
Abstract
Doxorubicin (DOX) is commonly used for the treatment of various types of cancer, however can cause serious side effects, including cardiotoxicity. The mechanisms involved in DOX-induced cardiac damage are complex and not yet fully understood. One mechanism is the disruption of cardiac metabolism, which can impair cardiac function. The mammalian target of rapamycin (mTOR) is a key regulator of cardiac energy metabolism, and dysregulation of mTOR signaling has been implicated in DOX-induced cardiac dysfunction. Natural compounds (NCs) have been shown to improve cardiac function in vivo and in vitro models of DOX-induced cardiotoxicity. This review article explores the protective effects of NCs against DOX-induced cardiac injury, with a focus on their regulation of mTOR signaling pathways. Generally, the modulation of mTOR signaling by NCs represents a promising strategy for decreasing the cardiotoxic effects of DOX.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Doxorubicin (DOX), an FDA-approved chemotherapy drug, is used to treat various types of cancers, including breast cancer and leukemia [1]. DOX can cause serious and potentially life-threatening side effects, such as cardiotoxicity [1]. The mechanisms involved in DOX-induced cardiac damage are complex and not yet fully understood [2], however, it is thought that may be caused by several factors, including reactive oxygen species (ROS) generation, mitochondrial dysfunction, calcium homeostasis disruption, and activation of apoptotic pathways [1, 3]. One of the mechanisms underlying the treatment with DOX is cardiac metabolism disruption [4]. DOX alters the expression of genes implicated in cardiac metabolism, which can impair cardiac function [5].
The mammalian target of rapamycin (mTOR) is a key regulator of cardiac energy metabolism by regulating fatty acid metabolism, glucose uptake and glycolysis, and mitochondrial function [6]. DOX has been shown induce cardiac dysfunction by the dysregulation of mTOR signaling [7]. It has also been suggested that DOX induces cardiac metabolism dysfunction in an mTOR-dependent manner [7, 8]. Recent research has proposed that the mTOR pathway could be a potential target for reducing the cardiotoxic effects of DOX [8, 9].
Several natural compounds (NCs) have been revealed to improve cardiac function in animal models of DOX-induced cardiotoxicity [10]. Some NCs can regulate mTOR expression, which may contribute to their cardioprotective effects against DOX treatment [11, 12]. The present study provides a review of the protective effects of NCs against DOX-induced cardiac injury, with a focus on their regulation of mTOR signaling pathways.
DOX-dependent Change in Cardiac Metabolism
The function and contraction of cardiomyocytes are sustained by oxidative phosphorylation of adenosine diphosphate (ADP) and the production of adenosine triphosphate (ATP) [5]. Mitochondria is responsible for supplying over 95% of the ATP molecules; the remaining 5% comes from the glycolysis process and the citric acid cycle [13]. Most of cardiac ATP (approximately 70%) is derived from fatty acids β-oxidation [14]. Glycolysis, a glucose metabolic pathway, produces a small amount of cardiac ATP. Ketone bodies, lactate, and amino acids contribute less to cardiac ATP generation in comparison with fatty acids and glucose [14]. Creatine phosphate (PCr) is a reservoir of high-energy phosphates in the myocardium to recycle ATP from ADP [15]. Therefore, the PCr/ATP ratio reflects the myocardial energy status [15]. Due to the continuous contractility, the myocardium consumes a high rate of ATP and depletes phosphate storage within a few seconds [14]. Since the cardiac function is dependent on mitochondrial ATP production, therefore ATP generation deficiency can alter myocardial contractility [14].
The cardiac metabolic change during DOX treatment has been reported that represents a compensatory response to imbalances in ATP demand and supply [16, 17]. DOX has been suggested that disrupt mitochondrial respiration and fatty acid oxidation in cardiomyocytes [18]. The mitochondrial accumulation of DOX is following its binding to cardiolipin located in the inner mitochondrial membrane [19]. The electron transport chain (ETC) is disrupted after DOX accumulation into the mitochondria and repressing mitochondrial complexes I and II [19]. ETC dysfunction is lead to decreasing in the cardiac ATP/ADP ratio and an increase in ROS generation, which damages the heart tissue [20]. DOX also affects glucose, amino acids, and fatty acids metabolism in the heart, resulting in decreased ATP production and cardiac dysfunction [4].
The overproduction of ROS in cardiomyocytes caused by DOX can have an impact on the metabolism of purine and pyrimidine nucleotides [21, 22]. This can occur through direct modifications to the nucleotides, inducing changes in DNA bases, activating DNA repair pathways, altering the availability of nucleotides, and influencing signaling pathways related to nucleotide metabolism [21]. As a result of the DNA damage, the metabolites of purine and pyrimidine can disrupt normal cardiac metabolism in various ways [21]. For example, they can affect the availability of nucleotides and interfere with the function of crucial enzymes involved in energy production [21, 22]. Additionally, DOX can affect the balance between pro-apoptotic BCL-2-associated X protein (BAX ) and anti-apoptotic B-cell lymphoma 2 (BCL-2) proteins, leading to structural modification of these proteins. BAX promotes cell death, while BCL-2 inhibits apoptosis [23]. The altered conformation of BCL-2 and BAX proteins caused by DOX can disrupt the normal regulation of apoptosis and have significant consequences on cardiac metabolism, including mitochondrial dysfunction, alterations in energy metabolism, and changes in nutrient utilization [21, 23].
mTOR-regulated Cardiac Metabolism
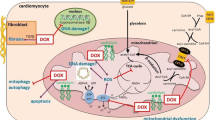
The mTOR is a protein kinase that phosphorylates the serine or threonine residues on cell proteins [24]. Phosphorylation is one of the primary mechanisms for regulating protein activity in signal transduction pathways [25]. The mTOR plays an essential role in regulating cardiac metabolism, cardiomyocyte growth, and survival [26]. Glycolysis is a metabolic pathway that generates ATP by converting glucose into pyruvate [27]. The mTOR activation promotes glycolysis by enhancing the expression and activity of key glycolytic enzymes, increasing the expression of glucose transporters, and facilitating glucose uptake into the heart cells [27, 28]. Lipid metabolism is another critical energy source in the heart, as it relies on the oxidation of fatty acids to generate ATP [27]. The mTOR regulates lipid metabolism by suppressing fatty acid synthesis and promoting fatty acid oxidation [27, 28]. The mTOR is the major component of two multiprotein complexes, mTOR complex 1 (mTORC1) and mTORC2 [24]. Each complex possesses distinct subunits that play a role in their specific functions [29] as depicted in Fig. 1. The regulatory-associated protein of mTOR (Raptor) plays a crucial role in the mTORC1 complex as its core component [29]. It is responsible for the cellular localization of mTORC1 and the recruitment of substrates [29, 30]. Additionally, Raptor is involved in conferring sensitivity of mTORC1 to rapamycin, a drug that inhibits its activity [29]. The core component of mTORC2 includes the stress-activated map kinase-interacting protein 1 (Sin1) and the rapamycin-insensitive companion of mTOR (Rictor) [29]. Sin1 plays a vital role in preserving the integrity of mTORC2 and facilitating the binding of substrates [30]. Rictor is responsible for conferring rapamycin insensitivity to mTORC2 [30]. Both complexes, mTORC1 and mTORC2, have two subunits in common: mTOR and the mammalian lethal with SEC13 protein 8 (mLST8) [30]. The mLST8 plays a critical role in stabilizing the mTOR kinase domain [30]. Moreover, the DEP domain-containing mTOR-interacting protein (Deptor) binds to both mTORC1 and mTORC2, acting as an endogenous inhibitor for both complexes [29, 30].

