
Abstract
Gestational diabetes mellitus (GDM) is defined as carbohydrate intolerance that begins or is first recognized during pregnancy. The prevalence of GDM is highly variable, depending on the population studied, and reflects the underlying pattern of diabetes in the population. GDM manifests by the second half of pregnancy and disappears following delivery in most cases, but is associated with the risk of subsequent diabetes development. Normal pregnancy induces carbohydrate intolerance to favor the availability of nutrients for the fetus, which is compensated by increased insulin secretion from the maternal pancreas. Pregnancy shares similarities with adiposity in metabolism to save energy, and both conditions favor the development of insulin resistance (IR) and low-grade inflammation. A highly complicated network of modified regulatory mechanisms may primarily affect carbohydrate metabolism by promoting autoimmune reactions to pancreatic β cells and affecting insulin function. As a result, diabetes development during pregnancy is facilitated. Depending on a pregnant woman’s genetic susceptibility to diabetes, autoimmune mechanisms or IR are fundamental to the development autoimmune or non-autoimmune GDM, respectively. Pregnancy may facilitate the identification of women at risk of developing diabetes later in life; autoimmune and non-autoimmune GDM may be early markers of the risk of future type 1 and type 2 diabetes, respectively. The most convenient and efficient way to discriminate GDM types is to assess pancreatic β-cell autoantibodies along with diagnosing diabetes in pregnancy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gestational diabetes mellitus (GDM) is defined as carbohydrate intolerance that begins or is first recognized during pregnancy. Its etiopathogenesis and prevalence have been studied since the 1960s [1]. The website of the National Center for Biotechnology Information lists more than 12,500 research articles and reviews published to date on GDM.
The prevalence of GDM is highly variable, depending on the population studied and diagnostic test and glycemic cutoff value used [2]. GDM development has been reported in association with 1 % of all pregnancies in Sweden [3] and in more than 20 or 30 % of pregnant women in Sardinia, Italy [4], and Asia [5]. The population-based prevalence of GDM generally reflects the underlying pattern of diabetes in the background population [4, 6]. The prevalence of diabetes in the general population of Europe is about 6–8 %; about 90 % of these cases are type 2 diabetes (T2D) and 10 % are type 1 diabetes (T1D), mostly autoimmune in origin [7].
GDM usually manifests by the second half of pregnancy, and it disappears after delivery in most cases. Normal pregnancy induces carbohydrate intolerance to mobilize more nutrients for fetal growth, which in turn creates an increased demand for insulin production by maternal pancreatic β cells. Failure to meet this increased need for insulin due to insufficient glucose update by insulin-sensitive tissues [causing insulin resistance (IR)] and/or insufficient secretion of insulin by β cells results in increased blood glucose levels. Many researchers previously considered the pathophysiology of GDM to be similar to that of IR-mediated T2D [2], and GDM has been considered a high-risk factor for T2D development later in life in up to 70 % of affected women [8]. More recently, however, growing evidence has indicated that GDM is similar to autoimmune T1D or latent autoimmune diabetes of adults (LADA) in a proportion of patients [9], as reviewed comprehensively by Lapolla and colleagues in 2009 [10]. In the worst-case scenario, half of affected patients will develop autoimmune diabetes a few years after index pregnancy [11]. In addition, discrimination between GDM and other types of diabetes that were previously undiagnosed or develop coincidentally at the time of pregnancy represents a diagnostic challenge during pregnancy [10].
This review describes recent perspectives on GDM, with particular focus on the autoimmune aspects of GDM and pregnancy. The population of patients with GDM appears to consist of at least four subpopulations (Fig. 1): pregnant women with (i) autoimmune or (ii) non-autoimmune GDM (considered to be “true” GDM), who are at risk for postpartum T1D and T2D, respectively, and those with existing (iii) T1D or (iv) T2D that is first diagnosed during pregnancy (missed diabetes). The contribution of pregnancy in the sense of “true” GDM is probably to identify women at risk of developing diabetes later in life [12]. The pathophysiological mechanisms underlying autoimmune and non-autoimmune GDM are likely distinct, but they may overlap [13, 14] and the type of diabetes developed may depend on which risk factors dominate. Still, importantly, these two types of GDM require different treatments and have different prognoses with respect to postpartum maternal health [15, 16]. Pancreatic β-cell autoantibodies during and after pregnancy are useful markers for discrimination of these conditions and prediction of the probability of autoimmune diabetes development later in life, respectively. The diagnostic or predictive value of certain β cell autoantibodies in cases of autoimmune GDM has not yet been determined, and the prediction of T2D development following GDM is even more problematic.

Putative changes in carbohydrate metabolism associated with pregnancy and aging in healthy and diabetes-prone women that contribute to the risk of diabetes. Healthy women (green line) who never develop diabetes still face increased insulin demand due to insulin resistance during pregnancy and weight gain later in life (e.g., due to menopause). However, the increased need for insulin is compensated by the pancreas, and the sum of diabetic risk factors never exceeds the threshold level. Women who are at risk of type 1 diabetes (T1D; red line) may exceed this threshold during pregnancy and develop autoimmune diabetes, which shows clinical improvement after delivery in many cases, but leads to T1D development 1–4 years postpartum in some cases. Similarly, women at risk of type 2 diabetes (T2D; blue line) may develop non-autoimmune diabetes during pregnancy and again 5–15 years postpartum. In some cases, the diagnosis of diabetes during pregnancy reflects the presence of this condition before pregnancy. Missed T1D (red dashed line) usually manifests earlier in life than missed T2D (blue dashed line), and both groups of patients remain diabetic postpartum. Diabetes first diagnosed during pregnancy is defined collectively as gestational diabetes mellitus (GDM)
Healthy Pregnancy: Changes in Glucose Metabolism and Immune Mechanisms
The somatogenic and lactogenic hormones of the placenta and maternal pituitary gland integrate the metabolic adaptations of pregnancy with the demands of fetal and neonatal development. Healthy pregnancy is associated with IR and activation of an inflammatory response (Fig. 2). IR is transient during pregnancy to facilitate the mobilization of maternal nutrients for fetal growth [17], and manifests as elevated postprandial glycemia, fasting hyperlipidemia (in the form of increased triglycerides, low-density lipoprotein particles, and free fatty acids), and accelerated ketosis [18]. Insulin sensitivity decreases by 50–70 % in normal and diabetic pregnancies; it returns to normal postpartum in women with normal glucose tolerance, but not in all cases of diabetic pregnancy [19]. Increased maternal obesity, hormones present in pregnancy [i.e., estrogen, progesterone, and prolactin], but especially placental growth hormone (GH) [17], human placental lactogen (hPL), leptin, and cortisol [20], as well as placental tumor necrosis factor α (TNF-α) [21], and magnesium cation (Mg2+) deficiency [22] may be responsible for increased IR in pregnancy.

