
Abstract
One class of molecules that are now coming to be recognized as essential for our understanding of the nervous system are the lysophospholipids. One of the major signaling lysophospholipids is lysophosphatidic acid, also known as LPA. LPA activates a variety of G protein-coupled receptors (GPCRs) leading to a multitude of physiological responses. In this review, I describe our current understanding of the role of LPA and LPA receptor signaling in the development and function of the nervous system, especially the central nervous system (CNS). In addition, I highlight how aberrant LPA receptor signaling may underlie neuropathological conditions, with important clinical application.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The human brain is an incredibly complex organ, with almost 100 billion neurons making a 100 trillion connections and an almost equal number of glial cells (Azevedo et al. 2009). Furthermore, the brain is not a static organ, but is undergoing cellular remodeling and synaptic modulation, which is the basis of our complex learning and memory. Even more amazing is that the brain develops de novo in every organism, ultimately from a single cell, the fertilized egg. The process of brain development and function requires the complex role of a large molecular repertoire. However, one signaling molecule that we are just beginning to learn its role, and which our understanding of that role is rapidly expanding, is the bioactive lipid lysophosphatidic acid (LPA).
Lysophosphatidic acid (LPA), also known as mono-acyl-sn-glycerol-3-phosphate, is a lysophospholipid that has a phosphoglycerol head group with a single fatty acid moiety. It is not a single chemical entity but represents a class of biological molecules with different fatty acid chain length and degrees of saturation. A variety of these different molecules are expressed biologically at different levels in different tissues, including brain (Sugiura et al. 1999; Yung et al. 2014), but one of the most abundant species is 18:1 oleoyl-LPA (1-acyl-2-hydroxy-sn-glycero-3-phosphate), which is also the primary species in laboratory use. The highest levels of LPA are produced by platelets during clotting (Eichholtz et al. 1993), which can reach 10–15 µM in serum (Yung et al. 2014). However, levels in plasma are lower, generally less than 1 µM, and even lower in cerebrospinal fluid (CSF); furthermore, tissue levels of LPA under normal physiological conditions are generally quite low, in the nanomolar to tens or low hundreds nanomolar range (Yung et al. 2014).
LPA is produced enzymatically primarily by two pathways, although there may be other biological mechanisms (Aoki et al. 2008, 2002; Pages et al. 2001). LPA can be produced from phosphatidic acid (PA) by phospholipase A (PLA)-type enzymes (Aoki et al. 2008). However, the majority of LPA is produced from lysophosphatidylcholine (LPC) by the action of the enzyme autotaxin, a secreted lysophospholipase D (Dennis et al. 2005; Perrakis and Moolenaar 2014; Herr et al. 2020), which is encoded by the ectonucleotide pyrophosphatase/phosphodiesterase 2 (Enpp2) gene. Mice heterozygous for Enpp2 (autotaxin) produce half the levels of LPA indicating that autotaxin activity is the major pathway of LPA production (van Meeteren et al. 2006; Fotopoulou et al. 2010). LPA production by autotaxin is essential in development as shown by early embryonic lethality of Enpp2/autotaxin null mice with major vascular and neural tube closure defects (Tanaka et al. 2006; van Meeteren et al. 2006). However, interestingly, autotaxin appears to be dispensable in adult as conditional genetic deletion of autotaxin in adult mice yielded animals that were viable without major complications, although LPA levels were reduced by 80% (Katsifa et al. 2015). The mechanism of action of autotaxin, as well as its role in development, physiology, and disease, including the CNS, has been reviewed elsewhere (Herr et al. 2020; Perrakis and Moolenaar 2014; van Meeteren and Moolenaar 2007).
LPA mediates its effects primarily by activating six known G protein-coupled receptors (GPCRs), with the protein products named LPA1 to LPA6 and with gene names LPAR1 to LPAR6 in humans and Lpar1 to Lpar6 in other species (Kihara et al. 2014), although historically other names have been used (see Table 1). LPA binds to and activates its receptors with high affinity, with binding constants and functional measurements in the nanomolar range, with the exception of LPA6 in which a higher functional activation (and no measurable binding constant) has been reported (Table 2). Furthermore, the different LPA receptors have different preferences among the different LPA types (chain-length and saturation) for binding as well as functional activation (Bandoh et al. 2000; Yanagida et al. 2009; Ray et al. 2020).
LPA receptors are classic seven-transmembrane GPCRs that activate heterotrimeric G proteins to transduce signals intracellularly. LPA receptors activate all four major classes of G alpha G protein subunits: pertussis-toxin sensitive Gi/o, Gs activation of adenylyl cyclase, Gq and intracellular calcium increase, and G12/13 mediated Rho/ROCK activation (Choi et al. 2010; Kihara et al. 2014; Yung et al. 2014). However, although the different LPA receptors activate multiple pathways, they do not each activate all pathways and the pathways are differentially activated by the different receptors (see Table 2).
Much of the initial work determining the physiological role of LPA has been done in vitro in cell culture systems, with a wide range of LPA concentrations, although some concentrations may not be physiological. Note that cell culture experiments can be complicated by the fact that there are high levels of LPA in serum, and many cells are cultured in 10% fetal bovine serum. More recently, there are now genetic null knockout mice for each of the LPA receptors. These have been valuable tools, as all are viable, although Lpar1 and Lpar4 null mice show partially penetrant lethality (Table 1). Furthermore, pharmacological antagonists of LPA receptors have been developed (see Archbold et al. 2014; Herr et al. 2020; Yung et al. 2014), although there are issues of specificity and aqueous solubility. Some of these have been very useful tools; the antagonist Ki16425, which blocks both receptors LPA1 and LPA3 (Ohta et al. 2003), has been used extensively, and a new LPA1-specific antagonist, AM095 (Swaney et al. 2011), is now also being used.
In addition to these validated and well-characterized LPA receptors, other GPCRs have been reported to respond to LPA (Tabata et al. 2007; Murakami et al. 2008; Kaya et al. 2020), although they have not been independently validated. Interestingly, LPA has been shown to bind to the TRPV1 channel and activate it (Nieto-Posadas et al. 2012; Canul-Sanchez et al. 2018). In addition, there are a family of putative LPA-interacting proteins, the Plasticity-Related Gene (PRG) family that appear to have a role in LPA signaling, although that role has not been clearly delineated (Brauer et al. 2003; Trimbuch et al. 2009; Strauss and Brauer 2013; Cheng et al. 2016; Vogt et al. 2016). There are five known PRG genes, PRG-1 to PRG-5, and by homology they are members of the lipid phosphate phosphatase (LPP) superfamily. Phosphatase activity toward LPA has been demonstrated for PRG-1 (Brauer et al. 2003), although less than other LPPs (Strauss and Brauer 2013); however, it is not clear if this phosphatase activity is essential to their function, although mutations in this domain can affect function. Interestingly, the effects of a PRG-1 null mutation were reversed by an Lpar2 null mutation (Trimbuch et al. 2009), and inhibition of the LPA producing enzyme autotaxin has been shown to correct the defects seen in PRG-1 heterozygous mice (Vogt et al. 2016). Thus, PRGs may have an effect on LPA signaling, including at the synapse, but that role is not well defined and may not be direct (Brosig et al. 2019; Strauss and Brauer 2013).
LPA and LPA receptor signaling have now been found to be connected with a wide variety of physiological and pathophysiological conditions, and this review does not attempt to cover them all. One major area of focus that will not be covered has been oncology, where LPA has been shown to promote cancer cell proliferation and migration (Benesch et al. 2018; Tigyi et al. 2019; Xu 2019; Lee et al. 2020). This review will focus on the roles of LPA and LPA receptor signaling in the development and function of the brain and central nervous system (CNS) as well as highlighting LPA’s role in neuropathological conditions. The physiological and pathophysiological role of LPA in a variety of other systems is beyond this review, and the reader is referred to other recent reviews (Choi et al. 2010; Choi and Chun 2013; Yung et al. 2014, 2015; Herr et al. 2020).
Brain Development
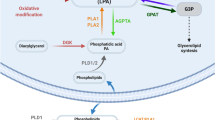
The importance of LPA in development (see Fig. 1) is most clearly delineated by targeted deletions in the autotaxin (Enpp2) gene, which eliminates most LPA production. Autotaxin null mice die during early embryonic development primarily due to vascular defects (van Meeteren et al. 2006; Fotopoulou et al. 2010; Koike et al. 2011). In addition, though, these autotaxin deficient mouse embryos fail to close the cranial neural tube, which forms the future brain, as well as possessing an abnormal “kinky” neural tube phenotype with increased neural tube apoptosis and decreased mitosis (van Meeteren et al. 2006; Fotopoulou et al. 2010; Koike et al. 2011). Examination of genetic markers further shows a requirement for autotaxin in establishing the midbrain-hindbrain boundary (Koike et al. 2011). This is similar to experiments using RNAi against autotaxin in chick in which autotaxin is required for proper formation of the diencephalon-mesencephalon boundary, with its loss perturbing regional identity genes (Ohuchi et al. 2010). Autotaxin is expressed in a variety of regions of the developing neural tube (Ohuchi et al. 2007; Fotopoulou et al. 2010), suggesting even further requirements for LPA in brain development that may not be seen in the knockout embryos due to early death.

Neurodevelopmental roles of LPA. Various aspects of CNS development are likely to be involved in LPA signaling through LPA receptors. a Based on autotaxin genetic null mice, LPA signaling is required for cranial neural tube closure. This involves multiple LPA receptors, with no specific LPA receptor null mice recapitulating this phenotype. Additionally, regionalization of the neural tube at the midbrain/hindbrain border requires LPA, but again the receptors have not yet been identified. b Various experiments demonstrate that LPA signaling through LPA1 and LPA2 are involved in cortical layer formation. Initial studies suggest that LPA1 is important for neuroprogenitor survival in the ventricular zone (VZ) and LPA2 later in their differentiation. Furthermore, LPA4 may also be involved in the migration of early cortical neurons to the layers of the cortical plate. c LPA has properties suggesting it could be guiding axons to their correct targets during development. LPA is repulsive to axonal growth cones and can cause them to collapse through a G12/13-Rho-ROCK pathway. However, the LPA receptors mediating these growth cone responses have not been elucidated. d Studies from genetic null mice, especially Lpar1 null animals, indicates a role for LPA in proper synaptic transmission, especially for glutamatergic synapses, and that could be developmental in origin. Lpar1 null mice show changes in glutamate, serotonin and GABA and a deficit in prepulse inhibition. Hippocampal CA1 pyramidal cells have more immature dendritic spines and reduced MMP-9 in Lpar1 null mice. LPA and LPA1 appear to be involved both presynaptically and postsynaptically
As might be expected, various LPA receptors are expressed in the developing as well as adult brain, and research is beginning to delineate some of their roles. The receptors LPA1, LPA2, LPA4, and LPA5 have been shown to be expressed in the early neural tube by in situ hybridization (Ohuchi et al. 2008). As specific brain regions form, LPA1, LPA2, LPA4, and LPA6 are found in developing and adult mouse neocortex, hippocampus, cerebellum, as well as the olfactory bulb (Suckau et al. 2019). The multiple receptors seem to have redundant functions, as genetic deletions of individual receptors do not show the severe early neural tube developmental defects that autotaxin deletion does. However, early perturbation of Lpar6 expression in Xenopus leads to defects in forebrain development, including reduced expression of telencephalon genes (Geach et al. 2014).
Neural Progenitors and Cortical Development
Based on known properties of LPA for cell proliferation and migration, roles of LPA have been found in neural progenitor proliferation, survival, and differentiation during early brain formation, especially the cortex. Much of the work has used LPA for in vitro treatment of cells. LPA induces changes in morphology of neuroblasts in culture (Fukushima et al. 2000, 2002). LPA has also been shown to increase rosette size from human neuroepithelial stem cells (Medelnik et al. 2018). In murine neurosphere cultures, LPA treatment leads to increased neuronal differentiation, which is blocked by the LPA1/3 antagonist Ki16425 (Fukushima et al. 2007). One of the more intriguing effects of LPA on cortical development is seen in ex vivo cortical cultures where LPA treatment leads to a thicker cortex and increased folding similar to gyri (Kingsbury et al. 2003). The thicker cortex was due to higher cell numbers that were related to reduced ventricular zone apoptosis, indicating greater survival of the neural progenitor cells, as well as increased mitosis and cell cycle exit, suggesting greater differentiation and migration.
The evidence for LPA signaling in neural progenitor cells in cortical development has been further strengthened by analysis of the LPA receptors, especially LPA1. Indeed, the first receptor to be identified as an LPA receptor, LPA1, was originally named vzg-1 as it was isolated from a ventricular zone neuroblast cell line (Hecht et al. 1996). LPA1 was found to be highly expressed in the ventricular zone of the cortex where the neural stem cells reside before differentiation and migration to the various cortical layers. The LPA-induced thickening of the cortex in the ex vivo cultures is blocked if the cultures are obtained from embryos with deletions in both Lpar1 and Lpar2 (Kingsbury et al. 2003), demonstrating the importance of these receptors in mediating the LPA response.