The subunits of mTORC1 and mTORC2 complexes
Deptor, DEP domain-containing mTOR-interacting protein; mLST8, mammalian lethal with SEC13 protein 8; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; mTORC2, mTOR complex 2; Raptor, regulatory-associated protein of mTOR; Rictor, rapamycin-insensitive companion of mTOR; Sin1, stress-activated map kinase-interacting protein 1.
The mTORC1 is mainly localized in the cytoplasm [31]. The activation of mTORC1 is regulated by levels of nutrients (amino acids, glucose, and fatty acids), growth factors, and hormones [6]. During nutrient-rich conditions, the lysosomal recruitment of mTORC1 by Ras-related GTP binding proteins (Rags) is the well-characterized mode of its activation [32]. Activated mTORC1 phosphorylates and inactivates the transcription factor EB (TFEB), a master regulator of lysosomal biogenesis and autophagy, and causes its binding to 14-3-3 protein at the lysosome surface [33]. However, the inactivation of mTORC1 under starvation conditions leads to the TFEB/14-3-3 complex dissociation and rapid transport of TFEB to the nucleus for regulating the expression of autophagosomal and lysosomal genes [34]. Therefore, TFEB regulation by mTORC1 provides a mechanism for coordinating lysosomal biogenesis and autophagy with the availability of nutrients within the cell [31]. During autophagy, damaged proteins or organelles are delivered to the lysosome via the autophagosomes, a double-membrane vesicle, for degradation and maintaining cellular homeostasis [35].
The main upstream regulators of mTORC1 activity are the protein kinase AKT and the adenosine monophosphate–activated protein kinase (AMPK) [36, 37]. AKT phosphorylates and inactivates the tuberous sclerosis complex 2 (TSC2), a mTORC1 negative regulator, leading to mTORC1 activation [37]. AMPK conserves cellular energy by the inhibition of mTORC1 during low energy states [36].
The mTORC2 is activated by growth factors, insulin, and cellular stress signals [38]. It is localized in both the cytoplasm and the cytoplasmic membrane and regulates a variety of cellular processes, including metabolism and cell survival [39]. The mTORC2 is known as a critical regulator of AKT and protein kinase C (PKC) [39]. AKT and PKC signaling modulates glucose and lipid metabolism through the phosphorylation of metabolic enzymes or control of various transcription factors [39, 40]. AMPK and sirtuin 1 (SIRT1) can reduce mTORC2 activity by regulating upstream signaling pathways [39, 41].
Overall, the mTOR protein plays a significant role in heart metabolism by regulating metabolism of fatty acid and the intake and use of glucose [6]. It functions as a cellular sensor, coordinating the cellular response to changes in nutrient availability [29]. When there is an abundance of nutrients, the mTOR signaling is activated, promoting anabolic processes such as protein synthesis and suppressing catabolic processes such as autophagy [29]. mTORC1 mainly promotes cell growth by enhancing protein synthesis [30]. It regulates glucose uptake glycolysis, and fatty acid synthesis, thereby providing energy to cells in the form of ATP [30]. By regulating these processes, mTORC1 ensures that the heart has the necessary energy to function properly [6]. mTORC2 regulates the glucose transporter proteins, which are responsible for carrying glucose into cells [30]. By controlling the activity of these transporters, mTORC2 can influence the amount of glucose that enters cells and is available for energy production [29]. Additionally, mTORC2 is implicated in the regulation of fatty acid oxidation, the process by which fatty acids are broken down to generate energy [29].
Myocardial ischemia, or energy stress in the heart, can lead to the inhibition of mTORC1 and trigger protective adaptations, such as autophagy, which help limit myocardial infarction [42]. It has been suggested that mTORC1 has different roles during ischemia and reperfusion [42, 43]. During ischemia-reperfusion injury, mTORC1 activation promotes a metabolic shift from fatty acid oxidation to glycolysis by the upregulation of glucose transporters and increasing glucose uptake into the myocardium [42]. This increased glucose availability favors glycolysis, which is a more efficient energy-generating pathway during reperfusion. mTORC1 activation also can inhibit the activity of key enzymes involved in fatty acid oxidation [42]. Inhibition of these enzymes shifts the metabolism away from fatty acid oxidation and towards glycolysis [42, 43]. Dysregulation of mTOR signaling pathways has been implicated in a variety of diseases, including metabolic and cardiovascular diseases [6, 44].
Targeting mTOR by Natural Compounds in the DOX-induced Cardiotoxicity
Numerous studies have shown the effects of DOX on cardiac mTOR expression [7, 8, 45]. Some studies have shown that DOX induces mTOR expression in cardiomyocytes, while others have reported a decrease in mTOR expression [45,46,47]. It has been suggested that the dysregulated mTOR expression may be involved in the development of DOX-induced cardiotoxicity [48]. Studies have explored how NCs can protect against DOX-induced cardiac injury by modulating the mTOR pathway [12, 47, 49] (Table 1).
The AKT/mTOR Pathway
Apigenin (API) is a flavonoid obtained from parsley, celery, and vine spinach [50]. It has various health benefits, including anti-inflammatory, antioxidant, antiviral, and anticancer effects [50]. API has been found to have cardioprotective properties, reducing blood pressure and the risk of atherosclerosis [51]. A study on mice investigated the potential protective effect of API against the cardiotoxic of DOX [52]. It was observed that API prevented DOX-induced apoptosis and autophagy, possibly through the activation of the AKT/mTOR pathway [52].
Curcumin (CUR) is a natural compound found in the turmeric (Curcuma longa L.) [53]. It possesses antitumor, antidiabetic, antihyperglycemic, antioxidant, and neuroprotective properties [54,55,56]. CUR has shown positive effects on cardiovascular health [56,57,58] and therapeutic efficacy against DOX-induced cardiomyopathy [49]. CUR reversed the down-regulation of AKT and mTORC1 caused by DOX in mice hearts and activated the AKT/mTOR signaling [49].
Luteolin-7-O-glucoside (LUTG) is a flavonoid found in Dracocephalum tanguticum Maxim [59]. It has various biological activities, such as anti-diabetic, anti-inflammatory, and anticancer effects [60,61,62]. LUTG has shown a potential protective effect in a cardiac hypertrophy model and protected against DOX-induced cardiotoxicity by down-regulating the AKT/mTOR pathway [63].
Neferine (NEF) is an alkaloid isolated from Nelumbo nucifera [64]. It possesses anti-diabetic, anti-cancer, and anti-microbial effects [65]. NEF has been reported to have potential therapeutic benefits for cardiovascular conditions, including atherosclerosis, hypertension, and heart failure [64, 66, 67]. NEF has shown a protective role in DOX-induced cardiotoxicity by inhibiting cardiac autophagy and increasing the expression of AKT and mTOR, thereby activating the AKT/mTOR pathway [68].
Wheat phenolics, such as ferulic acid (FA) and apigenin (AP) are natural compounds found in wheat and other cereal grains [69]. These phenolics have various biological activities, including antioxidant and anti-inflammatory effects [69, 70]. In a study investigating the potential protective effects against DOX-induced cardiotoxicity, the expressions of PI3K, p-AKT, and p-mTOR were found to be up-regulated in DOX-exposed cardiomyocytes treated with FA, AP, or whole wheat grain polyphenolic extract (WWGPE) [71]. The study suggests that WWGPE exhibited more effective cardioprotective effects compared to FA and AP, possibly due to the synergic interaction between the compounds [71]. These finding indicate that the protective effects of these wheat phenolics may be mediated through the activation of the PI3K/AKT/mTOR signaling pathway [71].