Overview of interactions between physiological adaptations during pregnancy (yellow) and conditions associated with the risk of type 1 (T1D, red) or type 2 (T2D, blue) diabetes in the manifestation of autoimmune or non-autoimmune gestational diabetes mellitus (GDM). Ab autoantibodies, HLA-G human leukocyte antigen G, CRP C-reactive protein, GH growth hormone, hPL human placental lactogen, IL interleukin, LIF leukemia inhibitory factor, Mg2+ magnesium cation, MCP-1 monocyte chemotactic peptide 1, RANTES regulated upon activation of normal T cells expressed and secreted, TNF-α tumor necrosis factor alpha, T-reg T regulatory lymphocytes, VIP vasoactive intestinal peptide
Placental GH [17] and hPL [23] control embryo growth, likely via the opposing actions of insulin and insulin-like growth factors, inducing IR in humans [24]. hPL and prolactin increase maternal food intake by inducing central leptin resistance [17]. Resistin is one of the adipocyte-derived signaling molecules called adipokines. Adipokines and/or their receptors are expressed in the placental tissue, contributing to maternal IR and ultimately to fetal growth. The levels of leptin and resistin in serum and placenta increase as pregnancy progresses, and high levels of both adipokines have been detected in umbilical plasma [25]. Conversely, the serum level of the insulin-sensitizing adipokine adiponectin decreases. However, recent studies have shown that TNF-α, but no other maternal or placental hormone (including cortisol and hPL), is the primary mediator of progressive IR during pregnancy [21]. TNF-α has also been found to be the link between IR and the physiological increase in inflammatory response in normal pregnancy [26] via the stimulation of hepatic C-reactive protein (CRP) production [27, 28]. TNF-α is a pro-inflammatory cytokine produced by many cell types, including macrophages, lymphocytes, fibroblasts, keratinocytes, adipocytes, and placental tissue [29], in response to inflammation, infection, and other environmental stresses. Interleukin (IL)-15, another pleiotropic inflammatory cytokine, is produced by multiple tissues, including the placenta, skeletal muscle, kidney, lung, heart, pancreas, monocytes/macrophages, and numerous cell types under various stimulatory conditions [30]. IL-15 is a powerful T cell growth factor facilitating the activation of autoreactive cluster of differentiation (CD)8(+) T cells by weak autoantigens [31]. A constant increase in IL-15 level is expected during the progression of a healthy pregnancy, as the fetus [32] and placenta [33] produce IL-15 in the processes of fetal immune response development and preparation for delivery. Nevertheless, pregnancy is characterized by intracellular Mg2+ depletion [34]. Mg2+ regulates ion channels and cellular processes, and serves as a cofactor for many essential metabolic reactions. It is involved in multiple steps of insulin signal transduction pathways, such as insulin secretion, binding, and receptor activity [35], suggesting that reduced intracellular Mg2+ decreases insulin receptor activity, impairs post-receptor action, and results in increased IR during pregnancy [34].
In normal pregnancy, IR is compensated by expansion of the functional β cell mass in the pancreas, preventing chronic hyperglycemia and GDM development [17]. This compensation is controlled by hPL and prolactin [17] and mediated by epigenetic regulation in islet cell genes. Rodent studies have revealed underlying changes in the expression of several islet microRNAs in compensatory β cell expansion. Decreased expression of miR-338-3p was associated with the promotion of islet cell proliferation and the protection of these cells from apoptosis [36]. β cell proliferation requires intact prolactin receptor signaling [37]. Any disturbance in β cell or insulin function, as expected in T1D and T2D, respectively, will challenge the compensatory mechanism underlying these physiological changes in pregnancy. The complexity of these mechanisms and inconsistent findings regarding metabolic changes in GDM have prevented us from drawing a comprehensive picture of diabetes-related metabolism in GDM [38]. For this reason, these aspects of GDM are not considered in detail in the present review. The role of microorganisms, including commensal microflora that may influence diabetes development during pregnancy and after delivery, is also not discussed. These factors are of general importance for diabetes development and have been discussed elsewhere [39, 40].
Pregnancy in Women at Risk of T1D may Result in Autoimmune GDM Development
In general, the risk of T1D is determined by the magnitude of autoimmunity against pancreatic β cells. In patients with GDM, general risk factors for T1D (reviewed elsewhere [10, 41, 42]) combine with pregnancy-related physiological changes in glucose metabolism and immune system regulation. The likelihood of GDM development is positively related to the number of risk factors present at the time of pregnancy (Fig. 2). Women with autoimmune GDM are commonly normal weight [4] and young (aged <30 years); they are more likely to require insulin treatment during pregnancy beyond dietary adjustment. They are more likely than women with non-autoimmune GDM to show pancreatic autoantibody positivity [12] and increased prevalence of human leukocyte antigen (HLA) DR3-DQ2/x or DR4-DQ8/x haplotypes [43]. In healthy pregnancy or pregnancy at risk of diabetes, anti-pancreatic β cell autoimmunity is the primary autoimmune reaction triggered, while other forms of autoimmunity are suppressed to avoid maternal immune reaction to the semi-allograft fetus. Pancreatic β cells may be preferentially targeted due to a combination of the embryo’s need for nutrients, which primarily involves the regulation of maternal carbohydrate metabolism, and the adaptation of maternal immune mechanisms to support the pregnancy, although this interpretation remains speculative.
Autoimmune GDM is associated with changes in chemokine concentrations in peripheral blood. The monocyte chemotactic peptide (MCP)-1 level is increased, and, the regulated upon activation, normal T cell expressed and secreted (RANTES) level is decreased in diabetic pregnancy [44]. MCP-1 serves as a pro-inflammatory activator of various immune cells [45], and RANTES may act as an immunomodulator suppressing the maternal allogeneic response [46]. Decreased expression of vasoactive intestinal peptide (VIP) at the implantation site has also been detected in prediabetic non-obese diabetic mice [47]. VIP is produced by trophoblast cells, where it induces a tolerogenic environment by increasing the expression of IL-10, transforming growth factor β, and Forkhead box P3 [48]. The expression level of VIP may serve as a link between placental trophoblasts, and the activation of autoimmune processes as a low VIP level activates proinflammatory leukemia inhibitory factor and T regulatory cells in contrast to VIP expression in healthy implantation [47]. Recent study findings suggest that certain subpopulations of T regulatory cells have reduced suppressive function in GDM [49]. These above-described mediators are needed to control excessive inflammation and moderate immune response, which are of vital importance for a successful and healthy pregnancy [50]. These changes augment inflammation during pregnancy, increasing the risks of T1D and unfavorable outcome of diabetic pregnancy; they may also activate adaptive immunity, further impairing pregnancy outcome.
