
Abstract
Mutations in the Fas gene (TNFRSF6) are the most common causes of Autoimmune Lymphoproliferative Syndrome (ALPS-FAS).
Purpose
In Argentina almost a third of patients with ALPS-FAS present a missense mutation affecting the extracellular cysteine rich domain 2 of Fas, p.Cys107Tyr (C107Y). This change was found in homozygous state in 2 patients from a consanguineous family, and heterozygously, in 3 other patients from 3 unrelated families. In these families, 12 relatives were identified as healthy carriers of the mutation. We sought to test the hypothesis that this mutation actually represents a single haplotype of TNFRSF6.
Methods
DNAs from ALPS-C107Y patients and their families, as well as from 150 Argentinean control subjects were sequenced for the known higher frequency single nucleotide polymorphisms (SNPs) of TNFRSF6. The C107Y-carriers were also genotyped at 5 microsatellites proximal to the Fas gene locus.
Results
All C107Y alleles presented a unique intragenic haplotype that could be restricted to this group. Extent of haplotype sharing and variability of microsatellite alleles in C107Y chromosomes support the presence of a single haplotype block including the mutation and encompassing 2.395 Mb.
Conclusions
A founder effect for C107Y has been evidenced in this work and the most common recent ancestor to the patients probably lived 350 years ago. This constitutes the first report of a founder event in ALPS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The autoimmune lymphoproliferative Syndrome (ALPS) is an immunologic disorder characterized by chronic massive, nonmalignant lymphadenopathy and splenomegaly, with an expansion of a normally rare population of T cells bearing α-β antigen receptors but lacking both CD4 and CD8 coreceptors, known as double negative T cells (DNT) [1–4]. According to a recent revision of the diagnostic criteria and classification for ALPS, a great number of ALPS patients harbor heterozygous germline or somatic mutations in Fas gene (TNFRSF6), ALPS-FAS and ALPS-sFAS respectively [5]. Fas (CD95/APO-1), a member of the tumor necrosis factor receptors (TNFR) superfamily, is a homotrimeric transmembrane receptor forming a complex that, triggered by a homotrimeric ligand (FasL), transmits an apoptosis signal via the intracellular domain, specially through the region termed “death domain” (DD). This pathway is crucial in lymphocyte homeostasis, serving to delete antigen-driven and autoreactive T cell clones [6–8].
The majority of patients presented mutations involving the region coding the intracellular domain of TNFRSF6, particularly the DD [9–11]. Although in lesser number, extracellular mutations in TNFRSF6 are found to display lower clinical penetrance of ALPS than those affecting the intracytoplasmic region [12–15]. Among the extracellular mutations, most of them are commonly nonsense, insertions, deletions, and splice site mutations with frameshift, with only a few number of missense changes [13].
Also very rare, so far only 3 patients have been identified harboring germline homozygous mutations in TNFRSF6 with a complete loss of Fas expression, an early onset and a severe phenotype of ALPS [1, 16–18]. Another report showed three siblings with combined different germinal mutations with slightly reduced expression of Fas and less severe clinical manifestations [19].
We report here 5 patients with ALPS-FAS phenotype carrying an extracellular missense mutation, 2 homozygous siblings and 3 heterozygous non related children, from 4 unrelated Argentinean families. In these patients the point mutation p.Cys107Tyr affecting the extracellular domain of Fas and leading to an impaired apoptosis assay, could be attributed to a single haplotype that probably originated from a single founder event.
Patients, Material and Methods
Patient Cohort
The samples in the study were collected from patients with suspicion of ALPS who were referred for molecular and functional studies to the Hospital Nacional de Pediatría J. P. Garrahan. Patients 1 and 2 were diagnosed and treated in the Immunology Unit in our Hospital; the rest of the patients were referred from Hospital Pedro Elizalde in Buenos Aires, and Hospital Sor Maria Ludovica in La Plata, Argentina. Informed consent was obtained from each patient or parental guardian prior to participation and the family members in accordance with the Declaration of Helsinki. The research protocol was approved by the Local Ethic Committee of our Institution.
Mutation Analysis
Genomic DNA was obtained by saline extraction from peripheral blood mononuclear cells (PBMCs). The patients were screened for the presence of mutations in the coding exons and exon/intron boundaries of TNFRSF6 through PCR amplification of genomic DNA followed by direct sequencing of the PCR products with primers and conditions as previously described [10].