However, the in vivo role of LPA receptors in cortical neural progenitor development and differentiation is less clear. In the original Lpar1 knockout and Lpar1/Lpar2 double knockout mice, there were no obvious differences in cortical development, including cortical thickness, cell counts, and proliferation (Contos et al. 2000, 2002). However, in a spontaneously derived variant of the original Lpar1 knockout line, termed the “Mãlaga variant” (or maLPA1-null), which had negligible perinatal lethality, cortical abnormalities were seen (Estivill-Torrus et al. 2008). Loss of LPA signaling through LPA1 in this mouse showed reduced cortical layers due to increased apoptosis and reduced neural progenitor cell proliferation due to early and aberrant differentiation of these progenitors. Intriguingly, in this same maLPA1-null mouse, in adult there is reduced neurogenesis in the hippocampus, especially when adult neurogenesis is enhanced through exercise and an enriched environment (Matas-Rico et al. 2008). However, the hippocampal dentate gyrus appears normal in these mice, suggesting that any developmental defect in the hippocampus was compensated. These studies indicate that LPA signaling through LPA1 can play an important role in neurogenesis, but there are other mechanisms that can compensate for loss of LPA1 in different strains or conditions. In addition, a potential role for LPA4 has been found in cortical neuron cultures treated with a Lpar4 shRNA that showed impaired transition to the bipolar morphology and disrupted radial glial migration (Kurabayashi et al. 2018).
Axon Outgrowth and Guidance
There is also a possible role for LPA in neural development in the processes of axon outgrowth and guidance to the proper targets. One of the earliest known effects of LPA on neurons is the demonstration that LPA, when applied uniformly to cultured neurons, causes a collapse of the growing tip of the axon, the growth cone, and then neurite retraction. This growth cone collapse and neurite retraction was seen in neuroblastoma and PC12 cell lines (Jalink et al. 1993; Tigyi et al. 1996a) as well as a variety of primary neuron cultures (Saito 1997; Campbell and Holt 2001; Sayas et al. 2002; Birgbauer and Chun 2010; Fincher et al. 2014). The effects of LPA have been demonstrated to proceed through signaling of Gα12/13 via Rho and ROCK to activate changes in the growth cone microfilament cytoskeleton (Jalink et al. 1994; Tigyi et al. 1996b; Kozma et al. 1997; Hirose et al. 1998; Kranenburg et al. 1999; Bito et al. 2000; Sayas et al. 2002; Yamazaki et al. 2008; Fincher et al. 2014), which also appears to involve a requirement for the proteasome (Campbell and Holt 2001, 2003). The roles of specific LPA receptors, however, have not been delineated. Analysis of retinal neuron cultures from Lpar1, Lpar2, and Lpar3 triple-null mice still showed a similar growth cone collapse response to LPA (Birgbauer and Chun 2010), suggesting extensive redundancy.
This property of LPA to cause growth cone collapse is intriguing as validated repulsive axon guidance cues were initially discovered by causing growth cone collapse in vitro, including the ephrins, semaphorins, etc. (see Kolodkin and Tessier-Lavigne 2011; Stoeckli 2017; Herrera et al. 2019). Indeed, this growth cone collapse in vitro, when confronted with a uniform pulse of a cue, is a simple assay for an axon guidance molecule (Kapfhammer and Raper 1987; Cox et al. 1990; Davies et al. 1990; Raper and Kapfhammer 1990; Luo et al. 1993); although artificial, this in vitro assay may mimic the in vivo encounter of a repulsive guidance cue on one edge of the growth cone, causing that edge to collapse and the growth cone to turn away from that region (Kapfhammer and Raper 1987; Fan and Raper 1995). However, growth cone collapse in this in vitro system does not define axon guidance, and further validation needs to be done in vivo. In this area, significant work is yet required to demonstrate that LPA serves as an axon guidance cue. This has been hindered by LPA receptor redundancy (as illustrated above) and viability of autotaxin null mice.
However, there are some suggestions that lysophospholipids, including LPA, may be involved in axon guidance. In Xenopus, the related signaling lysophospholipid S1P has been shown to be involved in retinal axon growth into the tectum. In an exposed brain preparation, application of S1P receptor agonists or antagonists perturbed entry of retinal ganglion cell axons into the tectum (Strochlic et al. 2008). Furthermore, another study suggests that LPA may be involved in guidance of thalamic axons to the cortex based on analysis of PRG-2 null mice (Cheng et al. 2016). In this study, embryonic thalamic axons grew aberrantly into the cortical plate in PRG-2 null animals, which was mimicked by autotaxin inhibitor treatment of the cortical plate. This led to imprecise innervation of the whisker barrels seen in adult animals. PRG-2 is a member by homology of the lipid phosphate phosphatase (LPP) superfamily, but the exact mechanism and relationship to LPA is not clear, and may not be direct (Brosig et al. 2019). In addition, other studies suggest the PRG genes PRG-1, PRG-3, and PRG-5 may be involved in axonal growth and retraction (Brauer et al. 2003; Broggini et al. 2010, 2016), although they may not be specific for LPA effects (Broggini et al. 2010, 2016).
Although much work has suggested that LPA inhibits axon outgrowth, there is a suggestion that LPA could induce neurite branching through LPA3, which is the one LPA receptor that does not signal through Rho/ROCK, and may signal through the novel GTPase Rnd2 (Furuta et al. 2012).
Glial Cells
In addition to effects on neurons and neural precursors, LPA may have effects on glial cell development. Oligodendrocytes, the myelinating cells of the CNS, and their precursor cells have been shown to express LPA receptors, especially LPA1, both in vitro and in vivo (Weiner et al. 1998; Handford et al. 2001; Cervera et al. 2002; Stankoff et al. 2002); they also respond to LPA in vitro (Moller et al. 1999; Yu et al. 2004). In general, no morphological effects on oligodendrocyte differentiation in vitro have been observed (Stankoff et al. 2002), but if endogenous autotaxin is inhibited, then LPA stimulates oligodendrocyte differentiation and myelin formation (Nogaroli et al. 2009). There are also changes in oligodendrocyte gene expression upon autotaxin inhibition, which appear to be mediated by the activity of histone deacetylases (Wheeler et al. 2015). In vivo, the effects of LPA signaling are less well characterized. None of the LPA receptor null mice show obvious oligodendrocyte or myelination defects under normal developmental conditions. However, in zebrafish, knockdown of autotaxin delayed or inhibited the differentiation of oligodendrocyte precursor cells in the hindbrain as determined by genetic markers (Yuelling et al. 2012).
In the PNS, Schwann cells are responsible for myelination, and they too express LPA receptors and are affected by LPA. Early experiments demonstrated that LPA enhances Schwann cell survival in vitro (Weiner and Chun 1999) and affects Schwann cell morphology (Weiner et al. 2001). In the Lpar1 null mouse, there was increased apoptosis of Schwann cells in the sciatic nerve, but no gross myelination defects with a majority of Schwann cells still intact (Contos et al. 2000). However, further analysis of the Lpar1 null mouse found thinner myelin around the sciatic nerve, and in culture these Lpar1 null Schwann cells had reduced migration in response to LPA (Anliker et al. 2013). Furthermore, in adult, analogous to a nerve injury model, LPA causes demyelination of the spinal dorsal root, which is myelinated by Schwann cells, in an Lpar1-dependent manner (Inoue et al. 2004; Nagai et al. 2010; Tsukahara and Ueda 2016).
Astrocytes also respond to LPA with a variety of signaling effects, including inhibition of glutamate uptake, although some of these responses vary depending on source and culture conditions (see Steiner et al. 2002). Astrocytes express LPA receptors LPA1 through LPA5, although LPA5 is barely detectable, and in vitro differentiation by DBcAMP changes the expression levels (Shano et al. 2008). Interestingly, LPA treatment of astrocytes in vitro produces a conditioned medium that promotes cortical neuron differentiation and neurite outgrowth (de Sampaio e Spohr et al. 2008, 2011). One of the components of this conditioned medium is laminin (de Sampaio e Spohr et al. 2011), which is well known to promote neurite outgrowth. This LPA effect on astrocytes is mediated by LPA1 and LPA2 on the astrocytes (de Sampaio e Spohr et al. 2008).
Microglial cells are the immune cells of the CNS and are responsible for clearance of debris and foreign material as well as mediating inflammation. Just as LPA and LPA receptors are significant in the immune system (Benesch et al. 2018; Choi et al. 2010; Herr et al. 2020), LPA and LPA receptors are important in microglial cell activation. Various LPA receptors are expressed on microglial cells, especially LPA1, LPA3, and LPA5, but the exact repertoire of these receptors varies with microglial source, culture conditions, and activation (Moller et al. 2001; Tham et al. 2003; Fujita et al. 2008; Plastira et al. 2016).
Neurodevelopmental Deficits
There are a number of deficits discovered in LPA receptor null animals that likely result from neurodevelopmental abnormalities and suggest roles for LPA and LPA receptor signaling in brain development. Many of these are related to glutamate signaling, an important brain signaling mechanism, especially for learning and memory (see Roza et al. 2019).
A series of studies demonstrate that Lpar1 null mice show abnormalities related to schizophrenia. These include the classic deficit in prepulse inhibition (Harrison et al. 2003) and changes in serotonin, glutamate, and GABA (Harrison et al. 2003; Roberts et al. 2005) as well as changes in hippocampal CaMKII and presynaptic SNARE complexes (Musazzi et al. 2011). Other behavioral characteristics similar to schizophrenia have also been noted in Lpar1 null mice (Castilla-Ortega et al. 2010). Interestingly, and seemingly contradictory, prenatal exposure of mice to LPA by intraventricular administration produced schizophrenia-like behavior such as prepulse inhibition, increased anxiety, reduced locomotor activity, and changes in genetic markers, which could be blocked in Lpar1 null mice or by co-administering the LPA1/3 antagonist Ki16425 (Mirendil et al. 2015). As schizophrenia is considered to be a neurodevelopmental disorder (Birnbaum and Weinberger 2017; Jaaro-Peled and Sawa 2020), these studies suggest deficits in LPA signaling through LPA1 lead to abnormalities of brain development. However, in a ketamine model of schizophrenia, direct treatment with autotaxin inhibitors reversed the schizophrenia-like symptoms, suggesting an acute role for LPA in this animal model (Thalman et al. 2018).
There is also evidence that LPA and LPA receptor signaling are required for development of mature synaptic connections, especially glutamatergic synapses (see Roza et al. 2019). Overexpression of LPA1 in cultured hippocampal neurons results in altered, likely more immature, dendritic spines, although it was independent of LPA signaling (Pilpel and Segal 2006). In Lpar1 null mice, there is a reduction of the major glutaminase isoform, KGA, in the prefrontal and motor cortex, which reduced total glutaminase activity, although there is likely compensation, as the levels of glutamate were similar to wild type (Penalver et al. 2017). Furthermore, in the hippocampus, genetic loss of Lpar1 results in more immature dendritic spines of CA1 pyramidal cells and also reduced matrix metalloproteinase 9 (MMP-9) in the hippocampus (Penalver et al. 2017), and MMP-9 has been shown to be involved in modulating synaptic plasticity (Dziembowska and Wlodarczyk 2012; Reinhard et al. 2015; Beroun et al. 2019). Since the hippocampus has a primary role in learning and memory, this suggests that LPA signaling through LPA1 may have significant neurodevelopmental impacts on synaptic transmission related to learning and memory. Indeed, analysis of Lpar1 null mice have found a variety of behavioral issues, including deficits in spatial learning and memory (Castilla-Ortega et al. 2010; Santin et al. 2009). Interestingly, LPA signaling may also have an acute effect on memory, as injection of LPA into the rat hippocampus after water maze training increased long-term memory as seen 48 h later (Dash et al. 2004). Molecularly, in a hippocampal progenitor cell line, LPA treatment stimulated CREB phosphorylation (Rhee et al. 2006), and CREB phosphorylation is involved in learning and memory, although also important in other physiological and developmental aspects.
Furthermore, Lpar1 null mice show increased anxiety in some, but not all, tests of anxiety (Castilla-Ortega et al. 2010; Santin et al. 2009; Tabbai et al. 2019), suggesting a possible role of LPA signaling in anxiety, although not conclusive and it could be a developmental deficit that is manifested in certain anxiety tests. Alternately, it could be a specific type of anxiety associated with depression (Moreno-Fernandez et al. 2017, 2018). There have also been other behavioral anomalies discovered in Lpar1 null mice, which could be developmental in origin (Santin et al. 2009; Castilla-Ortega et al. 2010; Roza et al. 2019). In addition, in zebrafish, genetic deletion of Lpar3 produced a variety of behavioral deficits, including increased anxiety and reduced short-term memory (Lin et al. 2020).
Although these deficits could be the result of acute requirement for LPA signaling, they could also be developmental in origin. With appropriate pharmacological reagents, the acute versus developmental role of LPA should be further explored, and has been in some instances. Lpar1 null mice display a mixed anxiety-depression phenotype (Moreno-Fernandez et al. 2017, 2018), but that phenotype is only partially recapitulated with intracerebroventricular injection of the LPA1/3 antagonist (Moreno-Fernandez et al. 2018). Although this could be due to partial antagonism, it also suggests anxiety-depression is partially due to developmental defects associated with loss of LPA1 and partially due to acute inhibition of LPA1. This may be relevant to human depression, as the levels of the enzyme autotaxin were reduced in human patients with major depressive disorder and were linked to depression severity (Itagaki et al. 2019), although the absolute levels of autotaxin enzyme may not be the rate limiting factor for LPA production in this case, as measurements of LPA levels did not show any difference in human patients with major depressive disorder (Gotoh et al. 2019) and thus did not correlate with reported changes in autotaxin levels. Thus, the lower levels of autotaxin may relate to reduced glial cells in depression (Wang et al. 2017), which could even be a developmental deficit. On the other hand, some studies have demonstrated direct effects of LPA on synaptic modulation. In CA1 pyramidal hippocampal neurons, LPA enhanced NMDA-evoked currents (Lu et al. 1999). In the hypoglossal motor system, LPA treatment led to depression of glutamate synaptic transmission as well as GABAergic transmission (Garcia-Morales et al. 2015).