Thus, in addition to the activation of innate immunity and inflammation, pregnancy may be accompanied by the activation of autoimmune reactions. GDM was previously thought not to be autoimmune in origin. This supposition was supported by similar frequencies of HLA DR2, DR3, and DR4 antigens in healthy pregnant women and those with GDM, as well as the low prevalence of markers of β cell autoimmune destruction in women with GDM [19]. However, adiposity is usually increased during pregnancy [20], and recent studies have shown that overnutrition under this condition results in the necrotic death of engorged adipocytes, leading to recruitment of classically activated macrophages to clear cellular debris [51]. These macrophages express molecules suitable for antigen presentation (major histocompatibility complex II, CD1d, costimulatory molecules, and CD11c), and necrotic cell-derived antigens can potentially be presented to T cells and B cells. This process activates adaptive immunity, resulting in clonal expansion of CD4+ T helper (Th) 1 cells and recruitment of CD8+ T cells. CD8+ T cells produce cytokines that increase the recruitment of macrophages into adipose tissue, and interferon-γ secreted by CD4+ Th1 cells increases their activation, establishing a vicious cycle of inflammation [51]. Furthermore, B cells can produce cytokines that affect T cell development, which in turn drives B cells to produce pathogenic antibodies [52]. In addition, B-cell responses can deplete T regulatory cells [53], resulting in adiposity-associated inflammation. Thus, inflammation initiated in adipose tissues propagates a vicious cycle that further worsens IR [52, 53]. Escalating IR is toxic to pancreatic β cells and further facilitates the development of pancreatic β-cell autoimmune responses [51]. The present authors have recently demonstrated an association between adiposity and pancreatic antibodies in diabetes (unpublished results).
Regardless of whether pregnancy is associated with increased adiposity, the antigenic load of the fetus has been postulated to trigger the diabetogenic process [54]. Membrane-bound or soluble (s) HLA-G expressed on or by the placental trophoblast cells is a central regulator of maternal immune mechanisms during pregnancy [55]. Interaction between HLA-G and nuclear factor kappa B (NF-κB) has been suggested to be central in events leading to GDM development. In general, HLA-G has anti-inflammatory and tolerogenic properties, including the induction of T regulatory cells, modulation of CD8+ T-cell cytotoxicity, and regulation of CD4+ T lymphocyte function [56]. These functions are partially shared with IL-10, which regulates the expression of HLA-G. However, the level of IL-10 is reduced in IR states [57]. In addition, HLA-G likely protects pancreatic islet cells [56]. Probably as a compensatory event, an increased plasma sHLA-G level is associated with the degree of glucose tolerance and is directly related to the levels and action of IL-6, a cytokine involved in subclinical inflammatory status in T2D, obesity, early stages of altered glucose tolerance, and several autoimmune diseases [58]. Under physiological conditions, activated CD14+ monocyte cells are primarily responsible for sHLA-G secretion into plasma in response to inflammatory stimuli [59].
As mentioned above, a constant increase in IL-15 level is expected during the progression of a healthy pregnancy due to fetal development. However, IL-15 production by mononuclear phagocytes and pancreatic β cells mediates inflammatory reactions in the pancreatic cells during the development of diabetes [60] by enabling autoreactive CD8+ T-cell responses to weak antigens; in other words, this process changes the avidity of autoreactive T cells [31]. Increased serum IL-15 levels are common to many autoimmune diseases in humans, including T1D [61], but also to pregnancies with preeclampsia [62] and other situations in which the maternal immune system reacts against the fetus [43]. The inflammatory properties of IL-15 are opposed by vitamin D in many organs, including the pancreatic islets [63] and placenta [64]. Pregnancy, however, is associated with increased need for vitamin D, primarily for fetal bone development. The maternal kidneys, and possibly the placenta, decidua, and fetal kidneys, try to meet this increased vitamin D requirement by providing the necessary 1-α hydroxylase activity; this enzyme hydroxylates vitamin D metabolite 25-hydroxycolecalciferol into biologically active 1α,25-dihydroxycolecalciferol [1,25(OH)2D3]. The result of this process is a several-fold increase in serum vitamin D level in healthy pregnancy compared with the pre-pregnancy level [65]. However, the prevalence of vitamin D deficiency is increased among pregnant women, despite sufficient exposure to sunlight [66]. These data collectively suggest the supportive effect of pregnancy for anti-pancreatic β-cell autoimmunity development (in addition to underlying genetic susceptibility) [10, 41, 42].
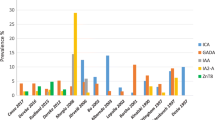
The prevalence and profile of pancreatic β-cell autoantibodies among women with GDM mirror the prevalence of T1D outside of pregnancy and depend on the profile of HLA DQ8/DR3/DR4 haplotypes in the background population [67]. The Finnish population is unique because of the high incidence rate of T1D, but similar frequency of T2D to other countries in Europe [68]. In this population, the prevalence of autoantibodies to islet cells (ICA), glutamic acid decarboxylase-65 (GADA), and insulinoma-associated protein 2 (IA-2A; 12.5, 5.9 and 4.7 %, respectively) is significantly higher among pregnant women with than among those without GDM, whereas the prevalence of antibodies to insulin (IAA) is similar (1 and 0.5 %, respectively) [12]. The incidence rate of T1D is also notably high in Sardinia, Italy [69], where 29, 14.5, and 3.2 % of women with GDM show positivity for IA-2A, IAA, and GADA, respectively. Almost 40 % of patients with GDM in Sardinia show positivity for at least one pancreatic autoantibody [4], in contrast to <9 % of patients in northern Italy (where GADA was detected in only 1.4 % of women with GDM) [70] and 6 % of patients in Sweden (where GADA was detected in <6 %, ICA in about 3 %, and IA-2A in about 2 % of women with GDM) [11]. Only one published study has assessed the prevalence of antibody to zinc transporter 8 (anti-ZnT8) in women with GDM, and the authors concluded that anti-ZnT8 detection would contribute about 2 % to autoantibody positivity among women with GDM and GADA and IA-2A negativity [71]. The presence of pancreatic β-cell autoantibodies has been known for some time to be associated with the need for insulin treatment during GDM [72]. Collectively, evidence is sufficient to prove that pancreatic autoantibody screening would be informative in cases of GDM, and that the pattern and prevalence of particular pancreatic autoantibodies are population dependent.
The post-GDM risk of T1D development also depends on the prevalence of T1D in the background population, which in turn determines the prevalence of anti-pancreatic autoimmunity among women with GDM (Fig. 1). These antibodies are present years before T1D onset and can occasionally be revealed in association with GDM. However, the profiles of different pancreatic β-cell autoantibodies may indicate whether GDM is an early manifestation of LADA or T1D [4, 73]. About 5 % of all post-GDM patients, but about 50 % of post-GDM patients with positivity to three pancreatic antibodies, develop T1D within 6 years postpartum in the Finnish population. However, the presence of ICA during GDM is the best single marker for the prediction of postpartum T1D development; more than 60 % of women with post-GDM T1D showed ICA positivity during GDM, whereas 56 % of such women showed GADA positivity, 38 % showed IA-2A positivity, and 0 % showed IAA positivity [12]. In Northern Italy, where <9 % of patients with GDM have any type of pancreatic autoantibody, the overall postpartum T1D rate was 3.5 % [70]. Similar results were obtained in a Swedish population, where autoimmune GDM was present in 6 % of patients with GDM and the overall rate of T1D or LADA was 3 % [11]. However, the diabetes rate among antibody-positive women with GDM in this population was 50 % [11]. Overall, post-GDM autoimmune diabetes can be predicted best by assessing all three antibodies (GADA, ICA, and IA-2A); this method has shown 67–100 % sensitivity, and the use of ICA or IA-2A in combination with GADA has shown 60–90 % sensitivity [74].
