
Abstract
Doxorubicin (DOX) is a chemotherapy drug known to induce metabolic changes in the heart, leading to potential heart toxicity. These changes impact various cellular functions and pathways such as disrupting the mechanistic target of rapamycin (mTOR) signaling pathway. The study aimed to investigate the effect of DOX on the mTOR pathway through an in vivo systematic review. Databases were searched on September 11, 2023. We finally included 30 in vivo studies that examined the mTOR expression in cardiac tissue samples. The present study has shown that the PI3K/AKT/mTOR, the AMPK/mTOR, the p53/mTOR signaling, the mTOR/TFEB pathway, the p38 MAPK/mTOR, the sestrins/mTOR, and the KLF15/eNOS/mTORC1 signaling pathways play a crucial role in the development of DOX-induced cardiotoxicity. Inhibition or dysregulation of these pathways can lead to increased oxidative stress, apoptosis, and other adverse effects on the heart. Strategies that target and modulate the mTOR pathways, such as the use of mTOR inhibitors like rapamycin, have the potential to enhance the anticancer effects of DOX while also mitigating its cardiotoxic side effects.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
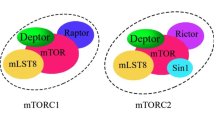
The mammalian target of rapamycin (mTOR) is a serine/threonine kinase that functions as an important regulator of cell growth, proliferation, metabolism, and survival in response to environmental and nutritional signals [1]. mTOR is a key component of two distinct complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), each playing specific roles in cellular regulation [2]. The mTORC1 regulates protein synthesis, cell growth, and autophagy [1]. In contrast, mTORC2 is involved in the regulation of cell survival, metabolism, and cytoskeleton organization [1]. It has been documented that the mTOR pathway plays a crucial role in the development and progression of cancer [3]. Doxorubicin (DOX) is a widely used chemotherapeutic agent with potent anticancer effects [4]. The anticancer effect of DOX may be mediated through its ability to inhibit the mTOR signaling pathways, and subsequently suppress cell growth and proliferation [5]. This inhibition of mTOR can be achieved through several mechanisms, such as the activation of the AMP-activated protein kinase (AMPK) and the inhibition of the phosphoinositide 3-kinase (PI3K)/AKT pathway [6]. AMPK is a main cellular energy sensor that negatively regulates the mTOR pathway in response to an energy-deficit condition, leading to enhanced ATP generation and the inhibition of energy-consuming processes [7, 8]. Several studies have revealed that DOX activates the AMPK pathway in various cancer cell lines and animal models [6, 9, 10]. AMPK activation is mediated following the inhibition of mitochondrial respiration, induction of oxidative stress, and disruption of calcium homeostasis due to DOX treatment [11, 12]. The PI3K/AKT pathway, as a central signaling cascade, is up-regulated in response to energy depletion and nutrient deprivation conditions [13]. Activated PI3K leads to the generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3), which in turn activates the serine/threonine kinase AKT [13]. Activated AKT can directly phosphorylate and inhibit the tuberous sclerosis complex (TSC2), a negative regulator of mTOR [14]. DOX treatment has been observed to decrease the phosphorylation and activation of key components in the PI3K/AKT pathway, including PI3K, AKT, and their downstream targets [14]. Down-regulating the PI3K/AKT signaling pathway by DOX is introduced as one of its mechanisms of anticancer activity [14]. Moreover, DOX induces autophagy, a cellular process that involves the degradation and recycling of damaged or unnecessary cellular components [15]. The mTOR pathway also regulates this process, and the induction of autophagy by DOX can contribute to its anticancer effects [15].
DOX has shown a significant effect on the cardiac mTOR pathway [1]. The impact of DOX on this pathway can lead to alterations in cardiac metabolism and energy balance, contributing to the development of cardiotoxicity [1, 16]. Several studies have investigated how DOX affects changes in the mTOR signaling pathway [1]. Some results suggested a decrease in mTOR protein activation in cardiac tissue, while others suggested an increase [1]. Moreover, some studies show that DOX increases cardiac autophagy, which mitigates the detrimental effects of DOX on cardiac cells [17]. Others reported a decrease in autophagic activity, which may lead to the accumulation of damaged proteins and organelles, contributing to cardiotoxicity [17]. Generally, the contrasts may appear from differences in experimental conditions, dosages, treatment duration, and the specific models used (in vivo and in vitro) [1, 17]. Therefore, this study aimed to systematically review the existing scientific literature on the involvement of the mTOR pathway in the cardiotoxic effects induced by DOX.
Material and Methods
Protocol and Registration
This study adhered to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guideline (Fig. 1). The protocol for the current study was registered with the International Prospective Register of Systematic Reviews (PROSPERO: CRD42023452058). Additionally, this research was approved by the Ethics Committee of Kermanshah University of Medical Sciences, Kermanshah, Iran (Approval Number: IR.KUMS.REC.1402.172).

PRISMA flow diagram
Search Strategy
On September 11, 2023, a systematic search of PubMed, Web of Science, and Scopus was conducted without restriction on date of coverage. The search was limited to literature published in the English language. The keywords used included “Doxorubicin,” “DOX,” “Adriamycin,” “Adriblastin,” “Adriablastin,” “Rubex,” “mTOR,” “mTOR complex,” “mammalian target of rapamycin,” “mTORC1,” and “mTORC2.”