Finally, studies of mice with genetic deletion of PRG-1 suggest a role for LPA signaling at excitatory glutamatergic synapses. PRG-1 null mice (Trimbuch et al. 2009) as well as heterozygous mice (Vogt et al. 2016) have deficits in excitatory synaptic transmission, and these are rescued by a Lpar2 homozygous null mutation or autotaxin inhibitors (Trimbuch et al. 2009; Vogt et al. 2016; Thalman et al. 2018).
LPA Signaling in Neuropathological Conditions
Neuropathic Pain
One of the best understood involvement of LPA and LPA receptor signaling is in the pathological condition of neuropathic pain (for other recent reviews, see Ueda 2019; Roza et al. 2019; Velasco et al. 2017; Herr et al. 2020). Neuropathic pain is characterized by an increased sensitivity to pain, which is manifest as hyperalgesia, whereby a mildly painful stimulus produces strong pain, and allodynia, whereby a normally nonpainful stimulus produces pain. Neuropathic pain is often long-lasting and involves a central sensitization. Neuropathic pain can be caused by nerve damage by injury or disease, including cancer and certain chemotherapy treatments. There are many animal models of different types of neuropathic pain (Jaggi et al. 2011), but one common model is the partial sciatic nerve ligation, or PSNL, which causes mechanical and thermal hyperalgesia and allodynia (Seltzer et al. 1990).
The role of LPA receptors in neuropathic pain has been defined by the lack of hyperalgesia and allodynia after partial sciatic nerve ligation in Lpar1 or Lpar3 null mice (Inoue et al. 2004; Ma et al. 2009). This effect was shown to involve acute LPA signaling and not a developmental defect through the use of an siRNA to Lpar1 (Inoue et al. 2004) as well as the LPA1/3 antagonist Ki16425 (Ueda et al. 2018). Furthermore, in autotaxin heterozygous mice, which have a 50% reduction in LPA production, the hyperalgesia and allodynia after PSNL was partially blocked (Inoue et al. 2008a, 2008c; Ma et al. 2009).
There has been significant work on the mechanism of LPA in neuropathic pain and the major players (Fig. 2), although some details are still not determined. The dorsal spinal cord is innervated by two types of pain-signaling fibers in the sciatic nerve: glutamatergic Aδ-fibers and Substance P secreting C-fibers. Intense stimulation of both is required to initiate the neuropathic pain state as inhibition of both the glutaminergic NMDA receptor and Substance P interacting Neurokinin-1 (NK-1) receptors, but neither alone, blocks hyperalgesia and allodynia (Inoue et al. 2008b; Ma et al. 2013; Nagai and Ueda 2011). This intense stimulation acts to produce LPA locally by a feed-forward mechanism involving microglia (Ma et al. 2010a), which results in stimulation of the phospholipase enzymes cytosolic phospholipase A(2), cPLA2, and calcium-independent phospholipase A(2), iPLA2, to produce increased lysophosphatidylcholine (LPC) (Ma et al. 2013, 2010a, 2010b, 2009). The LPC is acted upon by autotaxin (ATX) in the cerebrospinal fluid (CSF) to produce LPA locally that appears to be transported back along the dorsal root (Ma et al. 2013; Ueda 2019). This LPA in the dorsal root leads to demyelination in the dorsal root as well as upregulation of the α2δ1 subunit of the voltage-gated calcium channel in the dorsal root (Inoue et al. 2004; Fujita et al. 2007; Nagai et al. 2010; Xie et al. 2010; Tsukahara and Ueda 2016; Szepanowski et al. 2018). Centrally, protein kinase C γ-isoform (PKCγ) is upregulated and there appears to be neuronal sprouting in the spinal cord dorsal horn (Inoue et al. 2004). The end result is sensitization to pain that could be due to central sensitization from the neuronal sprouting with increased innervation and/or crosstalk (ephapses) between the demyelinated, and thus not insulated, fibers in the dorsal root (Ueda et al. 2013; Ueda 2019; Xie et al. 2008, 2010; Inoue et al. 2004; Fujita et al. 2007; Nagai et al. 2010; Tsukahara and Ueda 2016; Szepanowski et al. 2018; Ohsawa et al. 2013).

Model for the role of LPA in neuropathic pain. In a, the initiation events leading to LPA production are described, while b summarizes the responses to LPA and involvement of LPA in maintenance of the neuropathic pain response. Neuropathic pain development is thought to be initiated by an intense pain response that results in both Aδ fibers releasing glutamate (Glu) to activate NMDA receptors (NMDAR) and C fibers releasing Substance P (SP) to activate Neurokinin-1 (NK-1) receptors in the spinal cord. This may lead to activation of cytosolic phospholipase A(2), cPLA2, and calcium-independent phospholipase A(2), iPLA2, to catalyze the production of lysophosphatidylcholine (LPC) extracellularly which is converted to LPA by autotaxin (ATX) in the CSF. This LPA may bind to microglia, either through LPA1 or LPA3 receptors, which activate the microglia for a feed-forward production of additional LPA, possibly through microglial secretion of Interleukin-1β (IL-1β). In addition, LPA is transported to the dorsal root where it binds to LPA receptors on Schwann cells. As noted in b, LPA activation of Schwann cells causes demyelination in the dorsal root. Furthermore, the α2δ1 subunit of the voltage-gated calcium channel is upregulated in the dorsal root as well as protein kinase C γ-isoform (PKCγ) in the spinal cord. There is proposed to be cross-talk between Aβ and Aδ fibers that result in the severe pain response to innocuous stimuli; in addition, possible sprouting of Aβ and Aδ fibers in the spinal cord could lead to more intense stimulation. Furthermore, there is evidence of the involvement of LPA5 as well as astrocytes and microglia late in the maintenance of the neuropathic pain state. Finally, note that although this model is based on experimental evidence, some aspects, such as cellular locations of LPA receptors, have not been precisely determined yet
Intriguingly, this mechanism, including the hyperalgesia and allodynia, can be recapitulated by intrathecal injection of LPA or LPC into the spinal cord, which also requires microglia and a feed-forward mechanism of increased LPC and LPA production through activation of cPLA2 and iPLA2 and autotaxin, with a requirement for the 18:1 species of LPA (Ma et al. 2009, 2010b, 2013). This LPA-induced neuropathic pain and feed-forward mechanism requires both LPA1 and LPA3 since it is blocked in Lpar1 null mice and in Lpar3 null mice (Ma et al. 2009, 2013). Microglia are essential for this feed-forward mechanism, as well as activation of cPLA2 and iPLA2 to produce LPC for conversion to LPA by autotaxin (Ma et al. 2009, 2013). Production of LPC by cPLA2 and iPLA2 activation appears to occur in the spinal cord dorsal horn neurons since expression is highest there (Ma et al. 2013), but other possibilities cannot be ruled out. Furthermore, although microglia are required, their activation and signaling are not clear. Microglia do possess LPA receptors, although the specific repertoire is not clear. LPA1 has been shown to be expressed on rat microglia, while LPA3 has been shown to be expressed on mouse microglia (Moller et al. 2001; Tham et al. 2003; Fujita et al. 2008). Furthermore, culture conditions and activation can change microglial LPA receptor expression (Tham et al. 2003), so it could change in vivo too. Nonetheless, both LPA1 and LPA3 are required in the mouse model based on knockout mice studies, but which cell type requires them has not been determined. In addition, how microglia are involved is not yet known. Microglia produce various cytokines, including Interleukin-1β (IL-1β), and antibodies against IL-1β reduce neuropathic pain (Yano et al. 2013), which leads to the logical, but not yet proven, hypothesis that microglial IL-1β release induces the feed-forward LPA production and neuropathic pain.
There are other aspects to this pathway that we have gained insights into, but whose mechanism has not yet been defined. In initial experiments, the role of microglia was determined to be required early because early minocycline treatment, but not late (days 2–5) minocycline treatment, blocked the neuropathic pain response (Ma et al. 2010a). However, removal of microglia late, at days 8–12 by the microglial toxin Mac-1 saponin, did block the neuropathic pain (Ueda et al. 2018). Thus, there could be two windows for the requirement of microglia, or these two reagents that target microglia in different ways may have exposed two different requirements. There also appears to be an effect of astrocytes in neuropathic pain development late in the process since treatment with the astrocyte toxin L-AA on days 1–2 after PSNL partially, but significantly, reversed the hyperalgesia, but not the feed-forward LPA production (Ueda et al. 2018). This astrocyte activation appears to depend on LPA receptor signaling, as astrocyte cytokine upregulation and release is inhibited by the LPA1/3 antagonist Ki16425 (Ueda et al. 2018). Interestingly, although initial experiments confirmed the requirement of LPA receptors early in the development of neuropathic pain, repeated injections of the LPA1/3 antagonist Ki16425 late, at days 8–14, but not a single injection, reduced the neuropathic pain, although possibly not completely blocked by this late treatment (Ueda et al. 2018).
There has also been a role demonstrated for LPA5 in neuropathic pain, although the mechanism appears to be different. Lpar5 null mice do not develop mechanical allodynia after PSNL (Lin et al. 2012). However, unlike Lpar1 null and Lpar3 null mice, Lpar5 mice still showed demyelination of the dorsal root and upregulation of α2δ1 subunit of the voltage-gated calcium channel and astrocyte activation after PSNL (Lin et al. 2012). Lpar5 null mice did, though, show reduced phosphorylated CREB, indicative of a different pathway. Furthermore, in other models of neuropathic pain, Lpar5 null mice showed reduced cold allodynia in an acetone challenge test after a chronic constriction injury (CCI), but not mechanical allodynia, nor did they show any difference in a spared nerve injury (SNI) neuropathic pain model (Callaerts-Vegh et al. 2012). These data suggest that LPA5 is also involved in neuropathic pain, but in a quite distinct mechanism from LPA1 and LPA3.
Although much of the work on the mechanism of LPA and LPA receptor signaling in neuropathic pain has been done on the PSNL model, that is not the only model of neuropathic pain (Jaggi et al. 2011), and human neuropathic pain has many etiologies. Recent work has highlighted the role of LPA and LPA receptors in other neuropathic pain models. LPA1 and LPA3 are required in a late tissue plasminogen activator-induced central poststroke pain (Ueda et al. 2019). Human patients with osteoarthritis show elevated LPA levels, and blocking LPA1 and LPA3 with the antagonist Ki16425 reduced the pain and nerve damage in an animal model of osteoarthritis (McDougall et al. 2017). There is also evidence of LPA1 involvement in an animal model for fibromyalgia-like pain (Ueda 2019). In addition, in a clinically important chemotherapy model of paclitaxel-induced neuropathic pain, the allodynia was blocked in Lpar1 null and Lpar3 null mice, demonstrating a similar role in chemically induced neuropathic pain (Uchida et al. 2014). Even in an inflammatory orofacial pain model, Lpar1 null mice or treatment with the LPA1 antagonist AM095 reduced the pain, although it did not completely block it (Srikanth et al. 2018). Furthermore, demonstrating the clinical relevance of LPA in neuropathic pain, there was an association of higher LPA levels in CSF from human patients with neuropathic pain, and the level correlated with pain intensity (Kuwajima et al. 2018); however, there was no increase in autotaxin levels, again suggesting that autotaxin levels may not be the determining factor. Thus, in a variety of models and correlations in human patients, LPA and LPA receptor signaling are mechanistically involved in neuropathic pain.
Brain and Spinal Cord Injury
Recent work has implicated LPA in traumatic brain injury and spinal cord injury, both scenarios where the blood–brain barrier is damaged that could lead to a flood of LPA from serum. After traumatic brain injury in rat, autotaxin levels are highly upregulated after 2 days (Savaskan et al. 2007). However, in human patients, no difference in autotaxin or LPA receptor expression was seen immediately after traumatic brain injury, although there was reduced autotaxin levels later as well as increased LPAR2 expression (Frugier et al. 2011). On the other hand, LPA levels increased dramatically in human patients within the first 24 h after injury (Crack et al. 2014). Even more exciting, in a mouse model of mild traumatic brain injury, treatment with a monoclonal antibody against LPA (Lpathomab) significantly reduced lesion volume and cytokine levels and led to improved behavioral outcome (Crack et al. 2014).
Spinal cord injury is another significant medical problem with minimal treatment options, and recent results suggest a role for LPA signaling in this situation. Lpar2 and Lpar3 expression were upregulated in a mouse model of spinal cord injury (Goldshmit et al. 2010). Similar to neuropathic pain, spinal cord injury leads to demyelination, and LPA injection also induces demyelination (Santos-Nogueira et al. 2015). This demyelination is partially blocked in Lpar1 null mice or by application of the LPA1 antagonist AM095 (Santos-Nogueira et al. 2015). The LPA1 antagonist also has a small, but significant, effect on prevention of demyelination and recovery of function after spinal cord injury. LPA2 has also been implicated, as demyelination is partially reduced in Lpar2 null mice, which also show slightly improved functional recovery (Lopez-Serrano et al. 2019). These results have been partially linked to microglia, as LPA-activated microglia released ATP leading to oligodendrocyte death (Santos-Nogueira et al. 2015; Lopez-Serrano et al. 2019). Furthermore, in a different model, treatment with a LPA1 antagonist led to enhanced corticospinal tract sprouting after spinal cord injury (Fink et al. 2017), again suggesting a role for LPA and LPA receptors in spinal cord injury.