The mTOR/TFEB Pathway
Corosolic acid (CRA) is a natural triterpenoid isolated from the leaves of the banaba tree (Lagerstroemia speciosa) [72]. It possesses several health benefits such as anti-inflammatory, antioxidant, antidiabetic, and anticancer properties [72]. Studies have shown that CRA can reduce cardiac fibrosis in mice with heart failure and acute myocardial injury in diabetic rats [73, 74]. A study investigated the potential of CRA in improving myocardial injury caused by DOX [75]. It was found that CRA improved cardiac metabolism, ATP generation, and mitochondrial function [75]. It also increased the expression of AMPK and TFEB while reducing the expression of mTORC1 [75]. The study suggested that the cardioprotective effect of CRA against DOX-induced cardiotoxicity is related to the AMPK/mTORC1/TFEB pathway activation [75].
Dihydrotanshinone I (DHT) is a terpenoid compound derived from Salvia miltiorrhiza [76]. It has potential therapeutic effects in the treatment of cardiovascular diseases and cancer [76, 77]. In a study on DOX-induced cardiotoxicity, DHT was found to suppress the activation and nuclear localization of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [78], a transcription factor involved in the expression of inflammatory cytokines [79]. It also increased the nuclear expression of TFEB and decreased the cardiac ratio of p-mTOR/mTOR [78]. However, these effects were reversed by an mTOR agonist [78]. Therefore, the inhibition of mTOR expression by DHT may play a role in preventing DOX-related cardiac inflammation [78].
Tanshinone IIA (Tan-IIA) is the main lipophilic component extracted from Salvia miltiorrhiza [80]. Tan-IIA has anti-inflammatory, anti-oxidant, and immunomodulatory activities [81]. The cardiovascular pharmacology of Tan-IIA is characterized by its vasodilatory effect, antiarrhythmic effect, and inhibition of ischemia-reperfusion injury [82,83,84]. In a study on DOX-induced cardiotoxicity, Tan-IIA was found to restore the dynamic balance of autophagosome/autolysosome [85]. It reduced autolysosome accumulation and increased autophagosome formation, thereby promoting autophagy [85]. Tan-IIA up-regulated the expression of autophagy markers, including Beclin1 and the lysosomal-associated membrane proteins-1 (LAMP1), while decreasing the p-mTOR expression and increasing the TFEB expression [85]. These findings suggest that Tan-IIA protects against DOX-mediated cardiac damage by promoting autophagy through the inhibition of the mTOR/TFEB signaling pathway [85].
The AMPK/mTOR Pathway
Aspalathin (ASP), the primary polyphenol isolated from the rooibos plant (Aspalathus linearis), has various beneficial properties, antioxidant, anti-inflammatory, antidiabetic, antithrombotic, and anticancer effects [86, 87]. It has been shown to improve cardiovascular complications associated with diabetes [86]. In a study by Johnson et al. ASP prevented the decrease in cardiac ATP activity caused by DOX treatment [9]. While DOX treatment resulted in a slight increase in the p-mTOR/ t-mTOR ratio, Asp did not revered this effect [9]. Instead, ASP increased the expression of p-AMPK and autophagy in cardiomyocytes treated by DOX [9]. These findings suggest that the cardioprotective effects of ASP may not be mediated through the mTOR pathway [9].
Astragalus polysaccharide (APS) is derived from the roots of Astragalus membranaceus [88]. It exerts various pharmacological effects, including antidiabetic, anti-aging, antitumor, antibacterial, and antiviral properties [89]. ASP has shown a wide range of therapeutic effects in experimental cardiomyopathy, including cardiotoxicity caused by DOX and hypertrophic myocardium induced by isoproterenol [90, 91]. In a study on DOX-treated mice hearts, APS promoted mTOR activation and normalized autophagic flux [46]. The AMPK/mTOR pathway is considered one of the main targets of APS in protecting against DOX-induced impaired cardiac autophagy [46].
Dihydromyricetin (DHM) is a flavonoid found in the leaves of the Ampelopsis grossedentata [92]. It exhibits antioxidant, anti-inflammatory, mitochondrial dysfunction improvement, and autophagy regulation properties [93, 94]. Many benefits of DHM on the cardiovascular system have been reported [95]. In a study by Li et al. DHM pretreatment normalized left ventricular dysfunction in mice with DOX-induced cardiac injury [12]. DHM reversed DOX-induced inhibition of AMPK and induction of mTOR expression, suggesting that the cardioprotective effect of DHM involves activating the AMPK/mTOR pathway [12].
Scutellarin (SCU) is a polyphenolic flavonoid derived from Erigeron breviscapus [96]. It has antioxidant, anti-inflammatory, antiapoptotic, and neuroprotective properties [97, 98]. It has shown potential therapeutic benefits for cardiovascular diseases, such as heart failure and myocardial ischemia/reperfusion injury [96,97,98]. In rats with DOX-induced chronic cardiotoxicity, SCU treatment decreased AMPK expression and increased mTOR expression, preventing autophagy [99]. In another study, SCU reduced autophagy by increasing the expression of p-AKT and p-mTOR in cardiomyocytes treated by DOX [100]. SCU could potentially be a therapeutic option for preventing DOX-induced cardiotoxicity by inhibiting the AMPK/mTOR and the AKT-mTOR pathways [99, 100].
Thymoquinone (TQ) is a bioactive compound found in the seeds of Nigella sativa [101]. TQ has different pharmacological effects, such as antimicrobial, antihistamine, anti-inflammatory, antioxidant, and anticancer activities [102]. It has shown potential in preventing and treatment of myocardial ischemia/reperfusion injury and diabetic cardiomyopathy [101, 103]. TQ was found to have protective effects against DOX-induced cardiotoxicity in cardiomyocytes by decreasing the p-mTOR expression and increasing the p-AMPK expression [104]. This suggests that TQ induces cardiac autophagy through up-regulating the AMPK/mTOR pathway [104].
Glycyrrhizin (GL) is a glycoside derived from the Glycyrrhiza glabra root [105]. Many therapeutic activities of GL are determined, such as cardioprotective, neuroprotective, and hepatoprotective activities [106,107,108]. The protective effects of GL have been revealed in diabetic cardiomyopathy and isoproterenol-induced cardiac damage [109, 110]. LV et al. have revealed GL therapeutic strategy against DOX-induced cardiomyopathy [11]. Previous research have indicated that the high-mobility group box 1 (HMGB1) inhibitors down-regulated the AKT/mTOR pathway [111, 112]. GL is known as a direct HMGB1 antagonist [113]. In DOX-treated H9c2 cells, GL decreased the expressions of AKT, mTOR, and HMGB1 [11]. It also reduced the expressions of autophagy markers, including the protein light chain 3 (LC3) II and p62, and improved autophagy flux [11]. Overall, GL attenuated DOX-mediated cardiac autophagy by the down-regulation of the HMGB1-dependent AKT/mTOR pathway [11].
Resveratrol (RSV) is a polyphenol derived from grapes, berries, and peanuts [114]. It has anti-inflammatory, antioxidant, anti-cancer, neuroprotective, and cardioprotective properties [114]. Numerous studies have indicated that RSV can protect against heart failure, ischemia-reperfusion injury [115, 116], and DOX-induced cardiotoxicity [117, 118]. RSV was found to enhance DOX-mediated cardiac autophagy by reducing mTORC1 and increasing the E2 promoter binding factor 1 (E2F1) expression [117]. E2F1 is a transcription factor that is involved in regulating the expression of genes related to autophagy [119]. The up-regulation of the E2F1/mTORC1 pathway appears to contribute to the protective effect of RSV against cardiotoxicity [117]. Another study found that RSV activates AMPK, inhibits mTOR, and stimulates autophagy through the ULK1 complex [118]. ULK1 is a protein kinase involved in the process of autophagy and is stimulated by AMPK [120]. This suggests that RSV increases autophagy in cardiomyocytes via the activation of the AMPK/mTOR/ULK1 pathway [118].