T1D and LADA manifest rapidly after index pregnancy. About half of patients with post-GDM T1D develop the condition in the first year postpartum, and most other patients develop it within 4 years postpartum; only 2 % of patients develop T1D or LADA after this 4-year timepoint [11]. The first 2 years postpartum are thus most critical in terms of T1D development [74]. The timing of GDM manifestation also affects the probability of postpartum T1D development, with early pregnancy manifestation increasing this risk [15]. However, early pregnancy detection of diabetes may include patients with existing T1D that is not diagnosed before pregnancy [75].
In addition to pancreatic autoimmunity, GDM increases the risk of anti-thyroid antibody production. Anti-thyroid autoimmunity in GDM may be due to at least two conditions: pregnancy and a woman’s general risk of autoimmunity (Fig. 2). Pregnancy with GDM [76] and healthy pregnancy [77] may be associated with increased production of thyroid antibodies and altered thyroid function. Normally, the levels of circulating thyroid antibodies decrease during pregnancy, and small changes in thyroid hormonal level may contribute to energy conservation [78]. Anti-thyroid autoimmunity during pregnancy seems to be far more prevalent than GDM; anti-thyroid peroxidase (TPO) or thyroid globulin antibodies have been detected in about 10–16 % of pregnant women [76, 79] and more than 80 % of those who underwent routine GDM screening (only anti-TPO), whereas only 26 % of the latter patients had GDM [80]. These data suggest that thyroid autoimmunity is induced primarily by pregnancy, and that diabetes represents an additional risk (Fig. 2). Autoimmune thyroid disease has been associated with T2D with GADA positivity (our unpublished data) and with T1D [81]. Anti-TPO antibody detection during pregnancy is correlated positively with the level of thyroid-stimulating hormone (TSH) [80], indicating the presence of (subclinical) hypothyroiditis during pregnancy. However, a combination of high TSH and thyroid autoimmunity in early pregnancy increases the risk of GDM more than four times, as well as increasing the risk of adverse pregnancy outcome [79]. Hypothyroiditis is associated with decreased rates of insulin-stimulated glucose transport into cells, which can lead to IR [82].
Several studies have examined thyroid antibodies in GDM, but data on the presence of other organ-specific or non-specific autoantibodies are very limited. In 1991, Bech and colleagues [83] reported that parietal cell autoantibodies were present in 27 %, antibodies to TSH in 50 %, rheumatoid factor in about 18 %, and adrenal autoantibodies in 5 % of pregnant women with insulin-dependent diabetes. The limited data available suggest that pregnancy has no overall effect on autoimmunity development, despite the fostering of pancreatic β-cell or thyroid autoimmunity. This postulation is in agreement with the general understanding of immunomodulation during pregnancy to avoid unnecessary reaction to fetal allogeneic antigens.
Pregnancy in Women at Risk of T2D may Result in Non-Autoimmune GDM Development
The risk of T2D is defined by the magnitude of IR (Fig. 2) [2]. In non-pregnant women, adiposity is the most significant mediator of IR. It is associated with increased inflammation and changes in metabolic function. The prevalence of weight gain is increasing constantly, and up to 40 % of women and more than 20 % of girls are obese worldwide [84]. In pregnancy, weight gain intensifies physiological changes in carbohydrate metabolism and can lead to the development of non-autoimmune GDM (discussed by Carpenter [18]). Just as the prevalence of autoimmune GDM reflects that of T1D, the prevalence of non-autoimmune GDM mirrors the prevalence of T2D in the background population [12, 42]. As expected, the phenotype of pregnant women with non-autoimmune GDM is opposite to that described for autoimmune GDM [4, 12].
In adipose women who become pregnant or when pregnancy is associated with increased adiposity, adipocytes are responsible for the initiation in inflammation, which together with adipokines contributes to IR. In contrast to a reasonable increase in adipose tissue in healthy pregnancy, extensive adiposity increases adipose tissue-specific inflammation and induces adipose tissue hypoxia [85] due to altered angiogenesis [86]. In women with GDM, adipocytes produce significantly higher levels of the adipokine resistin compared with those in pregnant women with normal glucose tolerance and much more than the same cells in non-pregnant women [26]. In GDM, resistin and IL-6 levels are correlated positively [26]. Inflammatory cytokines, such as TNF-α and IL-6, are produced by adipocytes as well as by monocytes and macrophages, and their levels are increased in obese individuals. These cytokines activate transcription factor NF-κB to increase, in turn, the expression of resistin, mainly by macrophages but also by adipocytes, in humans [87]. High levels of resistin selectively inhibit hepatic insulin action on glucose metabolism, thereby increasing glucose production by the liver and contributing to IR. Thus, the physiological function of resistin is believed to be the maintenance of blood glucose during fasting [88]. Another important protein, the hormone adiponectin is secreted exclusively by white adipose tissue in inverse proportion to the extent of body fat mass. Adiponectin receptors are widely distributed in peripheral tissues and possess anti-diabetic, anti-atherogenic, and anti-inflammatory properties, among others [89]. Adiponectin also plays a key role in mediating IR and β cell dysfunction in the pathogenesis of diabetes [90]. Through ceramide deacylation stimulation, adiponectin promotes the survival of functional β cell mass, favoring insulin production to meet insulin demands [91]. Women with lower levels of adiponectin (adipose) in the first trimester of pregnancy thus have higher levels of IR and are more likely to develop GDM [92]. The increased level of CRP in GDM is also partly associated with increased adiposity [93] and IR [94]. These data indicate that adipocytes are capable of creating a low-grade inflammatory environment under conditions of adiposity.
However, chronic low-grade inflammation can be initiated by adipocytes in the absence of obesity, which can cause IR similar to that found in obese individuals. Our understanding of the origin of adipocytes has changed in the past 5 years. White and brown adipose tissues have been suggested to originate from distinct types of mesenchymal precursor or even from hematopoietic cells [95], explaining the central role of adipocytes in the initiation of inflammation. Research on rodents has shown that lean animals with chronic inflammatory disease develop IR at the level of muscle and liver tissue, and that this mechanism is mediated by lean adipose tissue [96]. In that experiment, adipose tissue was infiltrated by macrophages and the levels of MCP-1 and IL-6 were increased [96]. Another rodent study elegantly demonstrated that the infusion of free fatty acids into lean animals decreased insulin sensitivity in muscle cells [97]. Similarly, humans with some other chronic inflammatory conditions (chronic hepatitis C, rheumatoid arthritis, inflammatory lung disease) are at increased risk of diabetes because of elevated TNF-α [98] and IL-6 [99] levels. TNF-α elevation stimulates inhibitory phosphorylation of serine residues of the insulin receptor substrate family, promoting IR at the post-receptor level of insulin action [98]. Serum IL-6 elevation is also associated with diminished insulin secretion [99], indicating the role of inflammatory cytokines in promoting IR.
