
Abstract
Autoimmune lymphoproliferative syndrome (ALPS), a disorder characterized by immune dysregulation due to disrupted lymphocyte homeostasis, is mainly resulted from the mutations in FAS-mediated apoptotic pathway. In addition, other mutations of the genes such as Fas-ligand (FASLG), Caspase 10 (CASP10) and Caspase 8 (CASP8), NRAS and KRAS have also been observed in a small number of patients with ALPS or ALPS-related disorders. However, approximately 20-30 % of patients with ALPS have unidentified defect. Its clinical manifestations observed in multiple family members include unexplained lymphadenopathy, hepatosplenomegaly, autoimmune cytopenias such as thrombocytopenia, neutropenia, and anemia due to excessive production of antibodies by lymphocytes, elevated number of double-negative T (DNT) cells, and increased risk of lymphoma. As a very rare disease, ALPS was first characterized in the early 1990s. More than 300 families with hereditary ALPS have been reported till now; nearly 500 patients from these families have been studied and followed worldwide over the last 20 years. ALPS has historically considered as a primary immune defect presenting in early childhood, however, recent studies have shown that it may be more common than previous thought because adult onset presentation is increasingly becoming recognized and more adult ALPS patients are diagnosed. The new genetic and biological insights have improved the understanding of ALPS and a number of targeted therapeutic strategies such as mycophenolate mofetil, sirolimus, and pentostatin have been successfully applied in ALPS patients with promising treatment efficacy. This article comprehensively reviews the clinical and laboratory manifestations, new research advances in the molecular pathogenesis, diagnosis and treatments of this disorder.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autoimmune lymphoproliferative syndrome (ALPS), a typically genetic disorder associated with apoptosis, has very low incidence. The exact prevalence is still unknown. According to the revised criteria for the diagnosis and classification of ALPS from 2009 International Workshop at the National Institutes of Health (NIH), only approximately 500 patients with ALPS originating from more than 300 families have been investigated all over the world [1]. In 1967, five patients with lymphadenopathy, splenomegaly, and autoimmune cytopenia have been diagnosed as malignant lymphoma with similar characteristics by Canale and Smith [2] and named as Canale-Smith syndrome. In 1992, Sneller et al. [3] have found that ALPS exhibits similar manifestations from two lymphoproliferative mouse models caused by lpr and gld mutations as previously described in systemic lupus erythematosus (SLE). Subsequently, the homozygous mutations of FAS and FASLG genes in lpr and gld mice have been confirmed by Watanabe-Fukunaga and Takahashi et al. [4, 5]. Later, Rieux-Laucat et al. [6] and Fisher et al. [7] have documented that the onset cause of eight patients is due to the mutation of FAS gene. Therefore, this disease is officially named as ALPS responsible for the FAS-mediated defects of lymphocyte apoptosis. In the past few years, some ALPS patients are often misdiagnosed as other diseases such as idiopathic autoimmune disorder (Evans syndrome, ES), SLE, or histiocytic disorder. But, in recent years, significant research advances in pathophysiology, diagnostic criteria, and targeted therapeutic strategies of ALPS have already been achieved. Herein, the research advances in ALPS have been summarized and discussed in this article.
Clinical Manifestations
ALPS is often misdiagnosed due to variable phenotypic expressions and the overlap of symptoms with many other hematological disorders [8]. ALPS has been diagnosed in both sexes and in people with diverse racial backgrounds. It is a genetic disorder with the median age of 24 months for the first onset; however, the onset of ALPS patients at 3 weeks of age and 36 weeks of gestation in utero is recently reported [9]. With increasing awareness of this disease, adult ALPS patients are also now being diagnosed more frequently [10].
Lymphoproliferation is the most common clinical manifestation in ALPS accompanying with lymphadenopathy, hepatomegaly, or splenomegaly [11]. More than 80 % of patients with ALPS experience the prolonged period for the enlargement of palpable and non-tender lymph nodes [12]. The common manifestation with the involvement of lymphadenopathy can be observed in cervical, axillary, and inguinal chains, and sometimes in preauricular, submental, epitrochlear, mediastinal, and retroperitoneal nodes. Lymphoproliferation in ALPS must be chronic (>6 months), neoplastic and infectious etiology should be ruled out. If the isolated lymphadenopathy is present, it can affect at least two distinct nodal regions [13]. The moderate to massive splenomegaly is detected in more than 85 % of patients, and mild to moderate hepatomegaly is also common in ALPS patients [14]. Lymphadenopathy, hepatomegaly, and splenomegaly reveal a frequent improvement as the increase of age.