Beta-LAPachone (β-LAP), a quinone-containing compound, is extracted from the lapacho tree [121]. It has antioxidant, anti-inflammatory, anti-obesity, anticancer, antiviral, antimicrobial, nephroprotective, neuroprotective, and cardioprotective effects [121,122,123]. The potential of β-LAP to prevent DOX-mediated cardiotoxicity has been investigated [47]. β-LAP has been found to reduce histopathological injury and improve the cardiac function [47]. SIRT1 is a main energy sensor that requires the nicotinamide adenine dinucleotide (NAD+) as a substrate [124]. Therefore, the intracellular level of NAD+ regulates SIRT1 function [124]. β-LAP increased the cardiac NAD+/NADH ratio and up-regulated SIRT1 expression in DOX-exposed heart tissues [47]. Increased SIRT1 expression and activity is associated with the deacetylation of the liver kinase B1 (LKB1) [125], level of autophagy marker LKB1 was elevated in the heart tissues of mice treated with β-LAP [47]. LKB1, as a kinase, activates AMPK through its phosphorylation [126]. β-LAP up-regulated AMPK expression and down-regulated the cardiac expression of mTOR [47]. These findings suggested that β-LAP up-regulated the LKB1/AMPK/mTOR pathway [47].
Spinacetin (SP) is a flavonoid found in spinach (Spinacia oleracea L.) [127]. There are various health benefits associated with the consumption of SP, including cardiovascular protection, antiasthmatic properties, hypoglycemic activity, and anti-inflammatory effects [128, 129]. In the context of DOX-induced myocardiopathy, SP treatment increased the expression of AMPK and SIRT3, while decreasing mTOR expression [127]. SIRT3 is a deacetylase localized to the mitochondria, which plays a role in phospho-activation of AMPK and acts as a positive regulator of autophagy [130]. This led to the induction of autophagy in cardiomyocytes, suggesting that SP alleviates DOX-triggered cardiotoxicity by up-regulating the SIRT3/AMPK/mTOR pathway [127].
Conclusion
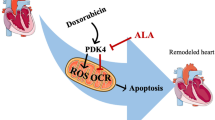
The regulation of the mTOR pathway is complex, and the effects of NCs on this pathway seem to differ depending on the stage of DOX-mediated cardiotoxicity progression. DOX may impact mTOR signaling in dissimilar ways depending on the stage of cardiotoxicity. According to findings, the AKT/mTOR and AMPK/mTOR signaling have been implicated in the protective effects of NCs against DOX-changed cardiac metabolism. Figure 2 effectively represents the protective effects of NCs against DOX-induced cardiotoxicity by targeting the mTOR pathways. In general, the use of NCs to modify mTOR signaling shows promise as a strategy to reduce the harmful effects of DOX on the heart in clinical settings. However, thorough clinical trials are necessary to establish the right dosages, treatment duration, and potential interactions with other medications. Nevertheless, further research is needed to gain a complete understanding of the safety and effectiveness of these compounds before they can be widely utilized in clinical practice.

The protective effects of natural compounds against DOX-induced cardiac metabolism dysfunction by targeting the AKT/mTOR and AMPK/mTOR pathways. AMPK, adenosine monophosphate–activated protein kinase; ATP, adenosine triphosphate; BAX, BCL-2-associated X protein; BCL-2, B-cell lymphoma 2; DOX, doxorubicin; LKB1, liver kinase B1; mTOR, mammalian target of rapamycin; PI3K, phosphoinositide 3-kinases; ROS, reactive oxygen species; TFEB, transcription factor EB; TSC2, tuberous sclerosis complex 2; ULK1, Unc-51-like kinase 1
Data Availability
Not applicable.
Code Availability
Not applicable.
Abbreviations
- β-lap:
-
Beta-LAPachone
- ADP:
-
Adenosine diphosphate
- AMPK:
-
Adenosine monophosphate–activated protein kinase
- AP:
-
Apigenin
- API:
-
Apigenin
- APS:
-
Astragalus polysaccharide
- ASP:
-
Aspalathin
- ATP:
-
Adenosine triphosphate
- BAX:
-
BCL-2-associated X protein
- BCL-2:
-
B-cell lymphoma 2
- CRA:
-
Corosolic acid
- CUR:
-
Curcumin
- Deptor:
-
DEP domain-containing mTOR-interacting protein
- DHM:
-
Dihydromyricetin
- DHT:
-
Dihydrotanshinone I
- DOX:
-
Doxorubicin
- E2F1:
-
E2 promoter binding factor 1
- ETC:
-
Electron transport chain
- FA:
-
Ferulic acid
- GL:
-
Glycyrrhizin
- HMGB1:
-
High-mobility group box 1
- LAMP1:
-
Lysosomal-associated membrane proteins-1
- LC3:
-
Protein light chain 3
- LKB1:
-
Liver kinase B1
- LUTG:
-
Luteolin-7-O-glucoside
- mLST8:
-
Mammalian lethal with SEC13 protein 8
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
MTOR complex 1
- mTORC2:
-
MTOR complex 2
- NAD+ :
-
Nicotinamide adenine dinucleotide
- NCs:
-
Natural compounds
- NEF:
-
Neferine
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- p-AKT:
-
Phosphorylated-AKT
- p-mTOR:
-
Phosphorylated-mTOR
- PCr:
-
Creatine phosphate
- PI3K:
-
Phosphoinositide 3-kinases
- PKC:
-
Protein kinase C
- Rags:
-
Ras-related GTP binding proteins
- Raptor:
-
Regulatory-associated protein of mTOR
- Rictor:
-
Rapamycin-insensitive companion of mTOR
- ROS:
-
Reactive oxygen species
- RSV:
-
Resveratrol
- SCU:
-
Scutellarin
- Sin1:
-
Stress-activated map kinase-interacting protein 1
- SIRT1:
-
Sirtuin 1
- SP:
-
Spinacetin
- t-AKT:
-
Total-AKT
- t-mTOR:
-
Total-mTOR
- Tan-IIA:
-
Tanshinone IIA
- TFEB:
-
Transcription factor EB
- TQ:
-
Thymoquinone
- TSC2:
-
Tuberous sclerosis complex 2
- ULK1:
-
Unc-51-like kinase 1
- WWGPE:
-
Whole wheat grain polyphenolic extract
References
Yarmohmmadi, F., Rahimi, N., Faghir-Ghanesefat, H., Javadian, N., Abdollahi, A., Pasalar, P., & Dehpour, A. R. (2017). Protective effects of agmatine on doxorubicin-induced chronic cardiotoxicity in rat. European Journal of Pharmacology, 796, 39–44. https://doi.org/10.1016/j.ejphar.2016.12.022
Okpara, E. S., Adedara, I. A., Guo, X., Klos, M. L., Farombi, E. O., & Han, S. (2022). Molecular mechanisms associated with the chemoprotective role of protocatechuic acid and its potential benefits in the amelioration of doxorubicin-induced cardiotoxicity: A review. Toxicology Reports, 9, 1713–1724. https://doi.org/10.1016/j.toxrep.2022.09.001
Rawat, P. S., Jaiswal, A., Khurana, A., Bhatti, J. S., & Navik, U. (2021). Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie, 139, 111708. https://doi.org/10.1016/j.biopha.2021.111708
Oxidative medicine and cellular longevity, 2022, 7176282. https://doi.org/10.1155/2022/7176282.
Journal of molecular and cellular cardiology, 69, 4–16. https://doi.org/10.1016/j.yjmcc.2014.01.007.