Opposite to T1D, the T2D-prone environment is characterized by a decreased serum IL-15 level. IL-15 is involved in the mechanisms that attenuate the deleterious effect of TNF-α to promote IR in muscle cells [100]. The actions of IL-15 are modulated by vitamin D in many regulations, including maturation of adipocytes capable of producing adipokines [101]. However, the vitamin D level in pregnant women is commonly very low [66], which also affects calcium metabolism. In the presence of a low calcium level, 1,25(OH)2D3 favors the expression of inflammatory cytokines (TNF-α, IL-6, IL-15) and inhibits the expression of anti-inflammatory cytokines (adiponectin) by adipocytes and muscle cells [102]. These data show that coincident vitamin D deficiency, pregnancy, and adiposity may further favor an inflammatory environment. The extent of inflammatory activation, in turn, encourages GDM development in women with pregnancy-associated IR [103]. Thus, excessive adiposity contributing to subclinical inflammation favors the development of GDM. However, as discussed above, escalating IR is toxic to pancreatic β cells, further facilitating the development of pancreatic β-cell autoimmune responses [13, 14, 51]. This effect closes the circle and suggests, at least theoretically, that a proportion of women with non-autoimmune GDM may develop autoimmune diabetes later in life or during subsequent pregnancy. Pancreatic β-cell autoantibody detection would be informative in this regard.
The prediction of T2D is far more difficult than the prediction of T1D by pancreatic β-cell autoantibody detection. In general, multiparity, the magnitudes of weight gain during and after pregnancy, low efficacy of IR maintenance, and postpartum insulin requirement are well-known markers of increased T2D risk [2]. However, an elevated fasting glucose level during pregnancy has been the most important risk factor for future T2D [8]. About 2 years after GDM, women continue to show significant deviations in several metabolic and inflammatory markers compared with women with normal glucose regulation during pregnancy. Importantly, higher serum triglyceride levels within the low–normal range (0.84–1.21 mmol/l) at 2–24 months after pregnancy with GDM have been associated with an increased risk of early impaired glucose intolerance [104]. Another study found that serum plasminogen-activator inhibitor-1 possessed similar prognostic value for impaired glucose intolerance and metabolic syndrome development by 12–24 weeks after delivery [105]. The overall risk of T2D within 5–16 years postpartum is 17–63 % for women with GDM [106]. The cumulative incidence of T2D increases markedly in the first 5 years after delivery and seems to plateau after 10 years (Fig. 1) [8].
Conclusions
There is enormous literature on both main types of diabetes -T1D and T2D, including the role of microflora [107, 108], inflammatory responses, including gestational influences [109–112], interventional studies in both humans and animal models [113–117], the role of vitamin D and other nutritional factors [118–121], and better definition of genetic control, autoantibodies, and the implications of offspring of the diabetic mother [122–124]. In this manuscript, we have focused on the research data that reflects the metabolic conditions of pregnancy and adiposity. Both of these conditions favor the development of two strongly associated changes: IR and low-grade inflammation. The established highly complicated network of regulatory mechanisms during pregnancy may primarily affect carbohydrate metabolism by promoting autoimmune reactions to pancreatic β cells and affecting insulin function, while suppressing other autoimmune mechanisms. As a result, diabetes development during pregnancy is facilitated. Depending on a pregnant woman’s genetic susceptibility, autoimmunity or IR is the fundamental mechanism causing autoimmune or non-autoimmune GDM, respectively. Pregnancy may facilitate the identification of women at risk of developing diabetes later in life; autoimmune and non-autoimmune GDM may be early markers of the risk of future T1D and T2D, respectively. Importantly, metabolic changes occurring in T2D may, in turn, promote pancreatic β-cell autoimmunity, and the development of T1D cannot be excluded in such cases. Thus, the most convenient and efficient way to discriminate GDM types and to predict autoimmune diabetes is to assess pancreatic β-cell autoantibodies along with diagnosing diabetes during and after pregnancy.
References
O’Sullivan JB (1961) Gestational diabetes. Unsuspected, asymptomatic diabetes in pregnancy. N Engl J Med 264:1082–1085
Ben-Haroush A, Yogev Y, Hod M (2004) Epidemiology of gestational diabetes mellitus and its association with type 2 diabetes. Diabet Med: J B Diabet Assoc 21:103–113
Sunehag A, Berne C, Lindmark G, Ewald U (1991) Gestational diabetes-perinatal outcome with a policy of liberal and intensive insulin therapy. Ups J Med Sci 96:185–198
Murgia C, Berria R, Minerba L et al (2006) Gestational diabetes mellitus in Sardinia: results from an early, universal screening procedure. Diabetes Care 29:1713–1714
Gunton JE, Hitchman R, McElduff A (2001) Effects of ethnicity on glucose tolerance, insulin resistance and beta cell function in 223 women with an abnormal glucose challenge test during pregnancy. Aust N Z J Obstet Gynaecol 41:182–186
King H (1998) Epidemiology of glucose intolerance and gestational diabetes in women of childbearing age. Diabetes Care 21(Suppl 2):B9–B13
Groop L, Pociot F (2014) Genetics of diabetes—are we missing the genes or the disease? Mol Cell Endocrinol 382:726–739
Kim C, Newton KM, Knopp RH (2002) Gestational diabetes and the incidence of type 2 diabetes: a systematic review. Diabetes Care 25:1862–1868
Collares CV, Evangelista AF, Xavier DJ et al (2013) Transcriptome meta-analysis of peripheral lymphomononuclear cells indicates that gestational diabetes is closer to type 1 diabetes than to type 2 diabetes mellitus. Mol Biol Rep 40:5351–5358
Lapolla A, Dalfra MG, Fedele D (2009) Diabetes related autoimmunity in gestational diabetes mellitus: is it important? Nutr Metab Cardiovasc Dis : NMCD 19:674–682
Nilsson C, Ursing D, Torn C, Aberg A, Landin-Olsson M (2007) Presence of GAD antibodies during gestational diabetes mellitus predicts type 1 diabetes. Diabetes Care 30:1968–1971
Jarvela IY, Juutinen J, Koskela P et al (2006) Gestational diabetes identifies women at risk for permanent type 1 and type 2 diabetes in fertile age: predictive role of autoantibodies. Diabetes Care 29:607–612
