
Abstract
The autoimmune lymphoproliferative syndrome (ALPS) or Canale–Smith syndrome is a recently described clinical entity consisting of chronic, non-malignant lymphadenopathy and hepatosplenomegaly together with hypergammaglobulinemia, positive autoantibodies and/or overt autoimmune diseases. It is caused by a genetic defect in the mechanism of programmed cell death (apoptosis) and is characterized by the presence of double-negative (TCR α/β CD4− CD8−) T lymphocytes (DNT). Although well known in pediatric patients, ALPS is an unusual diagnosis in adults. The oldest reported patient was aged 54. We describe another two adult patients in whom a presenting autoimmune disease led to the diagnosis of ALPS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
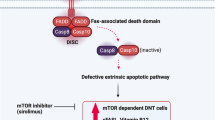
The autoimmune lymphoproliferative syndrome [1], (Canale–Smith) [2], is a recently described syndrome caused by the breakdown of lymphocyte homeostasis and immunologic tolerance, due to a genetic defect in the apoptosis mechanism. Patients with ALPS exhibit a clinical picture resembling that of lpr/gld (lyphoproliferative/generalized lymphoproliferative disease) mice, which are considered an animal model of an unusual form of systemic lupus erythematosus [3]. Characteristic double-negative T lymphocytes (DNT), not present in classic lupus, lacking CD4 and CD8 receptors, circulate in increased numbers (TCR α/β CD4− CD8−). The result is lymphadenopathy, hepatosplenomegaly, hypergammaglobulinemia, positive autoantibodies and/or overt autoimmune diseases. In ALPS, despite lymphoproliferation and an increased risk of lymphoma [4], no malignancy can be documented histopathologically.
The average age at presentation in ALPS patients is 1.8 years; however, several rare cases have been diagnosed in adulthood. The oldest reported patient was 54 years old. In the present report we describe another two adult patients with autoimmunity and non-malignant lymphoproliferation who fulfill the diagnostic criteria for ALPS.
Case reports
Patient 1
A 50-year-old woman presented with a 5-month history of weight loss and difficulty in swallowing. On clinical examination cervical lymphadenopathy, hepatosplenomegaly and an enlarged Waldeyer’s ring were found. Laboratory tests revealed Coombs’-positive autoimmune hemolytic anemia. The antinuclear antibodies, anti-dsDNA and antiU1-RNP were positive, and polyclonal hypergammmaglobulinemia (35.6%) together with 34% DNT cells were found. A CT scan revealed enlarged axillary, mediastinal and retroperitoneal lymph nodes, together with hepatomegaly and a 15-cm enlarged spleen. Bone marrow smear and bone marrow biopsy showed non-specific reactive changes. Upper gastrointestinal endoscopy was normal. Cervical lymph node biopsies and Waldeyer’s ring lymphatic tissue biopsy revealed follicular T-cell hyperplasia without signs of malignancy. Glucocorticosteroid therapy was started, resulting in remission of the autoimmune hemolytic syndrome. Six years later, without any medication, the patient is well, but lymphadenopathy and organomegaly are still present.
Patient 2
A 40-year-old woman presented with gingival hemorrhage and melena. On examination petechiae, generalized lymphadenopathy, hepatomegaly and splenomegaly were found. The Ht was 20%, the WBC 10 000/mm3 and the platelet count was 10 000/mm3. Bone marrow examination revealed normal cellularity and a large number of platelet-producing megakaryocytes. The immunofluorescence test for antiplatelet antibodies was positive, and immune thrombocytopenia was diagnosed. On serum electrophoresis the γ-globulins were 45.2%, with a polyclonal increase in all subclasses of immunoglobulins. The antinuclear antibodies were positive with a titer of 1:1280 and speckled fluorescence. The anti-ds-DNA were positive, the anti-ENA antibodies negative and the serum complement level was within normal limits. Lymphocyte phenotyping revealed 12% DNT and an increased percentage of lymphocytes (64%) expressing FAS death factor (CD95). Upper gastrointestinal endoscopy revealed Helicobacter pylori-negative hemorrhagic gastritis. Biopsies of the lymph nodes, pharyngeal lymphoid tissue and stomach showed reactive hyperplasia with a mixed polyclonal lymphocytic infiltration without signs of malignancy. Therapy with corticosteroids and human γ-globulin was started. Two years later the patient is asymptomatic, free of bleeding disorders with a normal platelet count, but with impressive persistent lymphadenopathy and hepatosplenomegaly.
