease. Clinica Chimica Acta. https://doi.org/10.1016/j.cca.2021.08.009.
Lee, Y., Lee, S., Chang, S. C., & Lee, J. (2019). Significant roles of neuroinflammation in Parkinson’s disease: Therapeutic targets for PD prevention. Archives of Pharmacal Research, 42(5), 416–425. https://doi.org/10.1007/s12272-019-01133-0.
Lee, S., Suh, Y. J., Lee, Y., Yang, S., Hong, D. G., Thirumalai, D., et al. (2021). Anti-inflammatory effects of the novel barbiturate derivative MHY2699 in an mptp‐induced mouse model of parkinson’s disease. Antioxidants, 10(11). https://doi.org/10.3390/antiox10111855.
Lee, S., Hong, D. G., Yang, S., Kim, J., Baek, M., Kim, S., et al. (2022). Anti-inflammatory effect of IKK‐Activated GSK‐3β inhibitory peptide prevented Nigrostriatal Neurodegeneration in the Rodent Model of Parkinson’s Disease. International Journal of Molecular Sciences, 23(2). https://doi.org/10.3390/ijms23020998.
Li, Q., & Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nature Reviews Immunology. https://doi.org/10.1038/nri.2017.125.
Li, Y., Chen, N., Wu, C., Lu, Y., Gao, G., Duan, C., et al. (2020). Galectin-1 attenuates neurodegeneration in Parkinson’s disease model by modulating microglial MAPK/IκB/NFκB axis through its carbohydrate-recognition domain. Brain, Behavior, and Immunity, 83, 214–225. https://doi.org/10.1016/j.bbi.2019.10.015.
Liu, T., Zhang, L., Joo, D., & Sun, S. C. (2017). NF-κB signaling in inflammation. Signal Transduct Target Ther, 2(April), https://doi.org/10.1038/sigtrans.2017.23.
Liu, B., Zhang, Y., Yang, Z., Liu, M., Zhang, C., Zhao, Y., & Song, C. (2021). ω-3 DPA protected neurons from Neuroinflammation by balancing Microglia M1/M2 polarizations through inhibiting NF-κB/MAPK p38 signaling and activating Neuron-BDNF-PI3K/AKT pathways. Marine Drugs, 19(11). https://doi.org/10.3390/md19110587.
Ma, X. L., Chen, C., & Yang, J. (2005). Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharmacologica Sinica, 26(4), 500–512. https://doi.org/10.1111/j.1745-7254.2005.00068.x.
Madhi, I., Kim, J. H., Shin, J. E., & Kim, Y. (2021). Ginsenoside re exhibits neuroprotective effects by inhibiting neuroinflammation via CAMK/MAPK/NF-κB signaling in microglia. Molecular Medicine Reports, 24(4), 1–10. https://doi.org/10.3892/mmr.2021.12337.
Marsili, L., Marconi, R., & Colosimo, C. (2017). Treatment Strategies in Early Parkinson’s Disease. Int Rev Neurobiol (1st ed., Vol. 132). Elsevier Inc. https://doi.org/10.1016/bs.irn.2017.01.002.
Mebratu, Y., & Tesfaigzi, Y. (2009). How ERK1/2 activation controls cell proliferation and cell death is subcellular localization the answer? Cell Cycle, 8(8), 1168–1175.
Mishima, K., Inoue, K., & Hayashi, Y. (2002). Overexpression of extracellular-signal regulated kinases on oral squamous cell carcinoma. Oral Oncology, 38(5), 468–474. https://doi.org/10.1016/S1368-8375(01)00104-X.
Novak, M. L., & Koh, T. J. (2013). Macrophage phenotypes during tissue repair. Journal of Leukocyte Biology, 93(6). https://doi.org/10.1189/jlb.1012512.
Oeckinghaus, A., Hayden, M. S., & Ghosh, S. (2011). Crosstalk in NF-κB signaling pathways. Nature Immunology, 12(8), 695–708. https://doi.org/10.1038/ni.2065.
Ohori, M. (2008). ERK inhibitors as a potential new therapy for rheumatoid arthritis. Drug News & Perspectives, 21(5). https://doi.org/10.1358/DNP.2008.21.5.1219006.
Olah, M., Biber, K., Vinet, J., & Boddeke, W. G. M., H (2011). Microglia phenotype diversity. CNS Neurol Disord Drug Targets, 10(1). https://doi.org/10.2174/187152711794488575.
Orihuela, R., McPherson, C. A., & Harry, G. J. (2016). Microglial M1/M2 polarization and metabolic states. British Journal of Pharmacology. https://doi.org/10.1111/bph.13139.