Stroke and Cerebral Ischemia
There is evidence supporting a role for LPA and LPA receptors in neuroinflammation following stroke and cerebral ischemia. Levels of LPA are increased in the plasma of human stroke patients (Li et al. 2008, 2010), most likely due to platelet activation from thrombosis seen in stroke (Fisher and Francis 1990). Reperfusion after stroke would then lead to high levels of LPA in the brain at the ischemic site. Recent work shows that the rodent model of stroke, a transient middle cerebral artery occlusion (tMCAO), which produces a transient focal cerebral ischemia, leads to increased brain levels of LPA (Wang et al. 2018; Zeng et al. 2020), and this is mostly blocked by an autotaxin inhibitor (Wang et al. 2018; Zeng et al. 2020). High levels of LPA have been shown to induce apoptosis in cortical neurons in vitro (Wang et al. 2018), although an effect of high lipids was not ruled out. In rats, intracerebroventricular injection of LPA leads to neurological damage (Zeng et al. 2020). In the tMCAO model, addition of exogenous LPA leads to a greater infarct size after reperfusion (Chi et al. 2020; Weiss et al. 2020). Significantly, the infarct size and apoptosis after tMCAO (without exogenous LPA added) was reduced (although not eliminated) by an autotaxin inhibitor (Wang et al. 2018; Zeng et al. 2020), suggesting an important role for LPA in cerebral ischemia and reperfusion. Both LPA1 and LPA5 appear to be involved, as treatment with the LPA antagonist AM095 or an shRNA against LPA1 (Gaire et al. 2019), or the LPA5 antagonist TCLPA5 (Sapkota et al. 2020), at the time of reperfusion significantly reduced damage, including infarct size, apoptosis, and neurological deficit, although the specificity of TCLPA5 does not appear to have been extensively tested (Kozian et al. 2012). Significantly, even treatment with TCLPA5 three hours after reperfusion reduced damage (Sapkota et al. 2020), suggesting important clinical application.
The role of LPA in cerebral ischemia appears to be related to neuroinflammation. LPA is known to activate microglia to a proinflammatory state (Ma et al. 2010a; Fujita et al. 2008; Plastira et al. 2016, 2017) Multiple LPA receptors are expressed in microglia (Tham et al. 2003; Fujita et al. 2008; Plastira et al. 2016) and may be involved. For instance, inhibition of LPA5 by TCLPA5 reduced various aspects of microglia activation by LPA (Plastira et al. 2016, 2017; Sapkota et al. 2020). In cerebral ischemia and reperfusion, there is significant neuroinflammation as seen by microglial activation and well as astrogliosis. After tMCAO, administration of an autotaxin inhibitor reduced microglial activation and release of proinflammatory cytokines (Zeng et al. 2020). Furthermore, inhibition of LPA1 by AM095 or shRNA treatment also reduced microglial activation and proinflammatory cytokines as well as astrogliosis (Gaire et al. 2019). In addition, LPA5 appears to be involved, as TCLPA5 treatment also reduced microglial activation and proinflammatory cytokines, although astrogliosis was not examined (Sapkota et al. 2020). Thus, LPA through LPA1 and LPA5 appears to be a major player in neuroinflammation produced by cerebral ischemia, and modulation of LPA or LPA receptors could be a promising treatment for stroke.
Multiple Sclerosis and Other Disorders
There are other neuropathological disorders that could be related to LPA. In multiple sclerosis (MS), it is thought that an autoimmune response leads to demyelination and eventually axonal damage. As mentioned above, LPA can cause demyelination, including oligodendrocyte death indirectly. Interestingly, LPA levels were reduced in serum, but not CSF, of multiple sclerosis patients as well as in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS (Schmitz et al. 2017). Furthermore, Lpar2 null mice have worse symptoms in EAE, while an LPA2 agonist improved outcomes from EAE in wild type mice (Schmitz et al. 2017); however, these effects were most likely related to the immune system and T-cell homing. In another neuropathological disorder, in a model of posthemorrhagic hydrocephalus, injection of LPA into the ventricle of embryonic mice kills ependymal cells and results in hydrocephalus, which is partially reduced in Lpar1 null animals as well as with LPA1 antagonist treatment (Lummis et al. 2019). There is also the suggestion that LPA and LPA receptor signaling may be involved in the pathogenesis of Alzheimer’s Disease based upon the evidence cited above on synaptic transmission and activation of microglia and astrocytes (for reviews, see Ramesh et al. 2018; Hao et al. 2020).
Glaucoma and Diabetic Retinopathy
Another important CNS region is the retina, with vision being a major medical issue. It has been estimated that in 2015, 36 million people worldwide were blind, with 216 million people having moderate to severe visual impairment (Bourne et al. 2017). One major medical problem is glaucoma in which elevated ocular pressure can lead to damage of the optic nerve, and increases in LPA, LPC, and autotaxin have been observed in human glaucoma patients (Honjo et al. 2018; Ho et al. 2020). In an elevated ocular pressure model in rat, the receptors LPA1 and LPA2 are upregulated with increased ocular pressure, and treatment with an LPA receptor agonist reduces the histological damage and leads to improvements in retinal electrophysiology (Savitz et al. 2006). Furthermore, in an in vitro model, an autotaxin inhibitor has been shown to block the fibrosis formed in the trabecular meshwork and to increase the aqueous outflow, which would reduce the intraocular pressure that builds up in glaucoma (Ho et al. 2020). Another major retinal disease is diabetic retinopathy, which often results in retinal ischemia and cell death. The levels of LPA are higher in retinal vitreous samples from diabetic retinopathy patients (Abu El-Asrar et al. 2013). In an oxygen-induced retinopathy model in rats, a shRNA directed to Lpar1 prevented retinal ganglion cell (RGC) loss (Yang et al. 2009). Other older studies found effects of LPA on ion currents in retinal glia and pigmented epithelium (Thoreson et al. 1997; Kusaka et al. 1998).
Conclusion
LPA and LPA receptor signaling have a vast influence on CNS development and physiology, including disease, with many of these effects still being worked out. One of the major tools for these studies are knockout mice for the different LPA receptors, which have been extremely useful, although developmental effects cannot be distinguished from acute requirements. To investigate acute effects, we need pharmacological agents, which are beginning to be developed more extensively. A number of compounds have been developed (see Archbold et al. 2014; Herr et al. 2020; Yung et al. 2014), but due to the lipid nature of LPA, it has been difficult to obtain water-soluble reagents that have receptor specificity and good in vivo pharmacology. There have been a couple of compounds that have been used experimentally, but others are needed. Much of the experimental work has used the LPA1/3 antagonist Ki16425, although it is not specific for a single LPA receptor (Ohta et al. 2003). A newer LPA1 antagonist, AM095, is now being used more in studies (Swaney et al. 2011). There are additional compounds in various stages of development. Due to the medical importance of LPA in various pathological states, some of these compounds are progressing to the clinic. For instance, Bristol-Meyers Squibb has a compound that has gone through phase 2 trials with efficacy for idiopathic pulmonary fibrosis (Palmer et al. 2018; Tager et al. 2008). As research goes forward, we will better understand the roles LPA and LPA receptor signaling have on the brain and its development and function, and how clinically this information can be used to advance medical treatment.
References
Abu El-Asrar, A. M., Mohammad, G., Nawaz, M. I., Siddiquei, M. M., Kangave, D., & Opdenakker, G. (2013). Expression of lysophosphatidic acid, autotaxin and acylglycerol kinase as biomarkers in diabetic retinopathy. Acta Diabetologica, 50(3), 363–371. https://doi.org/10.1007/s00592-012-0422-1
An, S., Bleu, T., Hallmark, O. G., & Goetzl, E. J. (1998). Characterization of a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. Journal of Biological Chemistry, 273(14), 7906–7910.
Anliker, B., Choi, J. W., Lin, M.-E., Gardell, S. E., Rivera, R. R., Kennedy, G., et al. (2013). Lysophosphatidic acid (LPA) and its receptor, LPA1, influence embryonic schwann cell migration, myelination, and cell-to-axon segregation. Glia, 61(12), 2009–2022. https://doi.org/10.1002/glia.22572
Aoki, J., Inoue, A., & Okudaira, S. (2008). Two pathways for lysophosphatidic acid production. Biochimica et Biophysica Acta, 1781(9), 513–518.
Aoki, J., Taira, A., Takanezawa, Y., Kishi, Y., Hama, K., Kishimoto, T., et al. (2002). Serum lysophosphatidic acid is produced through diverse phospholipase pathways. Journal of Biological Chemistry, 277(50), 48737–48744. https://doi.org/10.1074/jbc.M206812200
Archbold, J. K., Martin, J. L., & Sweet, M. J. (2014). Towards selective lysophospholipid GPCR modulators. Trends in Pharmacological Sciences, 35(5), 219–226. https://doi.org/10.1016/j.tips.2014.03.004
Azevedo, F. A., Carvalho, L. R., Grinberg, L. T., Farfel, J. M., Ferretti, R. E., Leite, R. E., et al. (2009). Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. The Journal of Comparative Neurology, 513(5), 532–541. https://doi.org/10.1002/cne.21974
Bandoh, K., Aoki, J., Hosono, H., Kobayashi, S., Kobayashi, T., Murakami-Murofushi, K., et al. (1999). Molecular cloning and characterization of a novel human G-protein-coupled receptor, EDG7, for lysophosphatidic acid. Journal of Biological Chemistry, 274(39), 27776–27785.
Bandoh, K., Aoki, J., Taira, A., Tsujimoto, M., Arai, H., & Inoue, K. (2000). Lysophosphatidic acid (LPA) receptors of the EDG family are differentially activated by LPA species. Structure-activity relationship of cloned LPA receptors. FEBS Letters, 478(1–2), 159–165. https://doi.org/10.1016/s0014-5793(00)01827-5
Benesch, M. G. K., MacIntyre, I. T. K., McMullen, T. P. W., & Brindley, D. N. (2018). Coming of age for autotaxin and lysophosphatidate signaling: clinical applications for preventing. Detecting and Targeting Tumor-Promoting Inflammation. Cancers (Basel). https://doi.org/10.3390/cancers10030073
Beroun, A., Mitra, S., Michaluk, P., Pijet, B., Stefaniuk, M., & Kaczmarek, L. (2019). MMPs in learning and memory and neuropsychiatric disorders. Cellular and Molecular Life Sciences, 76(16), 3207–3228. https://doi.org/10.1007/s00018-019-03180-8
Birgbauer, E., & Chun, J. (2010). Lysophospholipid receptors LPA1–3 are not required for the inhibitory effects of LPA on mouse retinal growth cones. Eye and Brain, 2010(2), 1–13.
Birnbaum, R., & Weinberger, D. R. (2017). Genetic insights into the neurodevelopmental origins of schizophrenia Nature reviews. Neuroscience, 18(12), 727–740. https://doi.org/10.1038/nrn.2017.125
Bito, H., Furuyashiki, T., Ishihara, H., Shibasaki, Y., Ohashi, K., Mizuno, K., et al. (2000). A critical role for a Rho-associated kinase, p160ROCK, in determining axon outgrowth in mammalian CNS neurons. Neuron, 26(2), 431–441.
Bourne, R. R. A., Flaxman, S. R., Braithwaite, T., Cicinelli, M. V., Das, A., Jonas, J. B., et al. (2017). Magnitude, temporal trends, and projections of the global prevalence of blindness and distance and near vision impairment: A systematic review and meta-analysis. Lancet Glob Health, 5(9), e888–e897. https://doi.org/10.1016/S2214-109X(17)30293-0
Brauer, A. U., Savaskan, N. E., Kuhn, H., Prehn, S., Ninnemann, O., & Nitsch, R. (2003). A new phospholipid phosphatase, PRG-1, is involved in axon growth and regenerative sprouting. Nature Neuroscience, 6(6), 572–578.
Broggini, T., Nitsch, R., & Savaskan, N. E. (2010). Plasticity-related gene 5 (PRG5) induces filopodia and neurite growth and impedes lysophosphatidic acid- and nogo-A-mediated axonal retraction. Molecular Biology of the Cell, 21(4), 521–537. https://doi.org/10.1091/mbc.E09-06-0506
Broggini, T., Schnell, L., Ghoochani, A., Mateos, J. M., Buchfelder, M., Wiendieck, K., et al. (2016). Plasticity Related Gene 3 (PRG3) overcomes myelin-associated growth inhibition and promotes functional recovery after spinal cord injury. Aging (Albany NY), 8(10), 2463–2487. https://doi.org/10.18632/aging.101066
Brosig, A., Fuchs, J., Ipek, F., Kroon, C., Schrotter, S., Vadhvani, M., et al. (2019). The axonal membrane protein PRG2 inhibits PTEN and directs growth to branches. Cell reports, 29(7), 2028–2040. https://doi.org/10.1016/j.celrep.2019.10.039
Callaerts-Vegh, Z., Leo, S., Vermaercke, B., Meert, T., & D’Hooge, R. (2012). LPA5 receptor plays a role in pain sensitivity, emotional exploration and reversal learning. Genes Brain Behav, 11(8), 1009–1019. https://doi.org/10.1111/j.1601-183X.2012.00840.x
Campbell, D. S., & Holt, C. E. (2001). Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron, 32(6), 1013–1026.
Campbell, D. S., & Holt, C. E. (2003). Apoptotic pathway and MAPKs differentially regulate chemotropic responses of retinal growth cones. Neuron, 37(6), 939–952.