Total RNA was extracted from PBMCs of patients with Trizol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. For patient 1, total RNA was isolated from T cells activated 9 days with phytohemagglutinin (PHA) (GIBCO). Fas cDNA PCR amplification was performed using primers and conditions as previously described [10].
Bioinformatics Analysis of Fas Amino Acid Substitutions
PolyPhen (Polymorphism Phenotyping) [20] is a bioinformatics method that predicts the possible impact of an amino acid substitution on the structure and function of a human protein by using physical and comparative considerations. The result is given as a score resulting for the protein change as “putative benign” (0.00–1.50), “possibly damaging” (1.51 to 2.00), and “probably damaging” (>2.01).
Single Nucleotide Polymorphisms and Microsatellites Analysis
We analyzed 6 SNPs (1 exonic (c.836C>T) and 5 intronic (−1377g>a, −691t>c, −671a>g, c.528(+46)c>t, c.699(+82)c>g) [21] and 5 extragenic microsatellites (UniSTS database) on genomic DNA. SNPs genotyping was performed with DYEnamic ET Terminator Cycle Sequencing Kit (Amersham Biosciences, Little Chalfont Buckinghamshire, UK) from PCR products as previously described [21]. Microsatellites, namely D10S1687, D10S541, D10S1739, D10S1753 and D10S185, were PCR-amplified with fluorescent-labeled specific forward primers (from UniSTS database, NCBI), run on an ABI PRISM 3130 Genetic Analyzer, and analyzed using the Gene Scan software (Applied Biosystem).
Control Group
Genomic DNA was extracted from 150 unrelated healthy Argentinean blood donors (300 chromosomes). These participants provided consent under a separate ethics protocol for healthy donors. We performed a Restriction Fragment Length Polymorphism assay (RFLP) for SNP c.528(+46)c>t by using NlaIII enzyme according to manufacturer’s recommendations (New England Biolabs, Ipswish, MA). The rest of SNPs were analyzed by direct sequencing from PCR products, as described above.
To define the haplotype in some controls, PCR products were cloned using TOPO TA cloning kit (Invitrogen) following the manufacturer’s instructions.
Age Estimation
In order to estimate the age to the most recent common ancestor of haplotypes carrying the C107Y mutation, a previously described likelihood method [22] was modified and applied on a set of five recombination markers located at variant distances at both sides of the disease mutation. The considerations of this method were introduced by Yancoski et al [23].
Results
Identification of Fas Mutation
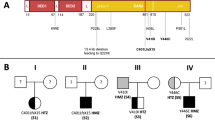
Germinal mutations in TNFRSF6 are frequently present in a proportion of patients with ALPS (ALPS-FAS). We found that almost 40 % of the Argentinean ALPS-FAS infants presented p.Cys107Tyr (c.514G>A), a germline substitution accounting for the most frequently observed TNFRSF6 mutation in this country. Actually, 5 patients (2 homozygous and 3 heterozygous) belonging to 4 unrelated families harbored this missense mutation affecting the sequence coding the cysteine rich domain 2 (CRD2) in the extracellular region of the protein (Fig. 1). C107Y was the only change revealed by direct sequencing of the 9 exons and the adjacent intronic regions of TNFRSF6.