Autoimmunity is the second most common clinical manifestation in ALPS patients. Systemic autoimmunity is the disease manifestation frequently requiring medical intervention. Autoimmunity usually affects over 70 % of patients. Many patients have multiple cytopenias such as Coombs positive autoimmune hemolytic anemia (AIHA) and immune mediated thrombocytopenia [10]. Autoimmune neutropenia is, however, uncommon. Teachey et al. have diagnosed 12 patients with ES through flow cytometric analysis for CD4−/CD8− (double negative) T cells (DNTs) and definitive test for ALPS, as well as defective Fas-mediated apoptosis in vitro. Six patients (50 %) with ES have an elevated number of DNTs, as the suggestive indicator of ALPS [15]. Same as lymphoproliferation, hepatosplenomegaly in autoimmune manifestations may also achieve improvement as the extension of the age. In addition, rashes especially urticarial, immune-mediated pulmonary fibrosis, and SLE are also reported as the common manifestations in ALPS [16]. Other autoimmune manifestations including autoimmune nephritis, hepatitis, gastritis, arthritis, and uveitis are infrequently observed [8].
ALPS patients have an increased risk of secondary malignancies. The risk is approximately 10–20 % and is most prevalent in FAS mutant ALPS [17]. Increased risk of cancers has also been observed in unaffected family members who may inherit the same gene mutation without the development of a clinical ALPS phenotype. Straus et al. have investigated 223 members from 39 families, and their risk of non-Hodgkin and Hodgkin lymphoma was 14 and 51 times higher than the expected risk, respectively. The median onset age of Hodgkin’s disease (HD) and non-Hodgkin lymphoma (NHL) was 11 and 21 years old, respectively. The type of lymphoma was reported as B-cell-derived lymphomas including HD, Burkitt’s lymphoma, follicular B lymphoma, and T-cell-rich B cell lymphoma [14].
Laboratory Findings
The elevation of T cell receptor (TCR) αβ+/CD4-/CD8- T cells in peripheral blood and lymphoid tissues is the most significant characteristic of ALPS patients, but some patients maybe have normal numbers. Patients with other autoimmune diseases such as SLE and autoimmune thrombocytopenic purpura (ITP) may have mild reactive elevation of TCR α/β+ DNT cells, but not exceeding 5 %. The population of TCR αβ+ DNT cells required for the diagnosis is higher than or equal to 1.5 % of total lymphocytes or 2.5 % of CD3+ T lymphocytes [18]. DNT cells in ALPS patients reveal the co-expression of CD45RA, CD57, CD27, CD28, perforin, and HLA-DR, but lack the expression of CD45RO and CD56 [19]. The population of DNT cells detected by flow cytometry in normal people can be varied among laboratories based on different gating, so it is important to set up the normal value for a particular laboratory. The origin of DNT cells is not very clear. It is thought to originate from CD8+ T cells or thymus-derived regulatory T (Treg) cells [20, 21].
The apoptotic assay of abnormal lymphocytes is previously thought to be the gold standard for the diagnosis of ALPS and documented in the required diagnostic criteria. Apoptotic assay measures the percentage of activated primary lymphocytes undergoing apoptosis after FAS activation (using recombinant Fas ligand/TCR re-stimulation/cytokine starvation) [22]. Approximately 50 % or less cell death than the control is considered as abnormal [18]; however, this test is labor intensive and expensive and is available in only few laboratories. Moreover, patients with somatic mutation in FAS and germline FASLG mutations have normal Fas-induced apoptosis assay [23]. So this test is no longer considered as the mandatory diagnosis of ALPS. But, this test is also useful for the diagnosis of patients without the mutation of FAS, FASLG, or CASP10 genes.