Szwed, A., Kim, E., & Jacinto, E. (2021). Regulation and metabolic functions of mTORC1 and mTORC2. Physiological Reviews, 101(3), 1371–1426. https://doi.org/10.1152/physrev.00026.2020
Jiao, Y., Li, Y., Zhang, J., Zhang, S., Zha, Y., & Wang, J. (2022). RRM2 alleviates doxorubicin-induced cardiotoxicity through the AKT/mTOR signaling pathway. Biomolecules, 12(2), 299. https://doi.org/10.3390/biom12020299
Molecular medicine reports, 23(4), 1–11. https://doi.org/10.3892/mmr.2021.11938.
Johnson, R., Shabalala, S., Louw, J., Kappo, A. P., & Muller, C. J. F. (2017). Aspalathin reverts Doxorubicin-Induced cardiotoxicity through increased autophagy and decreased expression of p53/mTOR/p62 signaling. Molecules (Basel Switzerland), 22(10), 1589. https://doi.org/10.3390/molecules22101589
Yarmohammadi, F., Rezaee, R., & Karimi, G. (2021). Natural compounds against doxorubicin-induced cardiotoxicity: A review on the involvement of Nrf2/ARE signaling pathway. Phytotherapy Research, 35(3), 1163–1175. https://doi.org/10.1002/ptr.6882
Lv, X., Zhu, Y., Deng, Y., Zhang, S., Zhang, Q., Zhao, B., & Li, G. (2020). Glycyrrhizin improved autophagy flux via HMGB1-dependent Akt/mTOR signaling pathway to prevent doxorubicin-induced cardiotoxicity. Toxicology, 441, 152508. https://doi.org/10.1016/j.tox.2020.152508
Phytomedicine: international journal of phytotherapy and phytopharmacology, 99, 154027. https://doi.org/10.1016/j.phymed.2022.154027.
Manolis, A. S., Manolis, A. A., Manolis, T. A., Apostolaki, N. E., Apostolopoulos, E. J., Melita, H., & Katsiki, N. (2021). Mitochondrial dysfunction in cardiovascular disease: Current status of translational research/clinical and therapeutic implications. Medicinal Research Reviews, 41(1), 275–313. https://doi.org/10.1002/med.21732
Garcia-Ropero, A., Santos-Gallego, C. G., Zafar, M. U., & Badimon, J. J. (2019). Metabolism of the failing heart and the impact of SGLT2 inhibitors. Expert Opinion on drug Metabolism & Toxicology, 15(4), 275–285. https://doi.org/10.1080/17425255.2019.1588886
Scientific Reports, 13(1), 8346. https://doi.org/10.21203/rs.3.rs-614231/v1.
Carvalho, R. A., Sousa, R. P. B., Cadete, V. J. J., Lopaschuk, G. D., Palmeira, C. M. M., Bjork, J. A., & Wallace, K. B. (2010). Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology, 270(2), 92–98. https://doi.org/10.1016/j.tox.2010.01.019
Journal of applied toxicology: JAT, 36(11), 1486–1495. https://doi.org/10.1002/jat.3307.
γ‐mediated metabolic reprogramming in cardiomyocytes. The Journal of pathology, 247(3), 320–332. https://doi.org/10.1002/path.5192.
Gorini, S., De Angelis, A., Berrino, L., Malara, N., Rosano, G., & Ferraro, E. (2018). Chemotherapeutic drugs and mitochondrial dysfunction: Focus on doxorubicin, trastuzumab, and sunitinib. Oxidative medicine and cellular longevity, 2018, 7582730. https://doi.org/10.1155/2018/7582730
Nolfi-Donegan, D., Braganza, A., & Shiva, S. (2020). Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biology, 37, 101674. https://doi.org/10.1016/j.redox.2020.101674
Russo, M., Della Sala, A., Tocchetti, C. G., Porporato, P. E., & Ghigo, A. (2021). Metabolic aspects of anthracycline cardiotoxicity. Current Treatment Options in Oncology, 22(2), 18. https://doi.org/10.1007/s11864-020-00812-1
Shi, S., Chen, Y., Luo, Z., Nie, G., & Dai, Y. (2023). Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Communication and Signaling: CCS, 21(1), 61. https://doi.org/10.1186/s12964-023-01077-5
Nature cancer, 1(3), 315–328. https://doi.org/10.1038/s43018-020-0039-1.
Chen, Y., & Zhou, X. (2020). Research progress of mTOR inhibitors. European Journal of Medicinal Chemistry, 208, 112820. https://doi.org/10.1016/j.ejmech.2020.112820
Getz, L. J., Runte, C. S., Rainey, J. K., & Thomas, N. A. (2019). Tyrosine phosphorylation as a widespread regulatory mechanism in prokaryotes. Journal of Bacteriology, 201(19), e00205–e00219. https://doi.org/10.1128/JB.00205-19
Sciarretta, S., Forte, M., Frati, G., & Sadoshima, J. (2018). New insights into the role of mTOR signaling in the cardiovascular system. Circulation Research, 122, 489–505. https://doi.org/10.1161/CIRCRESAHA.117.311147
Mao, Z., & Zhang, W. (2018). Role of mTOR in glucose and lipid metabolism. International Journal of Molecular Sciences, 19(7). https://doi.org/10.3390/ijms19072043
Molecular cell, 39(2), 171–183. https://doi.org/10.1016/j.molcel.2010.06.022.
Shi, F., & Collins, S. (2023). Regulation of mTOR signaling: Emerging role of cyclic nucleotide-dependent protein kinases and implications for cardiometabolic disease. International Journal of Molecular Sciences, 24(14). https://doi.org/10.3390/ijms241411497
Kaldirim, M., Lang, A., Pfeiler, S., Fiegenbaum, P., Kelm, M., Bönner, F., & Gerdes, N. (2022). Modulation of mTOR signaling in cardiovascular disease to target acute and chronic inflammation. Frontiers in Cardiovascular Medicine, 9, 907348. https://doi.org/10.3389/fcvm.2022.907348
Aging Cell, 19(4), e13126. https://doi.org/10.1111/acel.13126.
Science (New York, N.Y.), 366(6464), 468–475. https://doi.org/10.1126/science.aay0166.
Nature, 614(7948), 572–579. https://doi.org/10.1038/s41586-022-05652-7.
Dang, T. T., & Back, S. H. (2021). Translation inhibitors activate autophagy master regulators TFEB and TFE3. International Journal of Molecular Sciences, 22(21), 12083. https://doi.org/10.3390/ijms222112083
Deleyto-Seldas, N., & Efeyan, A. (2021). The mTOR–autophagy axis and the control of metabolism. Frontiers in Cell and Developmental Biology, 9, 655731. https://doi.org/10.3389/fcell.2021.655731
Sadria, M., & Layton, A. T. (2021). Interactions among mTORC, AMPK and SIRT: A computational model for cell energy balance and metabolism. Cell Communication and Signaling, 19(1), 1–17. https://doi.org/10.1101/2020.10.07.330308
Bazer, F. W., Seo, H., Wu, G., & Johnson, G. A. (2020). Interferon tau: Influences on growth and development of the conceptus. Theriogenology, 150, 75–83. https://doi.org/10.1016/j.theriogenology.2020.01.069
Jacinto, E. (2019). Amplifying mTORC2 signals through AMPK during energetic stress. Science Signaling, 12(585), eaax5855. https://doi.org/10.1126/scisignal.aax5855
Fu, W., & Hall, M. N. (2020). Regulation of mTORC2 signaling. Genes, 11(9), 1045. https://doi.org/10.3390/genes11091045
Nature Communications, 11(1), 575. https://doi.org/10.1038/s41467-020-14430-w.