Identification of C107Y mutation associated with ALPS-Fas. a DNA Sequencing. Direct sequencing of exon 3 with a reverse primer showed a homozygous change in patient 1: a missense mutation TGT(C)107TAT(Y) ( ) and the same change in heterozygous state in healthy carriers, as the mother of family 1. b C107Y leads to a normal-sized RT-PCR product. Total RNA was isolated from patient 1T cells, activated 9 days with PHA. The RT-PCR assay was performed with exonic primers spanning exon 3 to exon 9 of Fas cDNA (predicted size 572 bp). M: Marker, P1: Patient 1, C’ and C: controls. Sequencing of the RT-PCR product revealed TGT(C)107TAT(Y) (
) and the same change in heterozygous state in healthy carriers, as the mother of family 1. b C107Y leads to a normal-sized RT-PCR product. Total RNA was isolated from patient 1T cells, activated 9 days with PHA. The RT-PCR assay was performed with exonic primers spanning exon 3 to exon 9 of Fas cDNA (predicted size 572 bp). M: Marker, P1: Patient 1, C’ and C: controls. Sequencing of the RT-PCR product revealed TGT(C)107TAT(Y) ( ) without other abnormalities. c Pedigree of the 4 unrelated families. The patients are indicated by a red square or circle edge. The homozygous patients are shown in the fully black square or circle. The empty squares and circles indicate the wild type (wt) phenotype in the relatives. The incomplete squares or circles correspond to healthy heterozygous relatives
) without other abnormalities. c Pedigree of the 4 unrelated families. The patients are indicated by a red square or circle edge. The homozygous patients are shown in the fully black square or circle. The empty squares and circles indicate the wild type (wt) phenotype in the relatives. The incomplete squares or circles correspond to healthy heterozygous relatives
Clinical and immunological data of these patients are summarized in Table I. The changed residue is highly conserved between human and murine Fas [24], and also among TNFR superfamily members [25]. Actually, this amino acid substitution was neither reported as a SNP in the NCBI SNP database (dbSNP Short Genetic Variations) nor found in our healthy control group (data not shown). We also carried out bioinformatics analysis of C107Y mutation; PolyPhen [20] predicted that C107Y is “probably damaging” of altering local protein structures (score 3.608), and SIFT [26] another bioinformatics method, estimated an affected protein function.
PCR amplification of cDNA from the homozygous patients gave a normal-sized product, as was the case for the heterozygous patients (Fig. 1b and data not shown). Sequencing of the RT-PCR product revealed a wild-type sequence with the exception of the missense mutation c.514G>A (Fig. 1b). Therefore, this change had no effect at the mRNA level in terms of expression and splicing fidelity.
The consanguineous parents and the three healthy siblings of the patients from family 1 were also heterozygous for C107Y mutation, as well as 7 healthy relatives from the other three families (Fig. 1c).
Mutation C107Y Segregated with a Unique Gene Haplotype
Due to the high frequency of this missense mutation in our group of patients, we sought to establish whether the C107Y mutation was inherited among patients as a single shared allele. For this, we analyzed in the carrier group 6 SNPs of TNFRSF6 known to have a minor allele frequency (MAF) ≥5 % and being in linkage disequilibrium [21]. All patients and C107Y-healthy carriers shared the same SNP profile for these 6 SNPs (Table II), and the delineated haplotype, GTATCC, showed wild type alleles except for the minor allele t in the SNP c.528(+46)c>t. Among the haplotypes identified in our study (Table II), only GTATCC was not observed in the study carried out in Caucasians by Niemela et al [21]. Alike, none of the 300 chromosomes from our control group carried the haplotype GTATCC (data not shown), indicating that its frequency remains less or equal than 0.003 % in this population. Therefore, the probability that the mutation C107Y has occurred by chance, sharing the same set of polymorphic markers in the 4 affected families, is not larger than the probability of randomly sampling four times in a row the mutated haplotype, namely (0.01)4 = 10−8. Together, these data indicated that probably GTATCC haplotype is unique, and strongly suggested that the mutated allele has been originated from a single founder.
Founder Effect Analysis
To further confirm that C107Y represents a single ancestral event, we analyzed 5 microsatellites surrounding TNFRSF6 in chromosome 10. The genotyping of these microsatellites showed that the C107Y carriers shared a common haplotype block, encompassing 2.395 Mb. The alleles in D10S1687 and D10S185, more distant from the mutation, have shown recombination evidences (Fig. 2). Estimation of the age to the most common recent ancestor (MCRA) was undertaken from the information provided by these 5 extragenic recombination markers, applying a likelihood method based on that originally described by Genin et al. [22] and modified later [23]. For family 1, only one of the two alleles carrying the mutation was considered since the family was inbred.

Extent of the haplotype shared by the C107Y alleles. The five microsatellites studied in patients defined the common region. The brown arrow shows the extension of the common block. Brown-boxes: shared region. Dotted line: not shared region. Red Cross ( ): recombination points. Physical distances and references (#) are from UniSTS. Recombination frequencies for microsatellites from NCBI: D10S1687: 2,17, D10S541: 0, 804, D10S1739: 0,066, D10S1753: 1,76, D10S185: 4,685
): recombination points. Physical distances and references (#) are from UniSTS. Recombination frequencies for microsatellites from NCBI: D10S1687: 2,17, D10S541: 0, 804, D10S1739: 0,066, D10S1753: 1,76, D10S185: 4,685
The extent of the haplotype shared and the variability of microsatellite alleles in C107Y-chromosomes suggested that this mutation arose approximately 14 generations back, excluding a shared grandparent with a confidence of 99 %. Considering 25 years per generation, MCRA lived about 350 years ago (95 % confidence interval, 75 to 1,050 years ago).