Cytopenia is the one of the most common abnormal findings in laboratory due to autoimmune destruction or splenic sequestration, which is commonly observed in Coombs-positive hemolytic anemia and immune thrombocytopenia (together referred as Evans syndrome), while autoimmune neutropenia is less common. Autoimmune cytopenia may be difficult to distinguish from concomitant hypersplenism, and the examination of blood smears for hemolysis and measurement of autoantibodies may be helpful for establishing the distinction. Autoantibodies may be present in more than 92 % of patients and include positive Coombs’ direct antiglobulin, antiplatelet antibodies, antineutrophil antibodies, rheumatoid factor (RF), antinuclear antibodies (ANAs), and antiphospholipid antibodies. The titers of these antibodies are correlated with the number of DNT cells, which may be due to the elevated production of interleukin (IL)-10 that can induce the generation of anti-apoptotic protein Bcl-2 in both B and T cells and inhibit the cell death, and decreased production of IL-12 that can result in the decrease of T helper 1 (Th1) and the increase of T helper 2 (Th2) as well as the release of IL-4 and IL-5 in ALPS patients. Taken together, a series of these responses contribute to the production of antibodies and autoimmunity of ALPS [24].
Serum IL-10, IL-18, soluble FAS ligand (FasL), and vitamin B12 exhibit the common elevation in ALPS patients with FAS mutation and can be the useful biomarkers for the diagnosis of these patients [25, 26]. Recent studies have demonstrated that the presence of elevated TCR αβ+-DNT cells combined with high serum or plasma levels of either IL-10, IL-18, soluble FAS ligand (sFASL), or vitamin B12 can predict the mutation of germline or somatic FAS gene with the accurate rate of 85–97 % [25], which is confirmed by genetic analysis of Fas gene. Due to high specificity, these biomarkers have also documented as the diagnostic criteria [18], and the application of these biomarkers greatly facilitates the diagnosis in hospitals lacking the instruments of advanced genetic analysis or functional testing.
Hypergammaglobulinemia is also frequently present in ALPS patients [27]. Most patients have elevated IgG, IgA, or IgM. Nevertheless, only a small part of patients with ALPS (<10 %) have hypogammaglobulinemia and are susceptible to infection. In addition, 5–10 % of ALPS patients have common variable immunodeficiency disease (CVID) [28].
Histopathological findings in ALPS reveal the paracortical expansion due to the infiltration by polyclonal TCR α/β+ DNT cells accompanied by follicular hyperplasia and polyclonal plasmacytosis. Marked infiltration of DNT cells can lead to the architectural effacement of lymph nodes and lead to the erroneous diagnosis of T cell lymphoma in some patients. Moreover, although these DNT cells also express high proliferation index with mitosis and elevated expression of Ki-67, typically CD45RO negative, and express TIA-1 and CD57, which is opposite to lymphoma patients [18]. Similar with lymph nodes, the spleen also shows markedly expanded T cell areas dominated by DNT cells. Occasionally, the expansion of the splenic red pulps due to atypical T cell proliferation and extramedullary erythropoiesis in patients with anemia were reported [29]. In addition, the detection of TCR or BCR gene rearrangement does not show any monoclonal T or B cell population in ALPS.
Diagnostic and Classification Criteria and Differential Diagnosis
Since the investigators from NIH have established a triad of criteria for the diagnosis of ALPS in 1999 (Table 1) [30], important research advances have been achieved, which is a benefit for our understanding of this disease. In 2009, an international workshop from NIH has revised the criteria for the diagnosis and classification of ALPS and published the revised criteria in 2010 (Table 2) [18]. Uniformed and simplified diagnosis and classification of ALPS are highly desired, which will facilitate to the collaboration and data exchange between different clinicians and research centers all over the world. Based on these criteria, definitive diagnosis is based on the presence of two required criteria and one primary accessory criterion. A probable diagnosis is based on both required criteria plus one secondary accessory criterion. Compared with diagnostic criteria issued in 1999 [30], new diagnostic criteria do not require the apoptosis analysis of lymphocytes, because it is intensive resource during the accomplishment process and the variable results in different centers. In addition, apoptosis assay of lymphocytes is unable to identify patients with somatic FAS or germline FASLG mutations. Genetic information and other biomarkers such as mutated FAS, FASLG, or CASP10 and the levels of sFAS, IL-10, IL-18, and vitamin B12 with excellent prediction capacity of ALPS have been listed in the new criteria. The elevated number of CD3+TCRαβ+CD4−CD8− DNT cells has been set up as ≥1.5 % of total lymphocytes or 2.5 % of CD3+ lymphocytes. Moreover, histopathological examination and family history have not been used in the current diagnostic criteria. The addition of secondary accessory criteria may be helpful for supporting the diagnosis, even when both primary criteria may not meet the diagnostic requirements. Patients with probable ALPS should be treated and monitored in the same way as the patients with a definitive diagnosis from a clinical perspective, and are advised to be conducted a genetic or apoptosis assay-based diagnostic work whenever it is possible.