Journal of Diabetes Research, 2022. https://doi.org/10.1155/2022/1755563.
Shi, B., Ma, M., Zheng, Y., Pan, Y., & Lin, X. (2019). mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. Journal of Cellular Physiology, 234(8), 12562–12568. https://doi.org/10.1002/jcp.28125
Circulation research, 132(5), e148–e165. https://doi.org/10.1161/CIRCRESAHA.119.316388.
Daneshgar, N., Rabinovitch, P. S., & Dai, D. F. (2021). TOR signaling pathway in cardiac aging and heart failure. Biomolecules, 11(2), 168. https://doi.org/10.3390/biom11020168
Mohan, U. P., PB, T. P., Iqbal, S. T. A., & Arunachalam, S. (2021). Mechanisms of doxorubicin-mediated reproductive toxicity–a review. Reproductive Toxicology, 102, 80–89. https://doi.org/10.1016/j.reprotox.2021.04.003
Oncotarget, 8(3), 4837–4848. https://doi.org/10.18632/oncotarget.13596.
β-LAPachone ameliorates doxorubicin-induced cardiotoxicity via regulating autophagy and Nrf2 signalling pathways in mice. Basic & clinical pharmacology & toxicology, 126(4), 364–373. https://doi.org/10.1111/bcpt.13340.
Biochemical Pharmacology, 180, 114188. https://doi.org/10.1016/j.bcp.2020.114188.
Yu, W., Qin, X., Zhang, Y., Qiu, P., Wang, L., Zha, W., & Ren, J. (2020). Curcumin suppresses doxorubicin-induced cardiomyocyte pyroptosis via a PI3K/Akt/mTOR-dependent manner. Cardiovascular Diagnosis and Therapy, 10(4), 752. https://doi.org/10.21037/cdt-19-707
Wang, M., Firrman, J., Liu, L., & Yam, K. (2019). A review on flavonoid apigenin: Dietary intake, ADME, antimicrobial effects, and interactions with human gut microbiota. BioMed Research International, 2019, 7010467. https://doi.org/10.1155/2019/7010467
Thomas, S. D., Jha, N. K., Jha, S. K., Sadek, B., & Ojha, S. (2023). Pharmacological and molecular insight on the cardioprotective role of apigenin. Nutrients, 15(2), 385. https://doi.org/10.3390/nu15020385
Yu, W., Sun, H., Zha, W., Cui, W., Xu, L., Min, Q., & Wu, J. (2017). Apigenin attenuates adriamycin-induced cardiomyocyte apoptosis via the PI3K/AKT/mTOR pathway. Evidence-based complementary and alternative medicine: eCAM, 2017, 2590676. https://doi.org/10.1155/2017/2590676
Biomolecules, 11(3), 392. https://doi.org/10.3390/biom11030392.
Fu, Y. S., Chen, T. H., Weng, L., Huang, L., Lai, D., & Weng, C. F. (2021). Pharmacological properties and underlying mechanisms of curcumin and prospects in medicinal potential. Biomedicine & Pharmacotherapy, 141, 111888. https://doi.org/10.1016/j.biopha.2021.111888
Vafaeipour, Z., Razavi, B. M., & Hosseinzadeh, H. (2022). Effects of turmeric (Curcuma longa) and its constituent (curcumin) on the metabolic syndrome: An updated review. Journal of Integrative Medicine, 20(3), 193–203. https://doi.org/10.1039/D2FO02625B
Yarmohammadi, F., Hayes, A. W., & Karimi, G. (2021). Protective effects of curcumin on chemical and drug-induced cardiotoxicity: A review. Naunyn-Schmiedeberg’s Archives of Pharmacology, 394, 1341–1353. https://doi.org/10.1007/s00210-021-02072-8
Psychopharmacology, 240(5), 1179–1190. https://doi.org/10.1007/s00213-023-06357-z.
Zhang, Q., & Wu, L. (2022). In vitro and in vivo cardioprotective effects of curcumin against doxorubicin-induced cardiotoxicity: A systematic review. Journal of oncology, 2022, 7277562. https://doi.org/10.1155/2022/7277562
Yao, H., Zhou, L., Tang, L., Guan, Y., Chen, S., Zhang, Y., & Han, X. (2017). Protective effects of luteolin-7-O-glucoside against starvation-induced injury through upregulation of autophagy in H9c2 cells. Bioscience Trends, 11(5), 557–564. https://doi.org/10.5582/bst.2017.01111
Zang, Y., Igarashi, K., & Li, Y. (2016). Anti-diabetic effects of luteolin and luteolin-7-O-glucoside on KK-A y mice. Bioscience Biotechnology and Biochemistry, 80(8), 1580–1586. https://doi.org/10.1080/09168451.2015.1116928
Nutrients, 14(6), 1155. https://doi.org/10.3390/nu14061155.
Biomolecules, 10(4), 502. https://doi.org/10.3390/biom10040502.
Cardiovascular toxicology, 16(2), 101–110. https://doi.org/10.1007/s12012-015-9317-z.
Biomedicine & Pharmacotherapy, 158, 114203. https://doi.org/10.1016/j.biopha.2022.114203.
Asokan, S. M., Mariappan, R., Muthusamy, S., & Velmurugan, B. K. (2018). Pharmacological benefits of neferine-A comprehensive review. Life Sciences, 199, 60–70. https://doi.org/10.1016/j.lfs.2018.02.032
Guolan, D., Lingli, W., Wenyi, H., Wei, Z., Baowei, C., & Sen, B. (2018). Anti-inflammatory effects of neferine on LPS-induced human endothelium via MAPK, and NF-κβ pathways. Die Pharmazie-An International Journal of Pharmaceutical Sciences, 73(9), 541–544. https://doi.org/10.1691/ph.2018.8443
Lohanathan, B. P., Rathinasamy, B., Huang, C. Y., & Viswanadha, V. P. (2022). Neferine attenuates doxorubicin-induced fibrosis and hypertrophy in H9c2 cells. Journal of Biochemical and Molecular Toxicology, 36(7), e23054. https://doi.org/10.1002/jbt.23054
Bharathi Priya, L., Baskaran, R., Huang, C. Y., & Vijaya Padma, V. (2018). Neferine modulates IGF-1R/Nrf2 signaling in doxorubicin treated H9c2 cardiomyoblasts. Journal of Cellular Biochemistry, 119(2), 1441–1452. https://doi.org/10.1002/jcb.26305
Van Hung, P. (2016). Phenolic compounds of cereals and their antioxidant capacity. Critical Reviews in Food Science and Nutrition, 56(1), 25–35. https://doi.org/10.1080/10408398.2012.708909
Ma, D., Wang, C., Feng, J., & Xu, B. (2021). Wheat grain phenolics: A review on composition, bioactivity, and influencing factors. Journal of the Science of Food and Agriculture, 101(15), 6167–6185. https://doi.org/10.1002/jsfa.11428
Sahu, R., Dua, T. K., Das, S., De Feo, V., & Dewanjee, S. (2019). Wheat phenolics suppress doxorubicin-induced cardiotoxicity via inhibition of oxidative stress, MAP kinase activation, NF-κB pathway, PI3K/Akt/mTOR impairment, and cardiac apoptosis. Food and Chemical Toxicology: An International Journal Published for the British Industrial Biological Research Association, 125, 503–519. https://doi.org/10.1016/j.fct.2019.01.034
Zhao, J., Zhou, H., An, Y., Shen, K., & Yu, L. (2021). Biological effects of corosolic acid as an anti–inflammatory, anti–metabolic syndrome and anti–neoplasic natural compound. Oncology Letters, 21(2), 1. https://doi.org/10.3892/ol.2020.12345
International Journal of Molecular Medicine, 45(5), 1425–1435. https://doi.org/10.3892/ijmm.2020.4531.