Discussion
Among ALPS-FAS Argentinean patients an unexpected high number of them (5 out 12) harbored the missense mutation C107Y, lying in the region encoding the extracellular domain of the Fas receptor.
ALPS is a rare and complex genetic disorder due to the wide spectrum of clinical, immunological features and penetrance related to the mutation gene-site; although in the last years significant advances in our understanding of the disease has been done. Mutations located in the Fas extracellular domain (ECD) usually display lower clinical penetrance and recently it was reported that a second event would be necessary for disease expression in such cases. Indeed, somatic events affecting the second TNFRSF6 alleles were found associated with germline ECD mutations (novel somatic mutations or duplications of the mutant allele with loss of the wild-type allele) [27]. In this way, we looked for the presence of a second mutational event, and found a somatic mutation rending homozygous the change C107Y in DNT cells of patient 4 (data not shown).
Thereby, partial clinical penetrance was more frequently found for heterozygous germline TNFRSF6 mutations affecting ECD [11], as shown in our families presenting 12 healthy heterozygous relatives. On the contrary, a complete clinical penetrance was observed in the two C107Y-homozygous siblings, in keeping with the few reported cases from patients with germline homozygous mutations [16–18]. Indeed, they had a severe clinical presentation, with complete lack of Fas expression at the surface and no detectable apoptosis in the assay. However, homozygous patients from family 1 constitute the first reported cases with missense homozygous ECD changes, functionally expressing a Fas-null phenotype.
The physiopathological mechanism accounting for the C107Y heterozygous mutants, as for the rare patients carrying ECD missense mutations [2, 9] remains unknown. Haploinsufficiency has been proposed as a common disease mechanism in ALPS patients with extracellular FAS mutations [13, 14]. For instance, in preliminary studies by ectopic expression of the mutated protein carrying C107Y, we could observe a normally synthesized protein but a very low surface expression; because it is not achieving the cellular membrane (Simesen de Bielke unpublished data).
The finding of identical rare mutations in apparently unrelated patients from different regions in the country suggested that, despite the geographic separation, the families could share a common ancestry. By studying intragenic SNPs we discovered a unique profile, GTATCC in all C107Y-carriers. The most frequent haplotypes found in Caucasians by Niemela et al [21], appeared in our families in the alleles not associated with C107Y. According to the same study, Fas gene SNPs could reflect ethnic differences, as the African American group presented haplotypes not described in the Caucasians [21], thus it may be possible that the unique haplotype found in our ALPS Argentinean families could be privative. For instance, we could not exclude an Amerindian ancestry in these families. It should be interesting to analyze the presence of this haplotype in local populations, despite the difficulty to have access to a fully native population, like tribes or criollos (“local” origin).
In this work we also demonstrated a founder effect for C107Y and the MCRA to the patients probably lived 350 years ago (95 % CI, 75–1,050 years ago). This confidence interval would be narrowed if more data were available. It has been estimated that approximately 56 million people died as a result of the European colonization of America. This gave rise to a reduction of the Indian population size to only 10 % of its former level [28]. Also, massive transatlantic immigration starting in 1860 significantly modified the human genetic landscape of Argentina [29], rending difficult to identify a single most probable hypothesis concerning the ethnic origin of this founder effect. It is noteworthy that there is only one patient reported with C107Y from another country, but he was described as a novo mutation [26], supporting a founder event occurred in Argentina.
Founder mutations have been described for several diseases and for some primary immunodeficiencies, such as Hemophagocytic Lymphohistiocytosis [30, 31], Mendelian Susceptibility to Mycobacteria [23, 32], and FOXN1 deficiency in Nude SCID patients [33]. This study represents the first description of a founder event in patients with Autoimmune Lymphoproliferative Syndrome.
References
Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268(5215):1347–9.
Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober W, Lenardo MJ, Puck JM. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81(6):935–46.