There is an absolute requirement for the presence of lymphadenopathy and/or splenomegaly persistent for more than 6 months. If the isolated lymphadenopathy is present, it can affect at least two distinct nodal regions. Lymphadenopathy caused by neoplasia and infections must be excluded. In many cases, hepatomegaly may be present, but it is not a diagnostic criterion [31].
The new revised criteria for the classification of ALPS are shown in Table 3 [18]. Patients with germline homozygous, or heterozygous mutations in FAS, previously classified as ALPS type 0 and Ia, respectively, are now named as ALPS-FAS. Similarly, patients with somatic FAS mutations should be classified as ALPS-sFAS. In addition, patients harboring Fas ligand mutations should be classified as ALPS-FASLG, and patients with caspase-10 mutations should be classified as ALPS-CASP10. Patients who fulfill diagnostic criteria for ALPS without genetic diagnosis can be classified as ALPS-U (undetermined), instead of ALPS type III. Despite the lack of a genetic diagnosis, these patients have a clinical progression similar to other patients with ALPS. Approximately two thirds of ALPS patients have an identified genetic defect in FAS gene. Among them, a germline mutation in FAS is the most commonly identified genotype. Somatic FAS mutation accounts for 0.5 % of ALPS patients, but patients with the defects of FasL and caspase 10 genes are less than 1 %, respectively. ALPS-U accounts for approximately 20 % of ALPS patients [32, 33].
ALPS is a rare disease that has achieved some novel advances in pathogenic mechanisms with an increasing attention in recent years. Recently, a study from Argentina has first reported a founder event in ALPS. In addition, two patients with homozygous mutation from a consanguineous family and three patients with heterozygous mutation from three unrelated families have been reported to have a missense mutation affecting the extracellular cysteine-rich domain 2 of Fas, p.Cys107Tyr (C107Y), which has evidenced that this mutation represents a single haplotype of FAS gene [34]. CASP10 defect is a specific abnormality in patients with ALPS type IIa according to new diagnostic criteria. But, recent studies have shown that the suspected patients caused by CASP10 defect could not be diagnosed as ALPS. Tadaki et al. [35] have diagnosed 50 patients with systemic juvenile idiopathic arthritis (s-JIA) by single-nucleotide polymorphism (SNP) array analysis and confirmed a 13-kb intragenic deletion of CASP10 through RT-PCR in one patient with symptoms including lymphadenopathy and splenomegaly; however, TCRaβ+CD4⁄CD8 DNT cells in the peripheral blood are not at a high level and lymphocyte apoptosis induced by anti-Fas antibody is normal in this patient. The UNC13D gene encoding Munc13-4 is involved in the secretion of perforin and associated with the development of familial hemophagocytic lymphohistiocytosis (F-HLH) and is considered as a candidate risk gene of ALPS and DALD [36]. Recently, somatic mutations in KRAS gene have also been reported to be associated with a non-malignant syndrome of autoimmunity and breakdown of leukocyte homeostasis with normal TCR α/β+ DNT cells [37, 38] and can be designated as RALD. Another study has found that miRNAs may play an important role in the pathogenesis of ALPS. Guo et al. have reported that the enhanced expression of miR-146 may be involved in the pathogenesis of ALPS via the downregulation of Fas, suggesting that transgenic mouse line with the overexpression of miR-146a can develop spontaneous immunity mimicking human ALPS, including enlarged spleen and lymph nodes, inflammatory infiltration in the liver and lung, increased levels of DNT cells in peripheral blood, and increased serum immunoglobulin G level [39].
The diagnosis of ALPS is based on clinical observation, laboratory abnormalities such as the defect of Fas-mediated apoptosis in vitro, and elevated number of TCRαβ+ DNT cells in peripheral blood. The recommended flow chart of the diagnosis for the suspected patients with ALPS is shown in Fig. 1 [18].