Canul-Sanchez, J. A., Hernandez-Araiza, I., Hernandez-Garcia, E., Llorente, I., Morales-Lazaro, S. L., Islas, L. D., et al. (2018). Different agonists induce distinct single-channel conductance states in TRPV1 channels. Journal of General Physiology, 150(12), 1735–1746. https://doi.org/10.1085/jgp.201812141
Castilla-Ortega, E., Sanchez-Lopez, J., Hoyo-Becerra, C., Matas-Rico, E., Zambrana-Infantes, E., Chun, J., et al. (2010). Exploratory, anxiety and spatial memory impairments are dissociated in mice lacking the LPA1 receptor. Neurobiology of Learning and Memory, 94(1), 73–82. https://doi.org/10.1016/j.nlm.2010.04.003
Cervera, P., Tirard, M., Barron, S., Allard, J., Trottier, S., Lacombe, J., et al. (2002). Immunohistological localization of the myelinating cell-specific receptor LP(A1). Glia, 38(2), 126–136.
Cheng, J., Sahani, S., Hausrat, T. J., Yang, J. W., Ji, H., Schmarowski, N., et al. (2016). Precise somatotopic thalamocortical axon guidance depends on LPA-mediated PRG-2/radixin signaling. Neuron, 92(1), 126–142. https://doi.org/10.1016/j.neuron.2016.08.035
Chi, O. Z., Mellender, S. J., Kiss, G. K., Chiricolo, A., Liu, X., Patel, N., et al. (2020). Lysophosphatidic acid increased infarct size in the early stage of cerebral ischemia-reperfusion with increased BBB permeability. Journal of Stroke & Cerebrovascular Diseases, 29(10), 105029. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105029
Choi, J. W., & Chun, J. (2013). Lysophospholipids and their receptors in the central nervous system. Biochimica et Biophysica Acta, 1831(1), 20–32. https://doi.org/10.1016/j.bbalip.2012.07.015
Choi, J. W., Herr, D. R., Noguchi, K., Yung, Y. C., Lee, C.-W., Mutoh, T., et al. (2010). LPA receptors: Subtypes and biological actions. Annual Review of Pharmacology and Toxicology, 50(1), 157–186. https://doi.org/10.1146/annurev.pharmtox.010909.105753
Contos, J. J., Fukushima, N., Weiner, J. A., Kaushal, D., & Chun, J. (2000). Requirement for the lpA1 lysophosphatidic acid receptor gene in normal suckling behavior. Proceedings of the National Academy of Sciences of the United States of America, 97(24), 13384–13389.
Contos, J. J., Ishii, I., Fukushima, N., Kingsbury, M. A., Ye, X., Kawamura, S., et al. (2002). Characterization of lpa(2) (Edg4) and lpa(1)/lpa(2) (Edg2/Edg4) lysophosphatidic acid receptor knockout mice: signaling deficits without obvious phenotypic abnormality attributable to lpa(2). Molecular and Cellular Biology, 22(19), 6921–6929.
Cox, E. C., Muller, B., & Bonhoeffer, F. (1990). Axonal guidance in the chick visual system: posterior tectal membranes induce collapse of growth cones from the temporal retina. Neuron, 4(1), 31–37.
Crack, P. J., Zhang, M., Morganti-Kossmann, M. C., Morris, A. J., Wojciak, J. M., Fleming, J. K., et al. (2014). Anti-lysophosphatidic acid antibodies improve traumatic brain injury outcomes. J Neuroinflammation, 11, 37. https://doi.org/10.1186/1742-2094-11-37
Dash, P. K., Orsi, S. A., Moody, M., & Moore, A. N. (2004). A role for hippocampal Rho-ROCK pathway in long-term spatial memory. Biochemical and Biophysical Research Communications, 322(3), 893–898. https://doi.org/10.1016/j.bbrc.2004.08.004
Davies, J. A., Cook, G. M. W., Stern, C. D., & Keynes, R. J. (1990). Isolation from chick somites of a glycoprotein fraction that causes collapse of dorsal rool ganglion growth cones. Neuron, 2(1), 11.
de Sampaio e Spohr, T. C. L., Dezonne, R. S., Rehen, S. K., & Gomes, F. C. A. . (2011). Astrocytes treated by lysophosphatidic acid induce axonal outgrowth of cortical progenitors through extracellular matrix protein and epidermal growth factor signaling pathway. Journal of Neurochemistry, 119(1), 113–123. https://doi.org/10.1111/j.1471-4159.2011.07421.x
de Sampaio e Spohr, T. C., Choi, J. W., Gardell, S. E., Herr, D. R., Rehen, S. K., Gomes, F. C. A., , et al. (2008). Lysophosphatidic acid receptor-dependent secondary effects via astrocytes promote neuronal differentiation. Journal of Biological Chemistry, 283(12), 7470–7479. https://doi.org/10.1074/jbc.M707758200
Dennis, J., Nogaroli, L., & Fuss, B. (2005). Phosphodiesterase-Ialpha/autotaxin (PD-Ialpha/ATX): A multifunctional protein involved in central nervous system development and disease. Journal of Neuroscience Research, 82(6), 737–742.
Dziembowska, M., & Wlodarczyk, J. (2012). MMP9: A novel function in synaptic plasticity. International Journal of Biochemistry & Cell Biology, 44(5), 709–713. https://doi.org/10.1016/j.biocel.2012.01.023
Eichholtz, T., Jalink, K., Fahrenfort, I., & Moolenaar, W. H. (1993). The bioactive phospholipid lysophosphatidic acid is released from activated platelets. The Biochemical Journal, 291(Pt 3), 677–680.
Estivill-Torrus, G., Llebrez-Zayas, P., Matas-Rico, E., Santin, L., Pedraza, C., De Diego, I., et al. (2008). Absence of LPA1 signaling results in defective cortical development. Cerebral Cortex, 18(4), 938–950. https://doi.org/10.1093/cercor/bhm132
Fan, J., & Raper, J. A. (1995). Localized collapsing cues can steer growth cones without inducing their full collapse. Neuron, 14(2), 263–274.
Fincher, J., Whiteneck, C., & Birgbauer, E. (2014). G-protein-coupled receptor cell signaling pathways mediating embryonic chick retinal growth cone collapse induced by lysophosphatidic acid and sphingosine-1-phosphate. Developmental Neuroscience, 36(6), 443–453. https://doi.org/10.1159/000364858
Fink, K. L., Lopez-Giraldez, F., Kim, I. J., Strittmatter, S. M., & Cafferty, W. B. (2017). Identification of intrinsic axon growth modulators for intact CNS neurons after injury. Cell Reports, 18(11), 2687–2701. https://doi.org/10.1016/j.celrep.2017.02.058
Fisher, M., & Francis, R. (1990). Altered coagulation in cerebral ischemia. Platelet, thrombin, and plasmin activity. Archives of Neurology, 47(10), 1075–1079. https://doi.org/10.1001/archneur.1990.00530100037011
Fotopoulou, S., Oikonomou, N., Grigorieva, E., Nikitopoulou, I., Paparountas, T., Thanassopoulou, A., et al. (2010). ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol., 339(2), 451–464. https://doi.org/10.1016/j.ydbio.2010.01.007
Frugier, T., Crombie, D., Conquest, A., Tjhong, F., Taylor, C., Kulkarni, T., et al. (2011). Modulation of LPA receptor expression in the human brain following neurotrauma. Cellular and Molecular Neurobiology, 31(4), 569–577. https://doi.org/10.1007/s10571-011-9650-0
Fujita, R., Kiguchi, N., & Ueda, H. (2007). LPA-mediated demyelination in ex vivo culture of dorsal root. Neurochemistry International, 50(2), 351–355. https://doi.org/10.1016/j.neuint.2006.09.003
Fujita, R., Ma, Y., & Ueda, H. (2008). Lysophosphatidic acid-induced membrane ruffling and brain-derived neurotrophic factor gene expression are mediated by ATP release in primary microglia. Journal of Neurochemistry, 107(1), 152–160. https://doi.org/10.1111/j.1471-4159.2008.05599.x
Fukushima, N., Shano, S., Moriyama, R., & Chun, J. (2007). Lysophosphatidic acid stimulates neuronal differentiation of cortical neuroblasts through the LPA(1)-G(i/o) pathway. Neurochemistry International, 50(2), 302–307.
Fukushima, N., Weiner, J. A., & Chun, J. (2000). Lysophosphatidic acid (LPA) is a novel extracellular regulator of cortical neuroblast morphology. Dev. Biol., 228(1), 6–18.
Fukushima, N., Weiner, J. A., Kaushal, D., Contos, J. J., Rehen, S. K., Kingsbury, M. A., et al. (2002). Lysophosphatidic acid influences the morphology and motility of young, postmitotic cortical neurons. Molecular and Cellular Neuroscience, 20(2), 271–282.
Furuta, D., Yamane, M., Tsujiuchi, T., Moriyama, R., & Fukushima, N. (2012). Lysophosphatidic acid induces neurite branch formation through LPA3. Molecular and Cellular Neuroscience, 50(1), 21–34. https://doi.org/10.1016/j.mcn.2012.03.006
Gaire, B. P., Sapkota, A., Song, M.-R., & Choi, J. W. (2019). Lysophosphatidic acid receptor 1 (LPA1) plays critical roles in microglial activation and brain damage after transient focal cerebral ischemia. J Neuroinflammation, 16(1), 170. https://doi.org/10.1186/s12974-019-1555-8
Garcia-Morales, V., Montero, F., Gonzalez-Forero, D., Rodriguez-Bey, G., Gomez-Perez, L., Medialdea-Wandossell, M. J., et al. (2015). Membrane-derived phospholipids control synaptic neurotransmission and plasticity. PLoS Biology, 13(5), e1002153. https://doi.org/10.1371/journal.pbio.1002153
Geach, T. J., Faas, L., Devader, C., Gonzalez-Cordero, A., Tabler, J. M., Brunsdon, H., et al. (2014). An essential role for LPA signalling in telencephalon development. Development, 141(4), 940–949. https://doi.org/10.1242/dev.104901
Goldshmit, Y., Munro, K., Leong, S. Y., Pebay, A., & Turnley, A. M. (2010). LPA receptor expression in the central nervous system in health and following injury. Cell and Tissue Research, 341(1), 23–32. https://doi.org/10.1007/s00441-010-0977-5
Gotoh, L., Yamada, M., Hattori, K., Sasayama, D., Noda, T., Yoshida, S., et al. (2019). Lysophosphatidic acid levels in cerebrospinal fluid and plasma samples in patients with major depressive disorder. Heliyon, 5(5), e01699. https://doi.org/10.1016/j.heliyon.2019.e01699
Handford, E. J., Smith, D., Hewson, L., McAllister, G., & Beer, M. S. (2001). Edg2 receptor distribution in adult rat brain. NeuroReport, 12(4), 757–760.
Hao, Y., Guo, M., Feng, Y., Dong, Q., & Cui, M. (2020). Lysophospholipids and their G-coupled protein signaling in Alzheimer’s disease: From physiological performance to pathological impairment. Frontiers in Molecular Neuroscience, 13, 58. https://doi.org/10.3389/fnmol.2020.00058
Harrison, S. M., Reavill, C., Brown, G., Brown, J. T., Cluderay, J. E., Crook, B., et al. (2003). LPA1 receptor-deficient mice have phenotypic changes observed in psychiatric disease. Molecular and Cellular Neuroscience, 24(4), 1170–1179.
Hecht, J. H., Weiner, J. A., Post, S. R., & Chun, J. (1996). Ventricular zone gene-1 (vzg-1) encodes a lysophosphatidic acid receptor expressed in neurogenic regions of the developing cerebral cortex. Journal of Cell Biology, 135(4), 1071–1083.
Herr, D. R., Chew, W. S., Satish, R. L., & Ong, W.-Y. (2020). Pleotropic roles of autotaxin in the nervous system present opportunities for the development of novel therapeutics for neurological diseases. Molecular Neurobiology, 57(1), 372–392. https://doi.org/10.1007/s12035-019-01719-1
Herrera, E., Erskine, L., & Morenilla-Palao, C. (2019). Guidance of retinal axons in mammals. Seminars in Cell & Developmental Biology, 85, 48–59. https://doi.org/10.1016/j.semcdb.2017.11.027
Hirose, M., Ishizaki, T., Watanabe, N., Uehata, M., Kranenburg, O., Moolenaar, W. H., et al. (1998). Molecular dissection of the Rho-associated protein kinase (p160ROCK)- regulated neurite remodeling in neuroblastoma N1E–115 cells. Journal of Cell Biology, 141(7), 1625–1636.
Ho, L. T. Y., Osterwald, A., Ruf, I., Hunziker, D., Mattei, P., Challa, P., et al. (2020). Role of the autotaxin-lysophosphatidic acid axis in glaucoma, aqueous humor drainage and fibrogenic activity. Biochimica et Biophysica, 1866(1), 165560. https://doi.org/10.1016/j.bbadis.2019.165560
Honjo, M., Igarashi, N., Kurano, M., Yatomi, Y., Igarashi, K., Kano, K., et al. (2018). Autotaxin-lysophosphatidic acid pathway in intraocular pressure regulation and glaucoma subtypes. Investigative Ophthalmology & Visual Science, 59(2), 693–701. https://doi.org/10.1167/iovs.17-23218
Inoue, M., Ma, L., Aoki, J., Chun, J., & Ueda, H. (2008). Autotaxin, a synthetic enzyme of lysophosphatidic acid (LPA), mediates the induction of nerve-injured neuropathic pain. Molecular Pain, 4, 6.