Discussion
In this report we describe two patients who presented with clinical features of a severe autoimmune cytopenic syndrome (the first patient had autoimmune hemolytic anemia and the second patient autoimmune thrombocytopenia) associated with hypergammaglobulinemia, positive autoantibodies, hepatosplenomegaly and lymphadenopathy. An important characteristic of their disorder was the expansion of the unusual DNT lymphocytes. These two cases fulfill the diagnostic criteria of ALPS or Canale–Smith syndrome. The diagnosis is based on clinical criteria, although genetic studies proving the characteristic mutations in the apoptosis mechanism have not been performed.
The hallmark of these two ALPS cases is that the diagnosis was made in adulthood. ALPS is usually diagnosed in the first months of life, with an average age at presentation of 1.8 years [5], whereas adult cases have rarely been reported.
On the other hand, follow-up studies in individuals who share genetic apoptotic defects revealed that the clinical expression of ALPS might be highly variable. Penetrance of the disease seems to be in direct relation to the site of the mutations [6]. It is also known that several environmental factors or other, yet unknown, genetic factors may interact, resulting in a wide spectrum of disease signs and symptoms, which may become clinically relevant at any age.
Some patients are healthy and have only a subclinical ‘in vitro’ apoptotic defect, which can be found at any time during routine family genetic studies. In symptomatic patients the disease may go from a mild, insignificant lymphadenopathy and/or splenomegaly to a full-blown syndrome of lymphoproliferation and autoimmune phenomena. Thus, the diagnosis may be delayed, usually until the development of the first signs of autoimmune disease [7].
On the other hand, it has recently been reported that the risk of non-Hodgkin’s and Hodgkin’s lymphoma is 14.5 times higher in patients with apoptotic defects up to 48 years after lymphoproliferation is first documented [8]. Fas mutations were found in 11% of patients with non-Hodgkin’s lymphoma [9]; therefore, in some cases, lymphoma development, which can occur at any age, might represent the final step in the natural history of ALPS.
So, although ALPS is less often reported in adults, a wide spectrum of this disease may be present at any age and must be considered as a possible diagnosis in some characteristic cases.
In conclusion, ALPS should be suspected in patients with autoimmune features and chronic lymphoproliferation in whom neither systemic erythematosus lupus nor lymphoma can be diagnosed. The number of DNT should be determined, or genetic studies for defects in the apoptosis mechanism should be performed. It is possible that with a better diagnostic strategy, more underdiagnosed adult ALPS cases can be uncovered. ALPS in adults may be more common than previously believed.
Abbreviations
- ALPS:
-
Autoimmune lymphoproliferative syndrome
- DNT:
-
Double-negative lymphocytes
References
Straus SE, Sneller M, Lenardo MJ, Puck J, Strober W (1999) An inherited disorder of lymphocyte apoptosis: The autoimmune lymphoproliferative syndrome. Ann Intern Med 130:591–601
Drappa J, Vaishaw AK, Sullivan KE, Chu JL, Elkon KB (1996) Fas gene mutations in the Canale–Smith syndrome an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med 335:1643–1649
Sneller MC, Straus SE, Jaffe ES et al. (1992) A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest 90:334–341
Straus SE, Jaffe ES, Puck JM et al. (2001) The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 98:194–200
Jackson CE, Puck JM (1999) Autoimmune lymphoproliferative syndrome, a disorder of apoptosis. Curr Opin Pediatr 11:521–527
Rieux Laucat F, Blachere S, Danielan S et al. (1999) Lymphoproliferative syndrome with autoimmunity: a possible genetic basis for dominant expression of the clinical manifestations. Blood 94:2575–2582
Infante AJ, Britton HA, DeNapoliu T et al. (1998) The clinical spectrum in large kindred with autoimmune lymphoproliferative syndrome caused by a Fas mutation that impairs lymphocyte apoptosis. J Pediatr 133:629–633
Straus SE, Jaffe ES, Puck JM et al. (2001) The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood 98:194–200
Gronbaek K, Straten PT, Ralfkiaer E et al. (1998) Somatic Fas mutations in non-Hodgkin’s lymphoma: association with extranodal disease and autoimmuníty. Blood 92:3018–3024
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Deutsch, M., Tsopanou, E. & Dourakis, S.P. The autoimmune lymphoproliferative syndrome (Canale–Smith) in adulthood. Clin Rheumatol 23, 43–44 (2004). https://doi.org/10.1007/s10067-003-0830-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-003-0830-2