Alkholifi, F. K., Devi, S., Yusufoglu, H. S., & Alam, A. (2023). The cardioprotective effect of corosolic acid in the diabetic rats: A possible mechanism of the PPAR-γ pathway. Molecules, 28(3), 929. https://doi.org/10.3390/molecules28030929
Che, Y., Wang, Z., Yuan, Y., Zhou, H., Wu, H., Wang, S., & Tang, Q. (2022). By restoring autophagic flux and improving mitochondrial function, corosolic acid protects against dox-induced cardiotoxicity. Cell Biology and Toxicology, 38(3), 451–467. https://doi.org/10.1007/s10565-021-09619-8
International Journal of Molecular Sciences, 23(23), 15180. https://doi.org/10.3390/ijms232315180.
Wei, Y., Xu, M., Ren, Y., Lu, G., Xu, Y., Song, Y., & Ji, H. (2016). The cardioprotection of dihydrotanshinone I against myocardial ischemia–reperfusion injury via inhibition of arachidonic acid ω-hydroxylase. Canadian Journal of Physiology and Pharmacology, 94(12), 1267–1275. https://doi.org/10.1139/cjpp-2016-0036
κB inflammatory signaling axis: a novel therapeutic pathway of dihydrotanshinone I in doxorubicin-induced cardiotoxicity. Journal of experimental & clinical cancer research: CR, 39(1), 93. https://doi.org/10.1186/s13046-020-01595-x.
Yarmohammadi, F., Karbasforooshan, H., Hayes, A. W., & Karimi, G. (2021). Inflammation suppression in doxorubicin-induced cardiotoxicity: Natural compounds as therapeutic options. Naunyn-Schmiedeberg’s Archives of Pharmacology, 394(10), 2003–2011. https://doi.org/10.1007/s00210-021-02132-z
Fang, Z., Zhang, M., Liu, J., Zhao, X., Zhang, Y., & Fang, L. (2021). Tanshinone IIA: A review of its anticancer effects. Frontiers in Pharmacology, 11, 611087. https://doi.org/10.3389/fphar.2020.611087
Guo, R., Li, L., Su, J., Li, S., Duncan, S. E., Liu, Z., & Fan, G. (2020). Pharmacological activity and mechanism of tanshinone IIA in related diseases. Drug Design Development and Therapy, 14, 4735–4748. https://doi.org/10.2147/DDDT.S266911
Feng, J., Liu, L., Yao, F., Zhou, D., He, Y., & Wang, J. (2021). The protective effect of tanshinone IIa on endothelial cells: A generalist among clinical therapeutics. Expert Review of Clinical Pharmacology, 14(2), 239–248. https://doi.org/10.1080/17512433.2021.1878877
He, Z., Sun, C., Xu, Y., & Cheng, D. (2016). Reduction of atrial fibrillation by Tanshinone IIA in chronic heart failure. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie, 84, 1760–1767. https://doi.org/10.1016/j.biopha.2016.10.110
Frontiers in Pharmacology, 14, 1165212. https://doi.org/10.3389/fphar.2023.1165212.
Cancers, 11(7), 910. https://doi.org/10.3390/cancers11070910.
Chaudhary, S. K., Sandasi, M., Makolo, F., van Heerden, F. R., & Viljoen, A. M. (2021). Aspalathin: A rare dietary dihydrochalcone from aspalathus linearis (rooibos tea). Phytochemistry Reviews, 1–32. https://doi.org/10.1007/s11101-021-09741-9
Molecules, 24(9), 1713. https://doi.org/10.3390/molecules24091713.
Li, C., Liu, Y., Zhang, Y., Li, J., & Lai, J. (2022). Astragalus polysaccharide: A review of its immunomodulatory effect. Archives of Pharmacal Research, 45(6), 367–389. https://doi.org/10.1007/s12272-022-01393-3
Zheng, Y., Ren, W., Zhang, L., Zhang, Y., Liu, D., & Liu, Y. (2020). A review of the pharmacological action of Astragalus polysaccharide. Frontiers in Pharmacology, 11, 349. https://doi.org/10.3389/fphar.2020.00349
Liu, D., Chen, L., Zhao, J., & Cui, K. (2018). Cardioprotection activity and mechanism of astragalus polysaccharide in vivo and in vitro. International Journal of Biological Macromolecules, 111, 947–952. https://doi.org/10.1016/j.ijbiomac.2018.01.048
Zhang, Y., Zhou, Q., Ding, X., Wang, H., & Tan, G. (2021). HILIC-MS-based metabolomics reveal that astragalus polysaccharide alleviates doxorubicin-induced cardiomyopathy by regulating sphingolipid and glycerophospholipid homeostasis. Journal of Pharmaceutical and Biomedical Analysis, 203, 114177. https://doi.org/10.1016/j.jpba.2021.114177
Zhang, H., Caprioli, G., Hussain, H., Le, N. P. K., Farag, M. A., & Xiao, J. (2021). A multifaceted review on dihydromyricetin resources, extraction, bioavailability, biotransformation, bioactivities, and food applications with future perspectives to maximize its value. eFood, 2(4), 164–184. https://doi.org/10.53365/efood.k/143518
Frontiers in Pharmacology, 12, 3721. https://doi.org/10.3389/fphar.2021.794563.
Wang, Y., Wang, J., Xiang, H., Ding, P., Wu, T., & Ji, G. (2022). Recent update on application of dihydromyricetin in metabolic related diseases. Biomedicine & Pharmacotherapy, 148, 112771. https://doi.org/10.1016/j.biopha.2022.112771
Frontiers in pharmacology, 9, 1204. https://doi.org/10.3389/fphar.2018.01204.
Wang, L., & Ma, Q. (2018). Clinical benefits and pharmacology of scutellarin: A comprehensive review. Pharmacology & Therapeutics, 190, 105–127. https://doi.org/10.1016/j.pharmthera.2018.05.006
Huang, H., Geng, Q., Yao, H., Shen, Z., Wu, Z., Miao, X., & Shi, P. (2018). Protective effect of scutellarin on myocardial infarction induced by isoprenaline in rats. Iranian Journal of Basic Medical Sciences, 21(3), 267. https://doi.org/10.22038/ijbms.2018.26110.6415
Xu, L., Chen, R., Ma, X., Zhu, Y., Sun, G., & Sun, X. (2020). Scutellarin protects against myocardial ischemia-reperfusion injury by suppressing NLRP3 inflammasome activation. Phytomedicine, 68, 153169. https://doi.org/10.1016/j.phymed.2020.153169
Chemistry & biodiversity, 20(1), e202200450. https://doi.org/10.1002/cbdv.202200450.
Toxicology in vitro: an international journal published in association with BIBRA, 82, 105366. https://doi.org/10.1016/j.tiv.2022.105366.