Inoue, M., Ma, L., Aoki, J., & Ueda, H. (2008). Simultaneous stimulation of spinal NK1 and NMDA receptors produces LPC which undergoes ATX-mediated conversion to LPA, an initiator of neuropathic pain. Journal of Neurochemistry, 107(6), 1556–1565. https://doi.org/10.1111/j.1471-4159.2008.05725.x
Inoue, M., Rashid, M. H., Fujita, R., Contos, J. J., Chun, J., & Ueda, H. (2004). Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nature Medicine, 10(7), 712–718.
Inoue, M., Xie, W., Matsushita, Y., Chun, J., Aoki, J., & Ueda, H. (2008). Lysophosphatidylcholine induces neuropathic pain through an action of autotaxin to generate lysophosphatidic acid. Neuroscience, 152(2), 296–298. https://doi.org/10.1016/j.neuroscience.2007.12.041
Ishii, I., Contos, J. J., Fukushima, N., & Chun, J. (2000). Functional comparisons of the lysophosphatidic acid receptors, LP(A1)/VZG-1/EDG-2, LP(A2)/EDG-4, and LP(A3)/EDG-7 in neuronal cell lines using a retrovirus expression system. Molecular Pharmacology, 58(5), 895–902.
Itagaki, K., Takebayashi, M., Abe, H., Shibasaki, C., Kajitani, N., Okada-Tsuchioka, M., et al. (2019). Reduced serum and cerebrospinal fluid levels of autotaxin in major depressive disorder. International Journal of Neuropsychopharmacology, 22(4), 261–269. https://doi.org/10.1093/ijnp/pyz005
Jaaro-Peled, H., & Sawa, A. (2020). Neurodevelopmental factors in schizophrenia. Psychiatric Clinics of North America, 43(2), 263–274. https://doi.org/10.1016/j.psc.2020.02.010
Jaggi, A. S., Jain, V., & Singh, N. (2011). Animal models of neuropathic pain. Fundamental & Clinical Pharmacology, 25(1), 1–28. https://doi.org/10.1111/j.1472-8206.2009.00801.x
Jalink, K., Eichholtz, T., Postma, F. R., van Corven, E. J., & Moolenaar, W. H. (1993). Lysophosphatidic acid induces neuronal shape changes via a novel, receptor-mediated signaling pathway: Similarity to thrombin action. Cell Growth & Differentiation, 4(4), 247–255.
Jalink, K., van Corven, E. J., Hengeveld, T., Morii, N., Narumiya, S., & Moolenaar, W. H. (1994). Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. Journal of Cell Biology, 126(3), 801–810.
Kapfhammer, J. P., & Raper, J. A. (1987). Collapse of growth cone structure on contact with specific neurites in culture. Journal of Neuroscience, 7(1), 201–212.
Katsifa, A., Kaffe, E., Nikolaidou-Katsaridou, N., Economides, A. N., Newbigging, S., McKerlie, C., et al. (2015). The bulk of autotaxin activity is dispensable for adult mouse life. PLoS ONE, 10(11), e0143083. https://doi.org/10.1371/journal.pone.0143083
Kaya, B., Doñas, C., Wuggenig, P., Diaz, O. E., Morales, R. A., Melhem, H., et al. (2020). Lysophosphatidic acid-mediated GPR35 signaling in CX3CR1+ macrophages regulates intestinal homeostasis. Cell Reports, 32(5), 107979. https://doi.org/10.1016/j.celrep.2020.107979
Kihara, Y., Maceyka, M., Spiegel, S., & Chun, J. (2014). Lysophospholipid receptor nomenclature review: IUPHAR Review. British Journal of Pharmacology. https://doi.org/10.1111/bph.12678
Kingsbury, M. A., Rehen, S. K., Contos, J. J., Higgins, C. M., & Chun, J. (2003). Non-proliferative effects of lysophosphatidic acid enhance cortical growth and folding. Nature Neuroscience, 6(12), 1292–1299.
Koike, S., Yutoh, Y., Keino-Masu, K., Noji, S., Masu, M., & Ohuchi, H. (2011). Autotaxin is required for the cranial neural tube closure and establishment of the midbrain-hindbrain boundary during mouse development. Developmental Dynamics, 240(2), 413–421. https://doi.org/10.1002/dvdy.22543
Kolodkin, A. L., & Tessier-Lavigne, M. (2011). Mechanisms and molecules of neuronal wiring: A primer. Cold Spring Harbor perspectives in biology. https://doi.org/10.1101/cshperspect.a001727
Kotarsky, K., Boketoft, A., Bristulf, J., Nilsson, N. E., Norberg, A., Hansson, S., et al. (2006). Lysophosphatidic acid binds to and activates GPR92, a G protein-coupled receptor highly expressed in gastrointestinal lymphocytes. Journal of Pharmacology and Experimental Therapeutics, 318(2), 619–628.
Kozian, D. H., Evers, A., Florian, P., Wonerow, P., Joho, S., & Nazare, M. (2012). Selective non-lipid modulator of LPA5 activity in human platelets. Bioorganic & Medicinal Chemistry Letters, 22(16), 5239–5243. https://doi.org/10.1016/j.bmcl.2012.06.057
Kozma, R., Sarner, S., Ahmed, S., & Lim, L. (1997). Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Molecular and Cellular Biology, 17(3), 1201–1211.
Kranenburg, O., Poland, M., van Horck, F. P., Drechsel, D., Hall, A., & Moolenaar, W. H. (1999). Activation of RhoA by lysophosphatidic acid and Galpha12/13 subunits in neuronal cells: induction of neurite retraction. Molecular Biology of the Cell, 10(6), 1851–1857.
Kurabayashi, N., Tanaka, A., Nguyen, M. D., & Sanada, K. (2018). The LPA-LPA4 axis is required for establishment of bipolar morphology and radial migration of newborn cortical neurons. Development. https://doi.org/10.1242/dev.162529
Kusaka, S., Kapousta-Bruneau, N., Green, D. G., & Puro, D. G. (1998). Serum-induced changes in the physiology of mammalian retinal glial cells: role of lysophosphatidic acid. The Journal of physiology, 506(Pt 2), 445–458.
Kuwajima, K., Sumitani, M., Kurano, M., Kano, K., Nishikawa, M., Uranbileg, B., et al. (2018). Lysophosphatidic acid is associated with neuropathic pain intensity in humans: An exploratory study. PLoS ONE, 13(11), e0207310. https://doi.org/10.1371/journal.pone.0207310
Lee, C. W., Rivera, R., Dubin, A. E., & Chun, J. (2007). LPA(4)/GPR23 is a lysophosphatidic acid (LPA) receptor utilizing G(s)-, G(q)/G(i)-mediated calcium signaling and G(12/13)-mediated Rho activation. Journal of Biological Chemistry, 282(7), 4310–4317.
Lee, C. W., Rivera, R., Gardell, S., Dubin, A. E., & Chun, J. (2006). GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. Journal of Biological Chemistry, 281(33), 23589–23597.
Lee, S. C., Dacheux, M. A., Norman, D. D., Balazs, L., Torres, R. M., Augelli-Szafran, C. E., et al. (2020). Regulation of tumor immunity by lysophosphatidic acid. Cancers (Basel). https://doi.org/10.3390/cancers12051202
Li, Z. G., Yu, Z. C., Wang, D. Z., Ju, W. P., Zhan, X., Wu, Q. Z., et al. (2008). Influence of acetylsalicylate on plasma lysophosphatidic acid level in patients with ischemic cerebral vascular diseases. Neurological Research, 30(4), 366–369. https://doi.org/10.1179/174313208X300369
Li, Z. G., Yu, Z. C., Yu, Y. P., Ju, W. P., Wang, D. Z., Zhan, X., et al. (2010). Lysophosphatidic acid level and the incidence of silent brain infarction in patients with nonvalvular atrial fibrillation. International Journal of Molecular Sciences, 11(10), 3988–3998. https://doi.org/10.3390/ijms11103988
Lin, M.-E., Rivera, R. R., & Chun, J. (2012). Targeted deletion of LPA5 identifies novel roles for lysophosphatidic acid signaling in development of neuropathic pain. Journal of Biological Chemistry, 287(21), 17608–17617. https://doi.org/10.1074/jbc.M111.330183
Lin, Y. N., Audira, G., Malhotra, N., Ngoc Anh, N. T., Siregar, P., Lu, J. H., et al. (2020). A novel function of the lysophosphatidic acid receptor 3 (LPAR3) gene in Zebrafish on modulating anxiety, circadian rhythm locomotor activity, and short-term memory. International Journal of Molecular Sciences. https://doi.org/10.3390/ijms21082837
Lopez-Serrano, C., Santos-Nogueira, E., Francos-Quijorna, I., Coll-Miro, M., Chun, J., & Lopez-Vales, R. (2019). Lysophosphatidic acid receptor type 2 activation contributes to secondary damage after spinal cord injury in mice. Brain, Behavior, and Immunity, 76, 258–267. https://doi.org/10.1016/j.bbi.2018.12.007
Lu, W. Y., Xiong, Z. G., Lei, S., Orser, B. A., Dudek, E., Browning, M. D., et al. (1999). G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nature Neuroscience, 2(4), 331–338. https://doi.org/10.1038/7243
Lummis, N. C., Sánchez-Pavón, P., Kennedy, G., Frantz, A. J., Kihara, Y., Blaho, V. A., et al. (2019). LPA1/3 overactivation induces neonatal posthemorrhagic hydrocephalus through ependymal loss and ciliary dysfunction. Science Advances. https://doi.org/10.1126/sciadv.aax2011
Luo, Y., Raible, D., & Raper, J. A. (1993). Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell, 75(2), 217–227.
Ma, L., Nagai, J., Chun, J., & Ueda, H. (2013). An LPA species (18:1 LPA) plays key roles in the self-amplification of spinal LPA production in the peripheral neuropathic pain model. Molecular Pain, 9, 29. https://doi.org/10.1186/1744-8069-9-29
Ma, L., Nagai, J., & Ueda, H. (2010). Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. Journal of Neurochemistry, 115(3), 643–653. https://doi.org/10.1111/j.1471-4159.2010.06955.x
Ma, L., Uchida, H., Nagai, J., Inoue, M., Aoki, J., & Ueda, H. (2010). Evidence for de novo synthesis of lysophosphatidic acid in the spinal cord through phospholipase A2 and autotaxin in nerve injury-induced neuropathic pain. Journal of Pharmacology and Experimental Therapeutics, 333(2), 540–546. https://doi.org/10.1124/jpet.109.164830
Ma, L., Uchida, H., Nagai, J., Inoue, M., Chun, J., Aoki, J., et al. (2009). Lysophosphatidic acid-3 receptor-mediated feed-forward production of lysophosphatidic acid: An initiator of nerve injury-induced neuropathic pain. Mol Pain, 5, 64.
Matas-Rico, E., Garcia-Diaz, B., Llebrez-Zayas, P., Lopez-Barroso, D., Santin, L., Pedraza, C., et al. (2008). Deletion of lysophosphatidic acid receptor LPA1 reduces neurogenesis in the mouse dentate gyrus. Molecular and Cellular Neuroscience, 39(3), 342–355. https://doi.org/10.1016/j.mcn.2008.07.014
McDougall, J. J., Albacete, S., Schuelert, N., Mitchell, P. G., Lin, C., Oskins, J. L., et al. (2017). Lysophosphatidic acid provides a missing link between osteoarthritis and joint neuropathic pain. Osteoarthritis Cartilage, 25(6), 926–934. https://doi.org/10.1016/j.joca.2016.08.016
Medelnik, J. P., Roensch, K., Okawa, S., Del Sol, A., Chara, O., McHedlishvili, L., et al. (2018). Signaling-dependent control of apical membrane size and self-renewal in rosette-stage human neuroepithelial stem cells. Stem Cell Reports, 10(6), 1751–1765. https://doi.org/10.1016/j.stemcr.2018.04.018
Mirendil, H., Thomas, E. A., De Loera, C., Okada, K., Inomata, Y., & Chun, J. (2015). LPA signaling initiates schizophrenia-like brain and behavioral changes in a mouse model of prenatal brain hemorrhage. Transl Psychiatry, 5, e541. https://doi.org/10.1038/tp.2015.33
Mizuno, H., Kihara, Y., Kussrow, A., Chen, A., Ray, M., Rivera, R., et al. (2019). Lysophospholipid G protein-coupled receptor binding parameters as determined by backscattering interferometry. Journal of Lipid Research, 60(1), 212–217. https://doi.org/10.1194/jlr.D089938
Moller, T., Contos, J. J., Musante, D. B., Chun, J., & Ransom, B. R. (2001). Expression and function of lysophosphatidic acid receptors in cultured rodent microglial cells. Journal of Biological Chemistry, 276(28), 25946–25952.
Moller, T., Musante, D. B., & Ransom, B. R. (1999). Lysophosphatidic acid-induced calcium signals in cultured rat oligodendrocytes. NeuroReport, 10(14), 2929–2932.