The suggested flow chart of the diagnosis for patients with suspected ALPS [18]
ALPS patients have highly heterogeneous clinical phenotypes similar with malignant, infectious, autoimmune, rheumatologic conditions and other lymphoproliferative disorders. These disorders are often distinguishable by histopathology of tissue biopsy (bone marrow and/or lymph node) at initial onset. Patients with germline mutations of caspase 8 (CASP8) presenting manifestations of lymphadenopathy and/or splenomegaly, marginal elevation of DNT cells, and recurrent infections are named as caspase-8-deficient state (CEDS) now, although they are previously classified as ALPS type IIb [40]. These patients show defective activation of T, B, and NK cells, and consequent recurrent bacterial and viral infections, which is different with other ALPS cases. The patients with clinical syndromes of autoimmune phenomena, lymphocyte accumulation and/or splenomegaly, elevated or normal DNT cells, and somatic mutations in NRAS, previously designated as ALPS type IV, have been classified as Ras-associated lymphoproliferative disorder (RALD) now [41]. No nomenclature modifications are suggested for the ALPS-associated Dianzani autoimmune lymphoproliferative disease (DALD) [42], which is characterized as autoimmunity, lymphadenopathy and/or splenomegaly, normal DNT cells, and defective Fas-mediated apoptosis of lymphocytes in vitro, and the genetic defects are not clear [28, 43]. The last type of ALPS-related disorder, the X-linked lymphoproliferative disease (XLP1), is caused by mutations or deletions in SH2D1A gene. These patients frequently present with fulminant Epstein-Barr viral infection, hypogammaglobulinemia, or lymphoma [44]. Revised classification of ALPS-related disorders and other disorders needing to be differentially diagnosed is shown in Tables 4 and 5 [16, 18], which includes ES, hemophagocytic lymphohistiocytosis (HLH), Castleman’s disease, chronic Epstein-Barr virus (EBV) infection, juvenile myelomonocytic leukemia (JMML), s-JIA, and other lymphoproliferative disorders [39, 45, 46]. Diligent review of family history in both children and adults is helpful for the diagnosis of inheritable genetic disorders including ALPS.
Treatment
ALPS is an incurable disease, and its treatments mainly focus on the elimination of lymphadenopathy, autoimmune disease, lymphoma, and other concurrent diseases.
The initial therapy for autoimmune multilineage cytopenia is similar to that for other immune-mediated cytopenia and applies high-dose corticosteroids with or without intravenous injection of immunoglobulin (IVIG). High-dose pulse therapy with intravenous injection of methylprednisolone (starting at 5–10 mg/kg), followed by low-dose oral administration of prednisone (1–2 mg/kg) as the therapeutic maintenance, has been successfully applied in many patients. Autoimmune cytopenia reveals the excellent response to corticosteroids, and short courses of high-dose treatment are more effective at controlling these conditions. While corticosteroids are too toxic for the application in chronic diseases, many patients reveal the relapse after drug withdrawal. Unlike many patients with sporadic immune-mediated thrombocytopenia, ALPS associated with thrombocytopenia may not respond to intravenous injection of immunoglobulin G [19].
Rituximab is usually used in a subset of ALPS patients with refractory autoimmune cytopenia. However, 5–10 % of ALPS patients can develop as CVID, and this risk is increased with the application of rituximab. Moreover, the treatment efficacy of rituximab is generally transient, and many patients also reveal the disease relapse after its withdrawal [47, 48]. Therefore, the application of rituximab is not recommended in ALPS now.
Previously, splenectomy was performed in almost half of patients with ALPS to manage severe hypersplenism or refractory cytopenia or splenic rupture [1]. However, patients after splenectomy have a very high risk for the development of sepsis such as fatal pneumococcal bacteremia, even the combinatorial application of antibiotic prophylaxis and vaccination [27]. The reason may be lack of circulating CD27+ memory B lymphocyte population that can sustain protective levels of antibodies against pneumococcal polysaccharide antigens [1] or is related to anti-polysaccharide IgM antibody production for the specific defect in ALPS patients [49]. More than 50 % of ALPS patients relapse with severe cytopenia after splenectomy, which proves it to be a failed strategy only increasing risk of pneumococcal sepsis [33]. If patients have splenectomy prior to their diagnosis, it is recommended to provide long-term antibiotic prophylaxis against pneumococal sepsis using penicillin V or fluoroquinolones such as levofloxacin and to maintain the vaccination every 4 or 5 years. In addition, these patients should be educated about the importance of seeking medical care promptly for a significant febrile illness requiring intravenous antibiotics until bacterial sepsis is ruled out [1]. Now, splenectomy is recommended to be a final choice except in patients with uncontrolled hypersplenism and failure in other medical management and life-threatening cytopenia due to splenic sequestration. For these patients, partial splenectomy or splenic embolization may be a more suitable method instead of complete splenectomy [29].