Bidere N, Su HC, Lenardo M. Genetic disorders of programmed cell death in the immune system. Annu Rev Immunol. 2006;24:321–52.
Su HC, Lenardo MJ. Genetic defects of apoptosis and primary immunodeficiency. Immunol Allergy Clin N Am. 2008;28(2):329–51. ix.
Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, Rieux-Laucat F, Siegel RM, Su HC, Teachey DT, Rao VK. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116(14):e35–40.
Brunner T, Mogil RJ, LaFace D, Yoo NJ, Mahboubi A, Echeverri F, Martin SJ, Force WR, Lynch DH, Ware CF, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373:441–4.
Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature. 1995;373:438–41.
Ju ST, Panka DJ, Cui H, Ettinger R, el-Khatib M, Sherr DH, Stanger BZ, et al. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373:444–8.
Vaishnaw AK, Orlinick JR, Chu JL, Krammer PH, Chao MV, Elkon KB. The molecular basis for apoptotic defects in patients with CD95 (Fas/Apo-1) mutations. J Clin Invest. 1999;103:355–63.
Rieux-Laucat F, Blachère S, Danielian S, De Villartay JP, Oleastro M, Solary E, Bader-Meunier B, Arkwright P, Pondaré C, Bernaudin F, Chapel H, Nielsen S, Berrah M, Fischer A, Le Deist F. Lymphoproliferative syndrome with autoimmunity: a possible genetic basis for dominant expression of the clinical manifestations. Blood. 1999;94(8):2575–82.
Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, Dale JK, Fleisher TA, Middelton LA, Sneller MC, Lenardo MJ, Straus SE, Puck JM. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet. 1999;64:1002–14.
Roesler J, Izquierdo JM, Ryser M, Rösen-Wolff A, Gahr M, Valcarcel J, Lenardo MJ, Zheng L. Haploinsufficiency, rather than the effect of an excessive production of soluble CD95 (CD95{Delta}TM), is the basis for ALPS Ia in a family with duplicated 3’ splice site AG in CD95 intron 5 on one allele. Blood. 2005;106(5):1652–9.
Kuehn HS, Caminha I, Niemela JE, Rao VK, Davis J, Fleisher TA, Oliveira JB. FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome. J Immunol. 2011;186(10):6035–43.
Hsu AP, Dowdell KC, Davis J, Niemela JE, Anderson SM, Shaw PA, Rao VK, Puck JM. Autoimmune lymphoproliferative syndrome due to FAS mutations outside the signal-transducing death domain: molecular mechanisms and clinical penetrance. Genet Med. 2012;14(1):81–9.
Neven B, Magerus-Chatinet A, Florkin B, Gobert D, Lambotte O, De Somer L, Lanzarotti N, Stolzenberg MC, Bader-Meunier B, Aladjidi N, Chantrain C, Bertrand Y, Jeziorski E, Leverger G, Michel G, Suarez F, Oksenhendler E, Hermine O, Blanche S, Picard C, Fischer A, Rieux-Laucat F. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood. 2011;118(18):4798–807.
Le Deist F, Emile JF, Rieux-Laucat F, Benkerrou M, Roberts I, Brousse N, Fischer A. Clinical, immunological, and pathological consequences of Fas-deficient conditions. Lancet. 1996;348:719–23.
Kasahara Y, Wada T, Niida Y, Yachie A, Seki H, Ishida Y, Sakai T, Koizumi F, Koizumi S, Miyawaki T, Taniguchi N. Novel Fas (CD95/APO-1) mutations in infants with a lymphoproliferative disorder. Int Immunol. 1998;10(2):195–202.
van der Burg M, de Groot R, Comans-Bitter WM, den Hollander JC, Hooijkaas H, Neijens HJ, Berger RM, Oranje AP, Langerak AW, van Dongen JJ. Autoimmune lymphoproliferative syndrome (ALPS) in a child from consanguineous parents: a dominant or recessive disease? Pediatr Res. 2000;47(3):336–43.
Bettinardi A, Brugnoni D, Quiroos-Roldan E, Malagoli A, La Grutta S, Correra A, Notarangelo LD. Missense mutation in the Fas gene resulting in autoimmune lymphoproliferative syndrome: a molecular and immunological analysis. Blood. 1997;89(3):902–9.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Niemela JE, Hsu AP, Fleisher TA, Puck JM. Single nucleotide polymorphisms in the apoptosis receptor gene TNFRSF6. Mol Cell Probes. 2006;20(1):21–6.