Rai, S. N., Birla, H., Singh, S. S., Zahra, W., Patil, R. R., Jadhav, J. P., et al. (2017). Mucuna pruriens protects against MPTP intoxicated neuroinflammation in Parkinson’s disease through NF-κB/pAKT signaling pathways. Frontiers in Aging Neuroscience, 9(DEC), 1–14. https://doi.org/10.3389/fnagi.2017.00421.
Rai, S. N., Dilnashin, H., Birla, H., Singh, S. S., Zahra, W., Rathore, A. S., et al. (2019). The role of PI3K/Akt and ERK in Neurodegenerative disorders. Neurotoxicity Research, 35(3), 775–795. https://doi.org/10.1007/s12640-019-0003-y.
Shechter, R., Miller, O., Yovel, G., Rosenzweig, N., London, A., Ruckh, J., et al. (2013). Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by Remote Brain Choroid Plexus. Immunity, 38(3). https://doi.org/10.1016/j.immuni.2013.02.012.
Sugiura, R., Satoh, R., & Takasaki, T. (2021). Erk: A double-edged sword in cancer. Erk-dependent apoptosis as a potential therapeutic strategy for cancer. Cells, 10(10). https://doi.org/10.3390/cells10102509.
Tang, Y., & Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Molecular Neurobiology. https://doi.org/10.1007/s12035-014-9070-5.
Tran, K. A., Cheng, M. Y., Mitra, A., Ogawa, H., Shi, V. Y., Olney, L. P., et al. (2015). MEK inhibitors and their potential in the treatment of advanced melanoma: The advantages of combination therapy. Drug Design, Development and Therapy, 10, 43–52. https://doi.org/10.2147/DDDT.S93545.
Tysnes, O. B., & Storstein, A. (2017). Epidemiology of Parkinson’s disease. Journal of Neural Transmission (Vienna, Austria : 1996), 124(8), 901–905. https://doi.org/10.1007/s00702-017-1686-y.
Vila, M., Jackson-Lewis, V., Guégan, C., Wu, C., Teismann, D., Choi, P., D. K., et al. (2001). The role of glial cells in Parkinson’s disease. Current Opinion in Neurology, 14(4), 483–489. https://doi.org/10.1097/00019052-200108000-00009.
Wager, T. T., Chandrasekaran, R. Y., Hou, X., Troutman, M. D., Verhoest, P. R., Villalobos, A., & Will, Y. (2010). Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. Acs Chemical Neuroscience, 1(6), 420–434. https://doi.org/10.1021/cn100007x.
Yang, Q. qiao, & Zhou, J. (2019). Neuroinflammation in the central nervous system: Symphony of glial cells. Glia, 67(6), 1017–1035. https://doi.org/10.1002/glia.23571.
Funding
This work was supported by the National Research Foundation of Korea (NRF) (Grant no. NRF-2021R1A2C1010091) and by the 2022 BK21 FOUR Program of Pusan National University.
Author information
Authors and Affiliations
Contributions
Jaehoon Kim: Methodology, Validation, Formal analysis, Investigation, Writing-original draft, Visualization. Seulah Lee: Investigation, Visualization, Validation. Dong Geun Hong: Investigation, Visualization. Seonguk Yang: Investigation, Visualization. Cong So Tran: Resources. Jinsook Kwak: Resources. Min-Ju Kim: Software, Formal analysis, Validation. Thenmozhi Rajarathinam: Formal analysis, Resources. Ki Wung Chung: Data curation, Visualization. Young-Suk Jung: Data curation, Visualization. Akihito Ishigami: Data curation, Visualization. Seung-Cheol Chang: Data curation, Visualization. Haeseung Lee: Software, Data curation, Resources. Hwayoung Yun: Data curation, Resources. Jaewon Lee: Conceptualization, Writing-review & editing, Supervision, Project administration, Funding acquisition.
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no potential conflict of interest to declare.
Ethics Approval
The animal protocol employed in this study underwent prior review and received approval from the Animal Care Committee of Pusan National University Institutional Animal Care Committee (PNU-IACUC) under approval number PNU-2021-0232.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kim, J., Lee, S., Hong, D.G. et al. Amelioration of Astrocyte-Mediated Neuroinflammation by EI-16004 Confers Neuroprotection in an MPTP-induced Parkinson’s Disease Model. Neuromol Med 26, 1 (2024). https://doi.org/10.1007/s12017-023-08769-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12017-023-08769-8