Lu, Y., Feng, Y., Liu, D., Zhang, Z., Gao, K., Zhang, W., & Tang, H. (2018). Thymoquinone attenuates myocardial ischemia/reperfusion injury through activation of SIRT1 signaling. Cellular Physiology and Biochemistry, 47(3), 1193–1206. https://doi.org/10.1159/000490216
Tabassum, S., Rosli, N., Ichwan, S. J. A., & Mishra, P. (2021). Thymoquinone and its pharmacological perspective: A review. Pharmacological Research - Modern Chinese Medicine, 1, 100020. https://doi.org/10.1016/j.prmcm.2021.100020
Gur, F. M., & Aktas, I. (2021). The ameliorative effects of thymoquinone and beta-aminoisobutyric acid on streptozotocin-induced diabetic cardiomyopathy. Tissue and Cell, 71, 101582. https://doi.org/10.1016/j.tice.2021.101582
Liu, D., & Zhao, L. (2022). Thymoquinone–induced autophagy mitigates doxorubicin–induced H9c2 cell apoptosis. Experimental and Therapeutic Medicine, 24(5), 694. https://doi.org/10.3892/etm.2022.11630
Nagar, P. S., Rane, S., & Dwivedi, M. (2022). LC-MS/MS standardization and validation of glycyrrhizin from the roots of taverniera cuneifolia: A potential alternative source of glycyrrhiza glabra. Heliyon, 8(8), e10234. https://doi.org/10.1016/j.heliyon.2022.e10234
Thakur, V., Alcoreza, N., Delgado, M., Joddar, B., & Chattopadhyay, M. (2021). Cardioprotective effect of glycyrrhizin on myocardial remodeling in diabetic rats. Biomolecules, 11(4), 569. https://doi.org/10.3390/biom11040569
Translational stroke research, 11, 967–982. https://doi.org/10.1007/s12975-019-00772-1.
Mano, Y., Abe, K., Takahashi, M., Higurashi, T., Kawano, Y., Miyazaki, S., & Maeda-Minami, A. (2023). Optimal administration of glycyrrhizin avoids pharmacokinetic interactions with high-dose methotrexate and exerts a hepatoprotective effect. Anticancer Research, 43(4), 1493–1501. https://doi.org/10.21873/anticanres.16298
Jäger, T., Mokos, A., Prasianakis, N. I., & Leyer, S. (2022). Pore-level multiphase simulations of realistic distillation membranes for water desalination. Membranes, 11(4), 569. https://doi.org/10.3390/biom11040569
Naunyn-Schmiedeberg’s Archives of Pharmacology, 393, 979–989. https://doi.org/10.1007/s00210-019-01767-3.
Frontiers in Immunology, 11, 1104. https://doi.org/10.3389/fimmu.2020.01104.
κB and PI3K/Akt/mTOR pathways. Molecular immunology, 94, 7–17. https://doi.org/10.1016/j.molimm.2017.12.008.
Oh, H., Choi, A., Seo, N., Lim, J. S., You, J. S., & Chung, Y. E. (2021). Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on post-contrast acute kidney injury. Scientific Reports, 11(1), 1–12. https://doi.org/10.1038/s41598-021-94928-5
Malviya, V., Tawar, M., Burange, P., & Jodh, R. (2022). A brief review on resveratrol, 12(2), 157–162. https://doi.org/10.52711/2231-5659.2022.00027
Dyck, G. J. B., Raj, P., Zieroth, S., Dyck, J. R. B., & Ezekowitz, J. A. (2019). The effects of resveratrol in patients with cardiovascular disease and heart failure: A narrative review. International Journal of Molecular Sciences, 20(4), 904. https://doi.org/10.3390/ijms20040904
Li, T., Tan, Y., Ouyang, S., He, J., & Liu, L. (2022). Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene, 808, 145968. https://doi.org/10.1016/j.gene.2021.145968
Gu, J., Fan, Y. Q., Zhang, H. L., Pan, J. A., Yu, J. Y., Zhang, J. F., & Wang, C. Q. (2018). Resveratrol suppresses doxorubicin-induced cardiotoxicity by disrupting E2F1 mediated autophagy inhibition and apoptosis promotion. Biochemical Pharmacology, 150, 202–213. https://doi.org/10.1016/j.bcp.2018.02.025
Gu, J., Hu, W., Song, Z. P., Chen, Y. G., Zhang, D. D., & Wang, C. Q. (2016). Resveratrol-induced autophagy promotes survival and attenuates doxorubicin-induced cardiotoxicity. International Immunopharmacology, 32, 1–7. https://doi.org/10.1016/j.intimp.2016.01.002
Lei, Y., & Klionsky, D. J. (2023). Transcriptional regulation of autophagy and its implications in human disease. Cell Death & Differentiation, 1–14. https://doi.org/10.1038/s41418-023-01162-9
Rahman, M. A., Cho, Y., Nam, G., & Rhim, H. (2021). Antioxidant compound, oxyresveratrol, inhibits APP production through the AMPK/ULK1/mTOR-mediated autophagy pathway in mouse cortical astrocytes. Antioxidants, 10(3), 408. https://doi.org/10.3390/antiox10030408
Gomes, C. L., de Albuquerque Wanderley Sales, V., Gomes de Melo, C., Ferreira da Silva, R. M., Nishimura, V., Rolim, R. H., L. A., & Rolim Neto, P. J. (2021). Beta-lapachone: Natural occurrence, physicochemical properties, biological activities, toxicity and synthesis. Phytochemistry, 186, 112713. https://doi.org/10.1016/j.phytochem.2021.112713
β-Lapachone protects against doxorubicin-induced nephrotoxicity via NAD+/AMPK/NF-kB in mice. Naunyn-Schmiedeberg’s Archives of Pharmacology, 392, 633–640. https://doi.org/10.1007/s00210-019-01619-0.
Tabrizi, F. B., Yarmohammadi, F., Hayes, A. W., & Karimi, G. (2022). The modulation of SIRT1 and SIRT3 by natural compounds as a therapeutic target in doxorubicin-induced cardiotoxicity: A review. Journal of Biochemical and Molecular Toxicology, 36(1), e22946. https://doi.org/10.1002/jbt.22946
Chemico-biological interactions, 317, 108972. https://doi.org/10.1016/j.cbi.2020.108972.
Human Gene Therapy, 33(11–12), 598–613. https://doi.org/10.1089/hum.2021.176.
Frontiers in Pharmacology, 13, 870699. https://doi.org/10.3389/fphar.2022.870699.
Liu, D., & Zhao, L. (2022). Spinacetin alleviates doxorubicin-induced cardiotoxicity by initiating protective autophagy through SIRT3/AMPK/mTOR pathways. Phytomedicine: International Journal of Phytotherapy and Phytopharmacology, 101, 154098. https://doi.org/10.1016/j.phymed.2022.154098
Frontiers in Pharmacology, 9, 824. https://doi.org/10.3389/fphar.2018.00824.
Murcia, M. A., Jiménez-Monreal, A. M., Gonzalez, J., & Martínez-Tomé, M. (2020). Chapter 11 - Spinach. In A. K. B. T.-N. C. and A. P. of F. and V. Jaiswal (Ed.), (pp. 181–195). Academic Press. https://doi.org/10.1016/B978-0-12-812780-3.00011-8
Zhang, T., Liu, J., Tong, Q., & Lin, L. (2020). SIRT3 acts as a positive autophagy regulator to promote lipid mobilization in adipocytes via activating AMPK. International Journal of Molecular Sciences, 21(2), 372. https://doi.org/10.3390/ijms21020372
Acknowledgements
Authors are grateful to the Kermanshah University of Medical Sciences Office of Vice Chancellor for Research, Kermanshah, Iran.
Author information
Authors and Affiliations
Contributions
F.Y. and M.H. wrote the main manuscript text. F.Y. prepared figures. D.SH. edited the main manuscript text. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
All the authors are ready to publish the manuscript in ‘Cardiovascular Toxicology’ as per rule and regulations of the journal.
Competing Interests
The authors declare no competing interests.
Additional information
Communicated by Yajing Wang.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yarmohammadi, F., Hesari, M. & Shackebaei, D. The Role of mTOR in Doxorubicin-Altered Cardiac Metabolism: A Promising Therapeutic Target of Natural Compounds. Cardiovasc Toxicol 24, 146–157 (2024). https://doi.org/10.1007/s12012-023-09820-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-023-09820-7