Moreno-Fernandez, R. D., Nieto-Quero, A., Gomez-Salas, F. J., Chun, J., Estivill-Torrus, G., Rodriguez de Fonseca, F., et al. (2018). Effects of genetic deletion versus pharmacological blockade of the LPA1 receptor on depression-like behaviour and related brain functional activity. Disease Models & Mechanisms. https://doi.org/10.1242/dmm.035519
Moreno-Fernandez, R. D., Perez-Martin, M., Castilla-Ortega, E., Rosell Del Valle, C., Garcia-Fernandez, M. I., Chun, J., et al. (2017). maLPA1-null mice as an endophenotype of anxious depression. Transl Psychiatry, 7(4), e1077. https://doi.org/10.1038/tp.2017.24
Murakami, M., Shiraishi, A., Tabata, K., & Fujita, N. (2008). Identification of the orphan GPCR, P2Y(10) receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochemical and Biophysical Research Communications, 371(4), 707–712.
Musazzi, L., Di Daniel, E., Maycox, P., Racagni, G., & Popoli, M. (2011). Abnormalities in alpha/beta-CaMKII and related mechanisms suggest synaptic dysfunction in hippocampus of LPA1 receptor knockout mice. International Journal of Neuropsychopharmacology, 14(7), 941–953. https://doi.org/10.1017/S1461145710001240
Nagai, J., Uchida, H., Matsushita, Y., Yano, R., Ueda, M., Niwa, M., et al. (2010). Autotaxin and lysophosphatidic acid1 receptor-mediated demyelination of dorsal root fibers by sciatic nerve injury and intrathecal lysophosphatidylcholine. Molecular Pain, 6, 78. https://doi.org/10.1186/1744-8069-6-78
Nagai, J., & Ueda, H. (2011). Pre-emptive morphine treatment abolishes nerve injury-induced lysophospholipid synthesis in mass spectrometrical analysis. Journal of Neurochemistry, 118(2), 256–265. https://doi.org/10.1111/j.1471-4159.2011.07297.x
Nahum, S., Morice-Picard, F., Taieb, A., & Sprecher, E. (2011). A novel mutation in LPAR6 causes autosomal recessive hypotrichosis of the scalp. Clinical and Experimental Dermatology, 36(2), 188–194. https://doi.org/10.1111/j.1365-2230.2010.03944.x
Nieto-Posadas, A., Picazo-Juarez, G., Llorente, I., Jara-Oseguera, A., Morales-Lazaro, S., Escalante-Alcalde, D., et al. (2012). Lysophosphatidic acid directly activates TRPV1 through a C-terminal binding site. Nature Chemical Biology, 8(1), 78–85. https://doi.org/10.1038/nchembio.712
Nogaroli, L., Yuelling, L. M., Dennis, J., Gorse, K., Payne, S. G., & Fuss, B. (2009). Lysophosphatidic acid can support the formation of membranous structures and an increase in MBP mRNA levels in differentiating oligodendrocytes. Neurochemical Research, 34(1), 182–193. https://doi.org/10.1007/s11064-008-9772-z
Noguchi, K., Ishii, S., & Shimizu, T. (2003). Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. Journal of Biological Chemistry, 278(28), 25600–25606.
Ohsawa, M., Miyabe, Y., Katsu, H., Yamamoto, S., & Ono, H. (2013). Identification of the sensory nerve fiber responsible for lysophosphatidic acid-induced allodynia in mice. Neuroscience, 247, 65–74. https://doi.org/10.1016/j.neuroscience.2013.05.014
Ohta, H., Sato, K., Murata, N., Damirin, A., Malchinkhuu, E., Kon, J., et al. (2003). Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Molecular Pharmacology, 64(4), 994–1005.
Ohuchi, H., Fukui, H., Matsuyo, A., Tomonari, S., Tanaka, M., Arai, H., et al. (2010). Autotaxin controls caudal diencephalon-mesencephalon development in the chick. Developmental Dynamics, 239(10), 2647–2658. https://doi.org/10.1002/dvdy.22403
Ohuchi, H., Hamada, A., Matsuda, H., Takagi, A., Tanaka, M., Aoki, J., et al. (2008). Expression patterns of the lysophospholipid receptor genes during mouse early development. Developmental Dynamics, 237(11), 3280–3294.
Ohuchi, H., Hayashibara, Y., Matsuda, H., Onoi, M., Mitsumori, M., Tanaka, M., et al. (2007). Diversified expression patterns of autotaxin, a gene for phospholipid-generating enzyme during mouse and chicken development. Developmental Dynamics, 236(4), 1134–1143.
Pages, C., Simon, M. F., Valet, P., & Saulnier-Blache, J. S. (2001). Lysophosphatidic acid synthesis and release. Prostaglandins & Other Lipid Mediators, 64(1–4), 1–10.
Palmer, S. M., Snyder, L., Todd, J. L., Soule, B., Christian, R., Anstrom, K., et al. (2018). Randomized, double-blind, placebo-controlled, phase 2 trial of BMS-986020, a lysophosphatidic acid receptor antagonist for the treatment of idiopathic pulmonary fibrosis. Chest, 154(5), 1061–1069. https://doi.org/10.1016/j.chest.2018.08.1058
Pasternack, S. M., von Kugelgen, I., Aboud, K. A., Lee, Y. A., Ruschendorf, F., Voss, K., et al. (2008). G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nature Genetics, 40(3), 329–334.
Penalver, A., Campos-Sandoval, J. A., Blanco, E., Cardona, C., Castilla, L., Martin-Rufian, M., et al. (2017). Glutaminase and MMP-9 downregulation in cortex and hippocampus of LPA1 receptor null mice correlate with altered dendritic spine plasticity. Frontiers in Molecular Neuroscience, 10, 278. https://doi.org/10.3389/fnmol.2017.00278
Perrakis, A., & Moolenaar, W. H. (2014). Autotaxin: Structure-function and signaling. Journal of Lipid Research, 55(6), 1010–1018. https://doi.org/10.1194/jlr.R046391
Pilpel, Y., & Segal, M. (2006). The role of LPA1 in formation of synapses among cultured hippocampal neurons. Journal of Neurochemistry, 97(5), 1379–1392. https://doi.org/10.1111/j.1471-4159.2006.03825.x
Plastira, I., Bernhart, E., Goeritzer, M., DeVaney, T., Reicher, H., Hammer, A., et al. (2017). Lysophosphatidic acid via LPA-receptor 5/protein kinase D-dependent pathways induces a motile and pro-inflammatory microglial phenotype. Journal of Neuroinflammation, 14(1), 253. https://doi.org/10.1186/s12974-017-1024-1
Plastira, I., Bernhart, E., Goeritzer, M., Reicher, H., Kumble, V. B., Kogelnik, N., et al. (2016). 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. Journal of Neuroinflammation, 13(1), 205. https://doi.org/10.1186/s12974-016-0701-9
Ramesh, S., Govindarajulu, M., Suppiramaniam, V., Moore, T., & Dhanasekaran, M. (2018). Autotaxin-lysophosphatidic acid signaling in Alzheimer’s disease. International Journal of Molecular Sciences, 19(7), 1827.
Raper, J. A., & Kapfhammer, J. P. (1990). The enrichment of a neuronal growth cone collapsing activity from embryonic chick brain. Neuron, 2(1), 21–29.
Ray, M., Nagai, K., Kihara, Y., Kussrow, A., Kammer, M. N., Frantz, A., et al. (2020). Unlabeled lysophosphatidic acid receptor binding in free solution as determined by a compensated interferometric reader. Journal of Lipid Research, 61(8), 1244–1251. https://doi.org/10.1194/jlr.D120000880
Reinhard, S. M., Razak, K., & Ethell, I. M. (2015). A delicate balance: role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders. Frontiers in Cellular Neuroscience, 9, 280. https://doi.org/10.3389/fncel.2015.00280
Rhee, H. J., Nam, J. S., Sun, Y., Kim, M. J., Choi, H. K., Han, D. H., et al. (2006). Lysophosphatidic acid stimulates cAMP accumulation and cAMP response element-binding protein phosphorylation in immortalized hippocampal progenitor cells. NeuroReport, 17(5), 523–526. https://doi.org/10.1097/01.wnr.0000209011.16718.68
Roberts, C., Winter, P., Shilliam, C. S., Hughes, Z. A., Langmead, C., Maycox, P. R., et al. (2005). Neurochemical changes in LPA1 receptor deficient mice: A putative model of schizophrenia. Neurochemical Research, 30(3), 371–377. https://doi.org/10.1007/s11064-005-2611-6
Roza, C., Campos-Sandoval, J. A., Gomez-Garcia, M. C., Penalver, A., & Marquez, J. (2019). Lysophosphatidic acid and glutamatergic transmission. Frontiers in Cellular Neuroscience, 12, 138. https://doi.org/10.3389/fnmol.2019.00138
Saito, S. (1997). Effects of lysophosphatidic acid on primary cultured chick neurons. Neuroscience Letters, 229(2), 73–76.
Santin, L. J., Bilbao, A., Pedraza, C., Matas-Rico, E., Lopez-Barroso, D., Castilla-Ortega, E., et al. (2009). Behavioral phenotype of maLPA1-null mice: increased anxiety-like behavior and spatial memory deficits. Genes, Brain and Behavior, 8(8), 772–784. https://doi.org/10.1111/j.1601-183X.2009.00524.x
Santos-Nogueira, E., Lopez-Serrano, C., Hernandez, J., Lago, N., Astudillo, A. M., Balsinde, J., et al. (2015). Activation of lysophosphatidic acid receptor type 1 contributes to pathophysiology of spinal cord injury. Journal of Neuroscience, 35(28), 10224–10235. https://doi.org/10.1523/JNEUROSCI.4703-14.2015
Sapkota, A., Lee, C. H., Park, S. J., & Choi, J. W. (2020). Lysophosphatidic acid receptor 5 Plays a pathogenic role in brain damage after focal cerebral ischemia by modulating neuroinflammatory responses. Cells. https://doi.org/10.3390/cells9061446
Savaskan, N. E., Rocha, L., Kotter, M. R., Baer, A., Lubec, G., van Meeteren, L. A., et al. (2007). Autotaxin (NPP-2) in the brain: cell type-specific expression and regulation during development and after neurotrauma. Cellular and Molecular Life Sciences, 64(2), 230–243.
Savitz, S. I., Dhallu, M. S., Malhotra, S., Mammis, A., Ocava, L. C., Rosenbaum, P. S., et al. (2006). EDG receptors as a potential therapeutic target in retinal ischemia-reperfusion injury. Brain Research, 1118(1), 168–175. https://doi.org/10.1016/j.brainres.2006.05.060
Sayas, C. L., Avila, J., & Wandosell, F. (2002). Glycogen synthase kinase-3 is activated in neuronal cells by Galpha12 and Galpha13 by Rho-independent and Rho-dependent mechanisms. Journal of Neuroscience, 22(16), 6863–6875.
Schmitz, K., Brunkhorst, R., de Bruin, N., Mayer, C. A., Haussler, A., Ferreiros, N., et al. (2017). Dysregulation of lysophosphatidic acids in multiple sclerosis and autoimmune encephalomyelitis. Acta Neuropathologica Communications, 5(1), 42. https://doi.org/10.1186/s40478-017-0446-4
Seltzer, Z., Dubner, R., & Shir, Y. (1990). A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain, 43(2), 205–218. https://doi.org/10.1016/0304-3959(90)91074-s
Shano, S., Moriyama, R., Chun, J., & Fukushima, N. (2008). Lysophosphatidic acid stimulates astrocyte proliferation through LPA1. Neurochemistry International, 52(1–2), 216–220. https://doi.org/10.1016/j.neuint.2007.07.004
Srikanth, M., Chew, W. S., Hind, T., Lim, S. M., Hay, N. W. J., Lee, J. H. M., et al. (2018). Lysophosphatidic acid and its receptor LPA1 mediate carrageenan induced inflammatory pain in mice. European Journal of Pharmacology, 841, 49–56. https://doi.org/10.1016/j.ejphar.2018.10.005
Stankoff, B., Barron, S., Allard, J., Barbin, G., Noel, F., Aigrot, M. S., et al. (2002). Oligodendroglial expression of Edg-2 receptor: developmental analysis and pharmacological responses to lysophosphatidic acid. Molecular and Cellular Neuroscience, 20(3), 415–428.
Steiner, M. R., Urso, J. R., Klein, J., & Steiner, S. M. (2002). Multiple astrocyte responses to lysophosphatidic acids. Biochimica et Biophysica Acta, 1582(1–3), 154–160. https://doi.org/10.1016/s1388-1981(02)00150-6
Stoeckli, E. (2017). Where does axon guidance lead us? Research, 6, 78. https://doi.org/10.12688/f1000research.10126.1
Strauss, U., & Brauer, A. U. (2013). Current views on regulation and function of plasticity-related genes (PRGs/LPPRs) in the brain. Biochimica et Biophysica Acta, 1831(1), 133–138. https://doi.org/10.1016/j.bbalip.2012.08.010
Strochlic, L., Dwivedy, A., van Horck, F. P., Falk, J., & Holt, C. E. (2008). A role for S1P signalling in axon guidance in the Xenopus visual system. Development, 135(2), 333–342.
Suckau, O., Gross, I., Schrotter, S., Yang, F., Luo, J., Wree, A., et al. (2019). LPA1, LPA2, LPA4, and LPA6 receptor expression during mouse brain development. Developmental Dynamics, 248(5), 375–395. https://doi.org/10.1002/dvdy.23
Sugiura, T., Nakane, S., Kishimoto, S., Waku, K., Yoshioka, Y., Tokumura, A., et al. (1999). Occurrence of lysophosphatidic acid and its alkyl ether-linked analog in rat brain and comparison of their biological activities toward cultured neural cells. Biochimica et Biophysica Acta, 1440(2–3), 194–204.