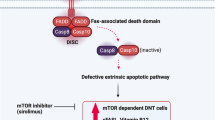
Mycophenolate mofetil (MMF), mammalian target of rapamycin (mTOR) inhibitors including sirolimus, and pentostatin have also been successfully applied in some children with refractory cytopenia [50–52]. MMF, a mycophenolic acid, is a reversible inhibitor of inosine monophosphate dehydrogenase (IMPDH) during purine (guanine) biosynthesis that is ultimately necessary for the growth of both T and B cells. Rao et al. [53] have analyzed 13 ALPS patients subjected to MMF treatment to reveal a measurable improvement in 80 % of patients with autoimmune diseases due to its characteristic of high treatment efficacy, convenient application, and high tolerance, thereby avoiding the application of splenectomy and/or steroids at the growth-impairing dose in children. However, these patients did not show an obvious improvement in lymphoproliferation or normalization of DNT cells although only partial patients reveal these therapeutic responses. The side effects of MMF include diarrhea and cytopenia as well as neutropenia. Therefore, the patients treated with MMF should be given close follow-up examination or observation. Sirolimus, an mTOR inhibitor, has also been successfully used to treat refractory cytopenia, and ALPS with other autoimmune manifestations in those cases with the failed treatment of initial corticosteroids and IVIG. Sirolimus has better treatment efficacy than conventional therapies, including MMF [53]. Sirolimus can cause the apoptosis of lymphocytes and the increase of Treg cells, so that it may be superior to MMF that inhibits lymphocyte proliferation without inducing the cell death of lymphocytes and the increase of Treg cells. Common toxicity observed in patients receiving sirolimus treatment includes hypercholesterolemia, hypertension, and mucositis [51]. The flow chart of the management recommend by NIH for patients with ALPS-associated chronic refractory cytopenia is shown in Fig. 2 [1].

Recommended flow chart of managements for patients with ALPS associated chronic refractory cytopenia [1]
Other targeted therapies are undergoing preclinical test and clinical trials. It is still not in consensus that pyrimethamine and sulfadoxine are effective in patients with ALPS, but their partial treatment efficacy is observed in some patients. Pentostatin, arsenic, and histone deacetylase (HDAC) inhibitor are also effective in ALPS patients and preclinical models of ALPS [52, 54, 55].
The only curative therapy for ALPS is hematopoietic stem cell transplantation (HCT). Bone marrow transplantation is required for patients with rare homozygous or compound heterozygous mutations for long-term survival. However, ALPS cases that experienced stem cell transplantation are extremely limited in currently published case reports. Long-term outcomes for the majority of ALPS patients treated with a single agent therapy are highly desired. As the development of new targeted therapies, bone marrow transplantation could be reserved only for those patients with highly refractory disease.
Conclusion
In summary, over the past decade, new insights of ALPS have been updated. More effective options for the management of refractory autoimmune complications have been applied in patients with ALPS to improve the life quality and relieve the clinical symptoms of these patients, which may lead to a new era of improved treatment outcome for ALPS.