Brooks-Worrell, B. M., Boyko, E. J. and Palmer, J. P. (2014), Impact of islet autoimmunity on the progressive beta-cell functional decline in type 2 diabetes. Diabetes Care
Lee MS (2014) Role of innate immunity in the pathogenesis of type 1 and type 2 diabetes. J Korean Med Sci 29:1038–1041
Bartha JL, Martinez-del-Fresno P, Comino-Delgado R (2001) Postpartum metabolism and autoantibody markers in women with gestational diabetes mellitus diagnosed in early pregnancy. Am J Obstet Gynecol 184:965–970
Borchers AT, Naguwa SM, Keen CL, Gershwin ME (2010) The implications of autoimmunity and pregnancy. J Autoimmun 34:J287–J299
Newbern D, Freemark M (2011) Placental hormones and the control of maternal metabolism and fetal growth. Curr Opin Endocrinol Diabetes Obes 18:409–416
Carpenter MW (2007) Gestational diabetes, pregnancy hypertension, and late vascular disease. Diabetes Care 30(Suppl 2):S246–S250
Kuhl C (1998) Etiology and pathogenesis of gestational diabetes. Diabetes Care 21(Suppl 2):B19–B26
Reece EA, Leguizamon G, Wiznitzer A (2009) Gestational diabetes: the need for a common ground. Lancet 373:1789–1797
Kirwan JP, Hauguel-De Mouzon S, Lepercq J et al (2002) TNF-alpha is a predictor of insulin resistance in human pregnancy. Diabetes 51:2207–2213
Bardicef M, Bardicef O, Sorokin Y, Altura BM, Altura BT, Resnick LM (1996) Perinatal cellular ion metabolism: 31P-nuclear magnetic resonance spectroscopic analysis of intracellular free magnesium and pH in maternal and cord blood erythrocytes. J Soc Gynecol Investig 3:66–70
Karabulut AK, Layfield R, Pratten MK (2001) Growth promoting effects of human placental lactogen during early organogenesis: a link to insulin-like growth factors. J Anat 198:651–662
Luthman M, Stock S, Werner S, Bremme K (1994) Growth hormone-binding protein in plasma is inversely correlated to placental lactogen and augmented with increasing body mass index in healthy pregnant women and women with gestational diabetes mellitus. Gynecol Obstet Investig 38:145–150
D’Ippolito S, Tersigni C, Scambia G, Di Simone N (2012) Adipokines, an adipose tissue and placental product with biological functions during pregnancy. BioFactors 38:14–23
Kuzmicki M, Telejko B, Szamatowicz J et al (2009) High resistin and interleukin-6 levels are associated with gestational diabetes mellitus. Gynecol Endocrinol : Off J Int Soc Gynecol Endocrinol 25:258–263
Kern PA, Ranganathan S, Li C, Wood L, Ranganathan G (2001) Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 280:E745–E751
Mohamed-Ali V, Pinkney JH, Coppack SW (1998) Adipose tissue as an endocrine and paracrine organ. Int J Obes Relat Metab Disord: J Int Assoc Study Obes 22:1145–1158
Yu J, Zhou Y, Gui J, Li AZ, Su XL, Feng L (2013) Assessment of the number and function of macrophages in the placenta of gestational diabetes mellitus patients. J Huazhong Univ Sci Technol Med Sci = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban 33:725–729
Fehniger TA, Caligiuri MA (2001) Interleukin 15: biology and relevance to human disease. Blood 97:14–32
Ramanathan S, Dubois S, Chen XL, Leblanc C, Ohashi PS, Ilangumaran S (2011) Exposure to IL-15 and IL-21 enables autoreactive CD8 T cells to respond to weak antigens and cause disease in a mouse model of autoimmune diabetes. J Immunol 186:5131–5141
Klimkiewicz-Blok D, Florjanski J, Zalewski J, Blok R (2012) Analysis of the concentrations of interleukin 15 in amniotic fluid in the second and the third trimesters of pregnancy. Adv Clin Exp Med: Off Organ Wroclaw Med Univ 21:75–79
Amash A, Huleihel M, Eyal S, Maor E, Myatt L, Holcberg G (2007) The expression of interleukin-15 and interleukin-18 by human term placenta is not affected by lipopolysaccharide. Eur Cytokine Netw 18:188–194
Nair AV, Hocher B, Verkaart S et al (2012) Loss of insulin-induced activation of TRPM6 magnesium channels results in impaired glucose tolerance during pregnancy. Proc Natl Acad Sci U S A 109:11324–11329
Barbagallo M, Dominguez LJ, Galioto A et al (2003) Role of magnesium in insulin action, diabetes and cardio-metabolic syndrome X. Mol Asp Med 24:39–52
Jacovetti C, Abderrahmani A, Parnaud G et al (2012) MicroRNAs contribute to compensatory beta cell expansion during pregnancy and obesity. J Clin Invest 122:3541–3551
Brelje TC, Parsons JA, Sorenson RL (1994) Regulation of islet beta-cell proliferation by prolactin in rat islets. Diabetes 43:263–273
Huynh, J., Xiong, G. and Bentley-Lewis, R. (2014), A systematic review of metabolite profiling in gestational diabetes mellitus. Diabetol
Borchers AT, Uibo R, Gershwin ME (2010) The geoepidemiology of type 1 diabetes. Autoimmun Rev 9:A355–A365
Cox, A. J., West, N. P. and Cripps, A. W. (2014), Obesity, inflammation, and the gut microbiota. Lancet. Diabetes Endocrinol
Papadopoulou A, Lynch KF, Shaat N et al (2011) Gestational diabetes mellitus is associated with TCF7L2 gene polymorphisms independent of HLA-DQB1*0602 genotypes and islet cell autoantibodies. Diabet Med: J B Diabet Assoc 28:1018–1027
Torn C, Gupta M, Sanjeevi CB, Aberg A, Frid A, Landin-Olsson M (2004) Different HLA-DR-DQ and MHC class I chain-related gene A (MICA) genotypes in autoimmune and nonautoimmune gestational diabetes in a Swedish population. Hum Immunol 65:1443–1450
Toth B, Haufe T, Scholz C et al (2010) Placental interleukin-15 expression in recurrent miscarriage. Am J Reprod Immunol 64:402–410
Wender-Ozegowska E, Michalowska-Wender G, Zawiejska A, Pietryga M, Brazert J, Wender M (2008) Concentration of chemokines in peripheral blood in first trimester of diabetic pregnancy. Acta Obstet Gynecol Scand 87:14–19
Sica A, Wang JM, Colotta F et al (1990) Monocyte chemotactic and activating factor gene expression induced in endothelial cells by IL-1 and tumor necrosis factor. J Immunol 144:3034–3038
Ramhorst RE, Garcia VE, Corigliano A, Rabinovich GA, Fainboim L (2004) Identification of RANTES as a novel immunomodulator of the maternal allogeneic response. Clin Immunol 110:71–80
Roca V, Calafat M, Larocca L et al (2009) Potential immunomodulatory role of VIP in the implantation sites of prediabetic nonobese diabetic mice. Reproduction 138:733–742
Hauk V, Azzam S, Calo G et al (2014) Vasoactive intestinal peptide induces an immunosuppressant microenvironment in the maternal-fetal interface of non-obese diabetic mice and improves early pregnancy outcome. Am J Reprod Immunol 71:120–130
Schober L, Radnai D, Spratte J et al (2014) The role of regulatory T cell (Treg) subsets in gestational diabetes mellitus. Clin Exp Immunol 177:76–85
Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010) FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10:490–500