Genin E, Tullio-Pelet A, Begeot F, Lyonnet S, Abel L. Estimating the age of rare disease mutations: the example of Triple-A syndrome. J Med Genet. 2004;41:445–9.
Yancoski J, Rocco C, Bernasconi A, Oleastro M, Bezrodnik L, Vrátnica C, Haerynck F, Rosenzweig SD. A 475 years-old founder effect involving IL12RB1: a highly prevalent mutation conferring Mendelian Susceptibility to Mycobacterial Diseases in European descendants. Infect Genet Evol. 2009;9(4):574–80.
Watanabe-Fukunaga R, Brannan CI, Itoh N, Yonehara S, Copeland NG, Jenkins NA, Nagata S. The cDNA structure, expression, and chromosomal assignment of the mouse Fas antigen. J Immunol. 1992;148(4):1274–9.
Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66(2):233–43.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Magerus-Chatinet A, Neven B, Stolzenberg MC, Daussy C, Arkwright PD, Lanzarotti N, Schaffner C, Cluet-Dennetiere S, Haerynck F, Michel G, Bole-Feysot C, Zarhrate M, Radford-Weiss I, Romana SP, Picard C, Fischer A, Rieux-Laucat F. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest. 2011;121(1):106–12.
Black FL. Why did they die? Science. 1992;258:1739–40.
Avena S, Via M, Ziv E, Pérez-Stable EJ, Gignoux CR, Dejean C, Huntsman S, Torres-Mejía G, Dutil J, Matta JL, Beckman K, Burchard EG, Parolin ML, Goicoechea A, Acreche N, Boquet M, Ríos Part Mdel C, Fernández V, Rey J, Stern MC, Carnese RF, Fejerman L. Heterogeneity in genetic admixture across different regions of Argentina. PLoS One. 2012;7(4):e34695.
Lee SM, Sumegi J, Villanueva J, Tabata Y, Zhang K, Chakraborty R, Sheng X, Clementi R, de Saint Basile G, Filipovich AH. Patients of African ancestry with hemophagocytic lymphohistiocytosis share a common haplotype of PRF1 with a 50delT mutation. J Pediatr. 2006;149(1):134–7.
Danielian S, Basile N, Rocco C, Prieto E, Rossi J, Barsotti D, Roche PA, Bernasconi A, Oleastro M, Zelazko M, Braier J. Novel syntaxin 11 gene (STX11) mutation in three Argentinean patients with hemophagocytic lymphohistiocytosis. Clin Immunol. 2010;30(2):330–7.
Sologuren I, Boisson-Dupuis S, Pestano J, Vincent QB, Fernández-Pérez L, Chapgier A, Cárdenes M, Feinberg J, García-Laorden MI, Picard C, Santiago E, Kong X, Jannière L, Colino E, Herrera-Ramos E, Francés A, Navarrete C, Blanche S, Faria E, Remiszewski P, Cordeiro A, Freeman A, Holland S, Abarca K, Valerón-Lemaur M, Gonçalo-Marques J, Silveira L, García-Castellano JM, Caminero J, Pérez-Arellano JL, Bustamante J, Abel L, Casanova JL, Rodríguez-Gallego C. Partial recessive IFN-gR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. 2011;20(8):1509–23.
Adriani M, Martinez-Mir A, Fusco F, Busiello R, Frank J, Telese S, Matrecano E, Ursini MV, Christiano AM, Pignata C. Ancestral founder mutation of the nude (FOXN1) gene in congenital severe combined immunodeficiency associated with alopecia in southern Italy population. Ann Hum Genet. 2004;68(Pt 3):265–8.
Acknowledgments
This research was supported by Agencia de Promocion Cientifica y Tecnologica grants PICT2004 21235 and PICT2008 0915.
Disclosures
The authors have no financial conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Simesen de Bielke, M.G., Yancoski, J., Rocco, C. et al. A Missense Mutation in the Extracellular Domain of Fas: The Most Common Change in Argentinean Patients with Autoimmune Lymphoproliferative Syndrome Represents a Founder Effect. J Clin Immunol 32, 1197–1203 (2012). https://doi.org/10.1007/s10875-012-9731-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-012-9731-y