References
Rao VK, Oliveira JB (2011) How I treat autoimmune lymphoproliferative syndrome. Blood 118:5741–5751
Canale VC, Smith CH (1967) Chronic lymphadenopathysimulating malignant Lymphoma.J. Paediatr 70:891–899
Sneller MC, Straus SE, Jaffe ES et al (1992) A novel lymphoproliferative⁄autoimmune syndrome resembling murinelpr⁄gld disease. J Clin Invest 90:334–341
Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Naqata S (1992) Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356:314–317
Takahashi T, Tanaka M, Brannan CI et al (1994) Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 76:969–976
Rieux-Laucat F, Le Deist F, Hivroz C et al (1995) Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 268:1347–1349
Fisher GH, Rosenberg FJ, Straus SE et al (1995) Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell 81:935–946
Worth A, Thrasher AJ H, Gaspar HB (2006) Autoimmune lymphoproliferative syndrome: molecular basis of disease and clinical phenotype. Br J Haematol 133:124–140
Hansford JR, Pal M, Poplawski N et al (2013) In utero and early postnatal presentation of autoimmune lymphoproliferative syndrome in a family with a novel FAS mutation. Haematologica 98:e38–e39
Deutsch M, Tsopanou E, Dourakis SP (2004) The autoimmune lymphoproliferative syndrome (Canale-Smith) in adulthood. Clin Rheumatol 23:43–44
der Werff V, ten Bosch J (2003) Autoimmune lymphoproliferative syndrome: etiology, diagnosis, and management. Paediatr Drugs 5:185–193
Teachey DT, Seif AE, Grupp SA (2010) Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS). Br J Haematol 148:205–216
Jackson CE, Puck JM (1999) Autoimmune lymphoproliferative syndrome, a Disorder of apoptosis. Curr Opin Pediatr 11:521–527
Straus SE, Jaffe ES, Puck JM et al (2001) The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 98:194–200
Teachey DT, Manno CS, Axsom KM et al (2005) Unmasking Evans syndrome: T-cell phenotype and apoptotic response reveal autoimmune lymphoproliferative syndrome (ALPS). Blood 105:2443–2448
Madkaikar M, Mhatre S, Gupta M, Ghosh K (2011) Advances in autoimmune lymphoproliferative syndromes. Eur J Haematol 87:1–9
Teachey DT (2012) New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome. Curr Opin Pediatr 24:1–8
Oliveira JB, Bleesing JJ, Dianzani U et al (2010) Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome: report from the 2009 NIH International Workshop. Blood 116:e35–e40
Bleesing JJ, Brown MR, Novicio C (2002) A composite picture of TcR alpha/ beta (+) CD4(−)CD8(+) T Cells (alpha/beta-DNTCs) in humans with autoimmune lymphoproliferative syndrome. Clin Immunol 104:21–30
Bleesing JJ, Brown MR, Dale JK et al (2001) TcR-alpha/beta (+) CD4(−)CD8(+)T cells in humans with the autoimmune lymphoproliferative syndrome express a novel CD45 isoform that is analogous to murine B220 and represents a marker of altered O-glycan biosynthesis. Clin Immunol 100:314–324
Fischer K, Voelkl S, Heymann J et al (2005) Isolation and characterization of human antigen-specific TCR alpha beta + CD4(−)CD8(+) double-negative regulatory T cells. Blood 105:2828–2835
Muppidi J, Porter M, Siegel RM (2004) Measurement of apoptosis and other forms of cell death. Curr Protoc Immunol. Chapter 3:Unit 3.17
Holzelova E, Vonarbourg C, Stolzenberg MC et al (2004) Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med 351:1409–1418
Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W (1999) An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med 130:591–601
Caminha I, Fleisher TA, Hornung RL et al (2010) Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol 125:946–949
Bowen RA, Dowdell KC, Dale JK et al (2012) Elevated vitamin B12 levels in autoimmune lymphoproliferative syndrome attributable to elevated haptocorrin in lymphocytes. Clin Biochem 45:490–492
Neven B, Magerus-Chatinet A, Florkin B et al (2011) A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood 118:4798–4807
Campagnoli MF, Garbarini L, Quarello P et al (2006) The broad spectrum of autoimmune lymphoproliferative disease:molecular bases, clinical features and long-term follow-up in 31 patients. Haematologica 91:538–541
Price S, Shaw PA, Seitz A et al (2014) Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood 123:1989–1999
Bleesing JJ, Straus SE, Fleisher TA (2000) Autoimmune lymphoproliferative syndrome. A human disorder of abnormal lymphocyte survival. Pediatr Clin North Am 47:1291–1310
Rao VK, Straus SE (2006) Causes and consequences of the autoimmune lymphoproliferative syndrome. Hematology 11:15–23
Chun HJ, Zheng L, Ahmad M et al (2002) Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419:395–399
Shah S, Wu E, Rao VK, Tarrant TK (2014) Autoimmune lymphoproliferative syndrome: an update and review of the literature. Curr Allergy Asthma Rep 14:462