Odegaard JI, Chawla A (2012) Connecting type 1 and type 2 diabetes through innate immunity. Cold Spring Harb Perspect Med 2:a007724
Winer DA, Winer S, Shen L et al (2011) B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17:610–617
Deiuliis J, Shah Z, Shah N et al (2011) Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE 6:e16376
Oztekin O (2007) New insights into the pathophysiology of gestational diabetes mellitus: possible role of human leukocyte antigen-G. Med Hypotheses 69:526–530
Le Bouteiller, P. (2014), HLA-G in human early pregnancy: control of uterine immune cell activation and likely vascular remodeling. Biomed J
Baricordi OR, Stignani M, Melchiorri L, Rizzo R (2008) HLA-G and inflammatory diseases. Inflamm Allergy Drug Targets 7:67–74
Urosevic M, Willers J, Mueller B, Kempf W, Burg G, Dummer R (2002) HLA-G protein up-regulation in primary cutaneous lymphomas is associated with interleukin-10 expression in large cell T-cell lymphomas and indolent B-cell lymphomas. Blood 99:609–617
Solini A, Muscelli E, Stignani M et al (2010) Soluble human leukocyte antigen-g expression and glucose tolerance in subjects with different degrees of adiposity. J Clin Endocrinol Metab 95:3342–3346
Mapp CE, Ferrazzoni S, Rizzo R et al (2009) Soluble human leucocyte antigen-G and interleukin-10 levels in isocyanate-induced asthma. Clin Exp Allergy : J Br Soc Allergy Clin Immunol 39:812–819
Chen J, Feigenbaum L, Awasthi P et al (2013) Insulin-dependent diabetes induced by pancreatic beta cell expression of IL-15 and IL-15Ralpha. Proc Natl Acad Sci U S A 110:13534–13539
Gupta S, Cerosaletti K, Long SA (2014) Renegade homeostatic cytokine responses in T1D: drivers of regulatory/effector T cell imbalance. Clin Immunol 151:146–154
Kalantar F, Rajaei S, Heidari AB et al (2013) Serum levels of tumor necrosis factor-alpha, interleukin-15 and interleukin-10 in patients with pre-eclampsia in comparison with normotensive pregnant women. Iran J Nurs Midwifery Res 18:463–466
Gysemans CA, Cardozo AK, Callewaert H et al (2005) 1,25-Dihydroxyvitamin D3 modulates expression of chemokines and cytokines in pancreatic islets: implications for prevention of diabetes in nonobese diabetic mice. Endocrinology 146:1956–1964
Liu NQ, Kaplan AT, Lagishetty V et al (2011) Vitamin D and the regulation of placental inflammation. J Immunol 186:5968–5974
Sanz-Salvador, L., Garcia-Perez, M. A., Tarin, J. J. and Cano, A. (2014), Endocrinology in pregnancy: bone metabolic changes during pregnancy: a period of vulnerability to osteoporosis and fracture. Eur J Endocrinol / Eur Fed Endocr Soc
Aydogmus, S., Kelekci, S., Aydogmus, H., et al. (2014), High prevalence of vitamin D deficiency among pregnant women in a Turkish population and impact on perinatal outcomes. J Matern Fetal Neonatal Med : Off J Eur Assoc Perinat Med, Fed Asia Ocean Perinat Soc, Int Soc Perinat Obstetricians 1–23.
Pihoker C, Gilliam LK, Hampe CS, Lernmark A (2005) Autoantibodies in diabetes. Diabetes 54(Suppl 2):S52–S61
Atkinson MA, Eisenbarth GS (2001) Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet 358:221–229
Songini M, Muntoni S (1991) High incidence of type I diabetes in Sardinia. Lancet 337:1047
Lapolla A, Fedele D, Pedini B et al (2002) Low frequency of autoantibodies to islet cell, glutamic acid decarboxylase, and second-islet antigen in patients with gestational diabetes mellitus: a follow-up study. Ann N Y Acad Sci 958:263–266
Dereke J, Nilsson C, Landin-Olsson M, Hillman M (2012) Prevalence of zinc transporter 8 antibodies in gestational diabetes mellitus. Diabet Med : J Br Diabet Assoc 29:e436–e439
Yu SH, Park S, Kim HS et al (2009) The prevalence of GAD antibodies in Korean women with gestational diabetes mellitus and their clinical characteristics during and after pregnancy. Diabetes Metab Res Rev 25:329–334
Palmer JP, Hampe CS, Chiu H, Goel A, Brooks-Worrell BM (2005) Is latent autoimmune diabetes in adults distinct from type 1 diabetes or just type 1 diabetes at an older age? Diabetes 54(Suppl 2):S62–S67
Fuchtenbusch M, Ferber K, Standl E, Ziegler AG (1997) Prediction of type 1 diabetes postpartum in patients with gestational diabetes mellitus by combined islet cell autoantibody screening: a prospective multicenter study. Diabetes 46:1459–1467
Wucher H, Lepercq J, Carette C et al (2011) Poor prognosis of pregnancy in women with autoimmune type 1 diabetes mellitus masquerading as gestational diabetes. Diabet Metab 37:47–51
Olivieri A, Valensise H, Magnani F et al (2000) High frequency of antithyroid autoantibodies in pregnant women at increased risk of gestational diabetes mellitus. Eur J Endocrinol / Eur Fed Endocr Soc 143:741–747
Weetman AP (2010) Immunity, thyroid function and pregnancy: molecular mechanisms. Nat Rev Endocrinol 6:311–318
Berghout A, Wiersinga W (1998) Thyroid size and thyroid function during pregnancy: an analysis. Eur J Endocrinol / Eur Fed Endocr Soc 138:536–542
Karakosta P, Alegakis D, Georgiou V et al (2012) Thyroid dysfunction and autoantibodies in early pregnancy are associated with increased risk of gestational diabetes and adverse birth outcomes. J Clin Endocrinol Metab 97:4464–4472
Agarwal MM, Dhatt GS, Punnose J, Bishawi B, Zayed R (2006) Thyroid function abnormalities and antithyroid antibody prevalence in pregnant women at high risk for gestational diabetes mellitus. Gynecol Endocrinol : Off J Int Soc Gynecol Endocrinol 22:261–266
Kordonouri O, Charpentier N, Hartmann R (2011) GADA positivity at onset of type 1 diabetes is a risk factor for the development of autoimmune thyroiditis. Pediatr Diabetes 12:31–33
Lambadiari V, Mitrou P, Maratou E et al (2011) Thyroid hormones are positively associated with insulin resistance early in the development of type 2 diabetes. Endocrine 39:28–32
Bech K, Hoier-Madsen M, Feldt-Rasmussen U, Jensen BM, Molsted-Pedersen L, Kuhl C (1991) Thyroid function and autoimmune manifestations in insulin-dependent diabetes mellitus during and after pregnancy. Acta Endocrinol (Copenh) 124:534–539
Hodgson E (2003) Combined spinal/epidural anesthesia. Middle East J Anesthesiol 17:103–112
Myre M, Imbeault P (2014) Persistent organic pollutants meet adipose tissue hypoxia: does cross-talk contribute to inflammation during obesity? Obes Rev : Off J Int Assoc Study Obes 15:19–28
Lemoine AY, Ledoux S, Larger E (2013) Adipose tissue angiogenesis in obesity. Thromb Haemost 110:661–668
Savage DB, Sewter CP, Klenk ES et al (2001) Resistin / Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes 50:2199–2202
Banerjee RR, Rangwala SM, Shapiro JS et al (2004) Regulation of fasted blood glucose by resistin. Science 303:1195–1198