Sumida, H., Noguchi, K., Kihara, Y., Abe, M., Yanagida, K., Hamano, F., et al. (2010). LPA4 regulates blood and lymphatic vessel formation during mouse embryogenesis. Blood, 116(23), 5060–5070. https://doi.org/10.1182/blood-2010-03-272443
Swaney, J. S., Chapman, C., Correa, L. D., Stebbins, K. J., Broadhead, A. R., Bain, G., et al. (2011). Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. Journal of Pharmacology and Experimental Therapeutics, 336(3), 693–700. https://doi.org/10.1124/jpet.110.175901
Szepanowski, F., Szepanowski, L. P., Mausberg, A. K., Kleinschnitz, C., Kieseier, B. C., & Stettner, M. (2018). Lysophosphatidic acid propagates post-injury Schwann cell dedifferentiation through LPA1 signaling. Neuroscience Letters, 662, 136–141. https://doi.org/10.1016/j.neulet.2017.10.023
Tabata, K., Baba, K., Shiraishi, A., Ito, M., & Fujita, N. (2007). The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochemical and Biophysical Research Communications, 363(3), 861–866.
Tabbai, S., Moreno-Fernandez, R. D., Zambrana-Infantes, E., Nieto-Quero, A., Chun, J., Garcia-Fernandez, M., et al. (2019). Effects of the LPA1 receptor deficiency and stress on the hippocampal LPA species in mice. Frontiers in Molecular Neuroscience, 12, 146. https://doi.org/10.3389/fnmol.2019.00146
Tager, A. M., LaCamera, P., Shea, B. S., Campanella, G. S., Selman, M., Zhao, Z., et al. (2008). The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nature Medicine, 14(1), 45–54. https://doi.org/10.1038/nm1685
Tanaka, M., Okudaira, S., Kishi, Y., Ohkawa, R., Iseki, S., Ota, M., et al. (2006). Autotaxin stabilizes blood vessels and is required for embryonic vasculature by producing lysophosphatidic acid. Journal of Biological Chemistry, 281(35), 25822–25830. https://doi.org/10.1074/jbc.M605142200
Thalman, C., Horta, G., Qiao, L., Endle, H., Tegeder, I., Cheng, H., et al. (2018). Synaptic phospholipids as a new target for cortical hyperexcitability and E/I balance in psychiatric disorders. Mol. Psychiatry, 23(8), 1699–1710. https://doi.org/10.1038/s41380-018-0053-1
Tham, C.-S., Lin, F.-F., Rao, T. S., Yu, N., & Webb, M. (2003). Microglial activation state and lysophospholipid acid receptor expression. International Journal of Developmental Neuroscience, 21(8), 431–443.
Thoreson, W. B., Khandalavala, B. N., Manahan, R. G., Polyak, I. A., Liu, J. L., & Chacko, D. M. (1997). Lysophosphatidic acid stimulates proliferation of human retinal pigment epithelial cells. Current Eye Research, 16(7), 698–702.
Tigyi, G., Fischer, D. J., Sebok, A., Marshall, F., Dyer, D. L., & Miledi, R. (1996). Lysophosphatidic acid-induced neurite retraction in PC12 cells: neurite-protective effects of cyclic AMP signaling. Journal of Neurochemistry, 66(2), 549–558.
Tigyi, G., Fischer, D. J., Sebok, A., Yang, C., Dyer, D. L., & Miledi, R. (1996). Lysophosphatidic acid-induced neurite retraction in PC12 cells: control by phosphoinositide-Ca2+ signaling and Rho. Journal of Neurochemistry, 66(2), 537–548.
Tigyi, G. J., Yue, J., Norman, D. D., Szabo, E., Balogh, A., Balazs, L., et al. (2019). Regulation of tumor cell - Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Advances in Biological Regulation, 71, 183–193. https://doi.org/10.1016/j.jbior.2018.09.008
Trimbuch, T., Beed, P., Vogt, J., Schuchmann, S., Maier, N., Kintscher, M., et al. (2009). Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell, 138(6), 1222–1235.
Tsukahara, R., & Ueda, H. (2016). Myelin-related gene silencing mediated by LPA1 - Rho/ROCK signaling is correlated to acetylation of NFkappaB in S16 Schwann cells. Journal of Pharmacological Sciences, 132(2), 162–165. https://doi.org/10.1016/j.jphs.2016.07.010
Uchida, H., Nagai, J., & Ueda, H. (2014). Lysophosphatidic acid and its receptors LPA1 and LPA3 mediate paclitaxel-induced neuropathic pain in mice. Molecular Pain, 10, 71. https://doi.org/10.1186/1744-8069-10-71
Ueda, H. (2019). Systems pathology of neuropathic pain and fibromyalgia. Biological &/and Pharmaceutical Bulletin, 42(11), 1773–1782. https://doi.org/10.1248/bpb.b19-00535
Ueda, H., Matsunaga, H., Olaposi, O. I., & Nagai, J. (2013). Lysophosphatidic acid: Chemical signature of neuropathic pain. Biochimica et Biophysica Acta, 1831(1), 61–73. https://doi.org/10.1016/j.bbalip.2012.08.014
Ueda, H., Neyama, H., Nagai, J., Matsushita, Y., Tsukahara, T., & Tsukahara, R. (2018). Involvement of lysophosphatidic acid-induced astrocyte activation underlying the maintenance of partial sciatic nerve injury-induced neuropathic pain. Pain, 159(11), 2170–2178. https://doi.org/10.1097/j.pain.0000000000001316
Ueda, H., Neyama, H., Sasaki, K., Miyama, C., & Iwamoto, R. (2019). Lysophosphatidic acid LPA1 and LPA3 receptors play roles in the maintenance of late tissue plasminogen activator-induced central poststroke pain in mice. Neurobiol Pain, 5, 100020. https://doi.org/10.1016/j.ynpai.2018.07.001
van Meeteren, L. A., & Moolenaar, W. H. (2007). Regulation and biological activities of the autotaxin-LPA axis. Progress in Lipid Research, 46(2), 145–160.
van Meeteren, L. A., Ruurs, P., Stortelers, C., Bouwman, P., van Rooijen, M. A., Pradere, J. P., et al. (2006). Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Molecular and Cellular Biology, 26(13), 5015–5022. https://doi.org/10.1128/MCB.02419-05
Velasco, M., O’Sullivan, C., & Sheridan, G. K. (2017). Lysophosphatidic acid receptors (LPARs): Potential targets for the treatment of neuropathic pain. Neuropharmacology, 113(Pt B), 608–617. https://doi.org/10.1016/j.neuropharm.2016.04.002
Vogt, J., Yang, J. W., Mobascher, A., Cheng, J., Li, Y., Liu, X., et al. (2016). Molecular cause and functional impact of altered synaptic lipid signaling due to a prg-1 gene SNP. EMBO Molecular Medicine, 8(1), 25–38. https://doi.org/10.15252/emmm.201505677
Wang, C., Zhang, J., Tang, J., Li, Y. Y., Gu, Y., Yu, Y., et al. (2018). Lysophosphatidic acid induces neuronal cell death via activation of asparagine endopeptidase in cerebral ischemia-reperfusion injury. Experimental Neurology, 306, 1–9. https://doi.org/10.1016/j.expneurol.2018.04.010
Wang, Q., Jie, W., Liu, J. H., Yang, J. M., & Gao, T. M. (2017). An astroglial basis of major depressive disorder? An overview. Glia, 65(8), 1227–1250. https://doi.org/10.1002/glia.23143
Weiner, J. A., & Chun, J. (1999). Schwann cell survival mediated by the signaling phospholipid lysophosphatidic acid. Proceedings of the National Academy of Sciences of the United States of America, 96(9), 5233–5238.
Weiner, J. A., Fukushima, N., Contos, J. J., Scherer, S. S., & Chun, J. (2001). Regulation of Schwann cell morphology and adhesion by receptor-mediated lysophosphatidic acid signaling. Journal of Neuroscience, 21(18), 7069–7078.
Weiner, J. A., Hecht, J. H., & Chun, J. (1998). Lysophosphatidic acid receptor gene vzg-1/lpA1/edg-2 is expressed by mature oligodendrocytes during myelination in the postnatal murine brain. The Journal of Comparative Neurology, 398(4), 587–598.
Weiss, H. R., Mellender, S. J., Kiss, G. K., Chiricolo, A., Liu, X., & Chi, O. Z. (2020). Lysophosphatidic acid reduces microregional oxygen supply/consumption balance after cerebral ischemia-reperfusion. Journal of Vascular Research, 57(4), 178–184. https://doi.org/10.1159/000506011
Wheeler, N. A., Lister, J. A., & Fuss, B. (2015). The autotaxin-lysophosphatidic acid axis modulates histone acetylation and gene expression during oligodendrocyte differentiation. Journal of Neuroscience, 35(32), 11399–11414. https://doi.org/10.1523/JNEUROSCI.0345-15.2015
Xie, W., Matsumoto, M., Chun, J., & Ueda, H. (2008). Involvement of LPA1 receptor signaling in the reorganization of spinal input through Abeta-fibers in mice with partial sciatic nerve injury. Molecular Pain, 4, 46. https://doi.org/10.1186/1744-8069-4-46
Xie, W., Uchida, H., Nagai, J., Ueda, M., Chun, J., & Ueda, H. (2010). Calpain-mediated down-regulation of myelin-associated glycoprotein in lysophosphatidic acid-induced neuropathic pain. Journal of Neurochemistry, 113(4), 1002–1011. https://doi.org/10.1111/j.1471-4159.2010.06664.x
Xu, Y. (2019). Targeting lysophosphatidic acid in cancer: the issues in moving from bench to bedside. Cancers (Basel). https://doi.org/10.3390/cancers11101523
Yamazaki, J., Katoh, H., & Negishi, M. (2008). Lysophosphatidic acid and thrombin receptors require both G alpha12 and G alpha13 to regulate axonal morphology in hippocampal neurons. Biological &/and Pharmaceutical Bulletin, 31(12), 2216–2222.
Yanagida, K., Ishii, S., Hamano, F., Noguchi, K., & Shimizu, T. (2007). LPA4/p2y9/GPR23 mediates rho-dependent morphological changes in a rat neuronal cell line. Journal of Biological Chemistry, 282(8), 5814–5824.
Yanagida, K., Masago, K., Nakanishi, H., Kihara, Y., Hamano, F., Tajima, Y., et al. (2009). Identification and characterization of a novel lysophosphatidic acid receptor, p2y5/LPA6. Journal of Biological Chemistry, 284(26), 17731–17741.
Yang, C., Lafleur, J., Mwaikambo, B. R., Zhu, T., Gagnon, C., Chemtob, S., et al. (2009). The role of lysophosphatidic acid receptor (LPA1) in the oxygen-induced retinal ganglion cell degeneration. Investigative Ophthalmology & Visual Science, 50(3), 1290–1298.
Yano, R., Ma, L., Nagai, J., & Ueda, H. (2013). Interleukin-1beta plays key roles in LPA-induced amplification of LPA production in neuropathic pain model. Cellular and Molecular Neurobiology, 33(8), 1033–1041. https://doi.org/10.1007/s10571-013-9970-3
Ye, X., Hama, K., Contos, J. J., Anliker, B., Inoue, A., Skinner, M. K., et al. (2005). LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature, 435(7038), 104–108.
Yu, N., Lariosa-Willingham, K. D., Lin, F. F., Webb, M., & Rao, T. S. (2004). Characterization of lysophosphatidic acid and sphingosine-1-phosphate-mediated signal transduction in rat cortical oligodendrocytes. Glia, 45(1), 17–27.
Yuelling, L. W., Waggener, C. T., Afshari, F. S., Lister, J. A., & Fuss, B. (2012). Autotaxin/ENPP2 regulates oligodendrocyte differentiation in vivo in the developing zebrafish hindbrain. Glia, 60(10), 1605–1618. https://doi.org/10.1002/glia.22381
Yung, Y. C., Stoddard, N. C., & Chun, J. (2014). LPA receptor signaling: Pharmacology, physiology, and pathophysiology. Journal of Lipid Research, 55(7), 1192–1214. https://doi.org/10.1194/jlr.R046458
Yung, Y. C., Stoddard, N. C., Mirendil, H., & Chun, J. (2015). Lysophosphatidic acid signaling in the nervous system. Neuron, 85(4), 669–682. https://doi.org/10.1016/j.neuron.2015.01.009
Zeng, X., Luo, Z., Wu, J., Zhou, J., Shan, Y., Zhu, Y., et al. (2020). Cerebral ischemia-reperfusion injury: Lysophosphatidic acid mediates inflammation by decreasing the expression of liver X receptor. Journal of Molecular Neuroscience, 70(9), 1376–1384. https://doi.org/10.1007/s12031-020-01554-3
Acknowledgements
I would like to thank Allison Reed and Thomas Gonzalez from the Birgbauer laboratory for critical reading of this manuscript. E. B. was supported in part by Grant P20GM103499 (SC INBRE) from the National Institute of General Medical Sciences, National Institutes of Health.
Funding
E.B. has been supported in part by Grant P20GM103499 (SC INBRE) from the National Institute of General Medical Sciences, National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Ethics Approval
This is a literature review article and no studies with human subjects and no studies with animals was performed by the author for this manuscript and is thus exempt.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Birgbauer, E. Lysophosphatidic Acid Signalling in Nervous System Development and Function. Neuromol Med 23, 68–85 (2021). https://doi.org/10.1007/s12017-020-08630-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-020-08630-2