Simesen de Bielke MG, Yancoski J, Rocco C et al (2012) A missense mutation in the extracellular domain of Fas: the most common change in Argentinean patients with autoimmune lymphoproliferative syndrome represents a founder effect. J Clin Immunol 32:1197–1203
Tadaki H, Saitsu H, Kanegane H et al (2011) Exonic deletion of CASP10 in a patient presenting with systemic juvenile idiopathic arthritis, but not with autoimmune lymphoproliferative syndrome type IIa. Int J Immunogenet 38:287–293
Aricò M, Boggio E, Cetica V et al (2013) Variations of the UNC13D gene in patients with autoimmune lymphoproliferative syndrome. PLoS One 8:e68045
Takagi M, Shinoda K, Piao J et al (2011) Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood 117:2887–2890
Niemela JE, Lu L, Fleisher TA et al (2011) Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood 117:2883–2886
Guo Q, Zhang J, Li J et al (2013) Forced miR-146a expression causes autoimmune lymphoproliferativesyndrome in mice via downregulation of Fas in germinal center B cells. Blood 121:4875–4883
Dowdell KC, Niemela JE, Price S et al (2010) Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood 115:5164–5169
Oliveira JB, Bidere N, Niemela JE et al (2007) NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A 104:8953–8958
Dianzani U, Bragardo M, DiFranco D et al (1997) Deficiency of the Fas apoptosis pathway without Fas gene mutations in pediatric patients with autoimmunity/lymphoproliferation. Blood 89:2871–2879
Ramenghi U, Bonissoni S, Migliaretti G et al (2000) Deficiency of the Fas apoptosis pathway without Fas gene mutations is a familial trait predisposing to development of autoimmune diseases and cancer. Blood 95:3176–3182
Snow AL, Marsh RA, Krummey SM et al (2009) Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest 119:2976–2989
Rudman Spergel A, Walkovich K, Price S et al (2013) Autoimmune lymphoproliferative syndrome misdiagnosed as hemophagocytic lymphohistiocytosis. Pediatrics 132:e1440–e1444
Nomura K, Kanegane H, Otsubo K et al (2011) Autoimmune lymphoproliferative syndrome mimicking chronic active Epstein-Barr virus infection. Int J Hematol 93:760–764
Rao VK, Price S, Perkins K et al (2009) Use of rituximab for refractory cytopenias associated with autoimmune lymphoproliferative syndrome (ALPS). Pediatric Blood Cancer 52:847–852
Cooper N, Davies EG, Thrasher AJ (2009) Repeated courses of rituximab for autoimmune cytopenias may precipitate profound hypogammaglobulinaemia requiring replacement intravenous immunoglobulin. Br J Haematol 146:120–122
Neven B, Bruneau J, Stolzenberg MC et al (2014) Defective anti-polysaccharide response and splenic marginal zone disorganization in ALPS patients. Blood 124:1597–1609
Arora S, Singh N, Chaudhary GK, John MJ (2011) Autoimmune lymphoproliferative syndrome: response to mycophenolate mofetil and pyrimethamine/sulfadoxine in a 5-year-old child. Indian J Hematol Blood Transfus 27:101–103
Teachey DT, Greiner R, Seif A et al (2009) Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol 145:101–106
Bajwa R, Savelli S, Gross T (2011) Pentostatin for treatment of refractory autoimmune lymphoproliferative syndrome. Pediatr Blood Cancer 57:336–337
Rao VK, Dugan F, Dale JK et al (2005) Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol 129:534–538
Bobé P, Bonardelle D, Benihoud K et al (2006) Arsenic trioxide: a promising novel therapeutic agent for lymphoproliferative and autoimmune syndromes in MRL/lpr mice. Blood 108:3967
Dowdell KC, Pesnicak L, Hoffmann V et al (2009) Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, diminishes lymphoproliferation in the Fas deficient MRL/lpr(−/−) murine model of autoimmune lymphoproliferative syndrome (ALPS). Exp Hematol 37:487–494
Acknowledgments
This work was financially supported by the grants of the Science and Technology Project from Education Department of Jiangxi province (GJJ14021) and the Natural Science Foundation of Jiangxi province (20142BAB205072)
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Author’ contributions
P.L. designed and wrote the manuscript. P.H. contributed to manuscript revision and final manuscript review. Y.Y., M.H., and H.P. contributed to manuscript revision. F.L. contributed to the design of the review and the approval of the final manuscript.
Pu Li and Ping Huang contributed equally to this work.
Rights and permissions
About this article

Cite this article
Li, P., Huang, P., Yang, Y. et al. Updated Understanding of Autoimmune Lymphoproliferative Syndrome (ALPS). Clinic Rev Allerg Immunol 50, 55–63 (2016). https://doi.org/10.1007/s12016-015-8466-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-015-8466-y