Palin MF, Bordignon VV, Murphy BD (2012) Adiponectin and the control of female reproductive functions. Vitam Horm 90:239–287
Retnakaran R, Hanley AJ, Raif N et al (2005) Adiponectin and beta cell dysfunction in gestational diabetes: pathophysiological implications. Diabetologia 48:993–1001
Tao C, Sifuentes A, Holland WL (2014) Regulation of glucose and lipid homeostasis by adiponectin: effects on hepatocytes, pancreatic beta cells and adipocytes. Best Pract Res Clin Endocrinol Metab 28:43–58
Lacroix, M., Battista, M. C., Doyon, M., et al. (2013), Lower adiponectin levels at first trimester of pregnancy are associated with increased insulin resistance and higher risk of developing gestational diabetes mellitus. Diabetes Care
Wolf M, Sandler L, Hsu K, Vossen-Smirnakis K, Ecker JL, Thadhani R (2003) First-trimester C-reactive protein and subsequent gestational diabetes. Diabetes Care 26:819–824
Morin-Papunen L, Rautio K, Ruokonen A, Hedberg P, Puukka M, Tapanainen JS (2003) Metformin reduces serum C-reactive protein levels in women with polycystic ovary syndrome. J Clin Endocrinol Metab 88:4649–4654
Sarjeant K, Stephens JM (2012) Adipogenesis. Cold Spring Harb Perspect Biol 4:a008417
Wu HT, Chang CK, Tsao CW et al (2009) Insulin resistance without obesity induced by cotton pellet granuloma in mice. Lab Investig; J Tech Methods Pathol 89:362–369
Yu C, Chen Y, Cline GW et al (2002) Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 277:50230–50236
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119
Penesova A, Rovensky J, Zlnay M et al (2005) Attenuated insulin response and normal insulin sensitivity in lean patients with ankylosing spondylitis. Int J Clin Pharmacol Res 25:107–114
Sanchez-Jimenez R, Alvarado-Vasquez N (2013) IL-15 that a regulator of TNF-alpha in patients with diabetes mellitus type 2. Med Hypotheses 80:776–777
Ding C, Gao D, Wilding J, Trayhurn P, Bing C (2012) Vitamin D signalling in adipose tissue. Br J Nutr 108:1915–1923
Sun X, Zemel MB (2007) Calcium and 1,25-dihydroxyvitamin D3 regulation of adipokine expression. Obesity 15:340–348
Retnakaran R, Hanley AJ, Raif N, Connelly PW, Sermer M, Zinman B (2003) C-reactive protein and gestational diabetes: the central role of maternal obesity. J Clin Endocrinol Metab 88:3507–3512
Sokup A, Goralczyk B, Goralczyk K, Rosc D (2012) Triglycerides as an early pathophysiological marker of endothelial dysfunction in nondiabetic women with a previous history of gestational diabetes. Acta Obstet Gynecol Scand 91:182–188
Morimitsu LK, Fusaro AS, Sanchez VH, Hagemann CC, Bertini AM, Dib SA (2007) Fibrinolytic dysfunction after gestation is associated to components of insulin resistance and early type 2 diabetes in Latino women with previous gestational diabetes. Diabetes Res Clin Pract 78:340–348
Hanna FW, Peters JR (2002) Screening for gestational diabetes; past, present and future. Diabet Med: J B Diabet Assoc 19:351–358
Peng J, Narasimhan S, Marchesi JR, Benson A, Wong FS, Wen L (2014) Long term effect of gut microbiota transfer on diabetes development. J Autoimmun 53:85–94
Munyaka PM, Khafipour E, Ghia JE (2014) External influence of early childhood establishment of gut microbiota and subsequent health implications. Front Pediatr 2:109
Van Belle TL, Pagni PP, Liao J et al (2014) Beta-cell specific production of IL6 in conjunction with a mainly intracellular but not mainly surface viral protein causes diabetes. J Autoimmun S0896-8411(14):00041–00049. doi:10.1016/j.jaut.2014.02.002
Chen, P., Wang, S., Ji, J., et al. (2014), Risk factors and management of gestational diabetes. Cell Biochem Biophys Oct 1.
Skupien, J., Cyganek, K. and Malecki, M. T. (2014), Diabetic pregnancy: an overview of current guidelines and clinical practice. Curr Opin Obstet Gynecol Sep 27.
Sarikonda G, Pettus J, Phatak S et al (2014) CD8 T-cell reactivity to islet antigens is unique to type 1 while CD4 T-cell reactivity exists in both type 1 and type 2 diabetes. J Autoimmun 50:77–82
Stefan M, Zhang W, Concepcion E, Yi Z, Tomer Y (2014) DNA methylation profiles in type 1 diabetes twins point to strong epigenetic effects on etiology. J Autoimmun 50:33–37
Kachapati K, Bednar KJ, Adams DE et al (2013) Recombinant soluble CD137 prevents type one diabetes in nonobese diabetic mice. J Autoimmun 47:94–103
Huang X, Wu H, Lu Q (2014) The mechanisms and applications of T cell vaccination for autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol 47:219–233
Gravano DM, Hoyer KK (2013) Promotion and prevention of autoimmune disease by CD8+ T cells. J Autoimmun 45:68–79
Peacock AS, Bogossian F, McIntyre HD, Wilkinson S (2014) A review of interventions to prevent type 2 diabetes after gestational diabetes. Women Birth S1871-5192(14):00088–2. doi:10.1016/j.wombi.2014.09.002
Selmi C (2012) Autoimmunity in 2011. Clin Rev Allergy Immunol 43:194–206
Agmon-Levin N, Theodor E, Segal RM, Shoenfeld Y (2013) Vitamin D in systemic and organ-specific autoimmune diseases. Clin Rev Allergy Immunol 45:256–266
Morton, S., Kirkwood, S. and Thangaratinam, S. (2014), Interventions to modify the progression to type 2 diabetes mellitus in women with gestational diabetes: a systematic review of literature. Curr Opin Obstet Gynecol 2014 Oct 21.
Zhuang T, Han H, Yang Z (2014) Iron, oxidative stress and gestational diabetes. Nutrients 6:3968–3980
Bour-Jordan H, Thompson HL, Giampaolo JR, Davini D, Rosenthal W, Bluestone JA (2013) Distinct genetic control of autoimmune neuropathy and diabetes in the non-obese diabetic background. J Autoimmun 45:58–67
Mitanchez D, Yzydorczyk C, Siddeek B, Boubred F, Benahmed M, Simeoni U (2014) The offspring of the diabetic mother—short- and long-term implications. Best Pract Res Clin Obstet Gynaecol S1521-6934(14):00163–1. doi:10.1016/j.bpobgyn.2014.08.004
Boettler T, Pagni PP, Jaffe R, Cheng Y, Zerhouni P, von Herrath M (2013) The clinical and immunological significance of GAD-specific autoantibody and T-cell responses in type 1 diabetes. J Autoimmun 44:40–48
Funding
This study was supported by the Estonian Research Council (institutional research funding IUT20-43), by European Union (the European Regional Developmental Fund), and by Enterprise Estonia (grant no EU30020).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Haller-Kikkatalo, K., Uibo, R. Clinical Recommendations for the Use of Islet Cell Autoantibodies to Distinguish Autoimmune and Non-Autoimmune Gestational Diabetes. Clinic Rev Allerg Immunol 50, 23–33 (2016). https://doi.org/10.1007/s12016-014-8461-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-014-8461-8