Eligibility Criteria
Two reviewers (S.G. and Z.V.) independently assessed the eligibility of the studies, and any disagreement were resolved by a third reviewer (F.Y.). All studies were evaluated on the PICO principles: participants (P): animals; intervention (I): DOX treatment; comparison (C): control group; outcome (O): cardiac mTOR expression.
The inclusion criteria were as follows: (1) all animal studies that utilized DOX as an intervention; (2) studies with a vehicle-treated control group or an untreated group; and (3) studies that measured cardiac mTOR expression. There were no limitations regarding the route, dosage, or duration of treatment, and no restrictions were placed on the types of study designs eligible for inclusion.
The exclusion criteria were as follows: (1) studies that were not conducted on animals; (2) in vitro, ex vivo, or human studies; (3) studies that did not measure cardiac mTOR expression: (4) studies without a control group; and (5) studies that did not utilize DOX as an intervention. Additionally, review papers, letters to the editors, book chapters, posters, oral presentations, conference abstracts, nonpeer-reviewed papers, and editorials were also excluded.
Data Extraction and Quality Assessment
The data were independently extracted by two reviewers (Z.V. and M.H.) and included: (1) the first author names and publication year; (2) species, sex, age, weight, and sample size of the animals; (3) control group; (4) DOX cumulative dose, route of administration, and duration of treatment; and (5) outcomes, including mTOR, AMPK, and AKT expressions, as well as ATP and AMP/ATP ratio. The quality assessment of the included studies was conducted using SYRCLE’s risk of bias tool by two reviewers (S.S. and M.H.), with any disparities resolved by a third reviewer (F.Y.).
Results
Literature Search and Basic Characteristics of the Included Studies
Initially, 1879 publications were obtained through searches of the databases (PubMed, Scopus, and Web of Science). After removing duplicates, 834 articles underwent screening. Following a review of titles and abstracts, 786 articles were excluded. Subsequently, 48 articles were identified for further consideration based on full text. Ultimately, 17 articles were excluded due to irrelevance, resulting in 31 related studies included in the qualitative synthesis (Fig. 1). The characteristics of the included studies are summarized in Table 1. The majority of the studies (22 studies) were conducted in mice, while the remaining 9 involved rats.
Risk of Bias of Included Studies
The included studies were assessed for risks of bias using the SYRCLE’s risk of bias tool, which covered aspects such as random group allocation, baseline similarities, blinded group allocation, random housing, blinded interventions, random outcome assessment, blinded outcome assessment, incomplete outcome data, selective outcome reporting, and conflict of interest. Two review authors (S.G. and S.S.) independently evaluated the risk of bias, categorizing responses as “yes,” “no,” or “unclear”. The findings, as detailed in Table 2, revealed that the majority of included studies exhibited a predominant unclear risk of bias.
Discussion and Conclusion
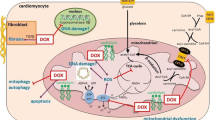
The mTOR protein plays a crucial role in the anticancer effects of DOX as well as its cardiotoxic side effects. DOX can inhibit the mTOR pathway, leading to suppression of cell growth and proliferation in cancer cells. However, prolonged or dysregulated activation of the mTOR pathway can lead to cardiac apoptosis and autophagy, contributing to DOX-induced cardiotoxicity. While autophagy is typically viewed as a protective process that sustains cell viability by recycling damaged cellular components, it can become dysregulated, leading to harmful effects on the cardiac cell [18]. Autophagic cell death has been identified as a contributor to DOX cardiotoxicity [19]. Furthermore, mitophagy, defined as the autophagic elimination of damaged mitochondria [20,21,22], is influenced by DOX in a way that exacerbates cardiotoxicity [19]. Several research papers have demonstrated that the downregulation of the PI3K/AKT/mTOR pathway, characterized by decreased phosphorylation of AKT and mTOR, is implicated in the development of DOX-induced cardiac apoptosis and autophagy [23,24,25,26,27,28,29,30]. It has also been reported that the downregulation or impairment of the AKT and mTOR expression may be a key mechanism by which DOX induces cardiomyopathy [31,32,33,34]. The brain-derived neurotrophic factor (BDNF) is a neurotrophin that binds to the tropomyosin receptor kinase B (TrkB) and activates the AKT/mTOR signaling cascade in cardiomyocytes [35]. It has been suggested that BDNF can protect against DOX-induced cardiotoxicity by activating the AKT pathway and promoting the subsequent phosphorylation of mTOR [36]. Insulin-like growth factor 1 (IGF-1) is a hormone that binds to and activates the IGF-1 receptor, leading to the upregulation of the PI3K/AKT cascade and subsequent activation of the mTORC1 complex [37]. In a study by Shati, the activation of mTOR by IGF-1 reduced death in the DOX-treated cardiac myocytes [38]. Generally, the dysregulation of the PI3K/AKT/mTOR signaling pathway appears to be the main mechanism underlying the cardiotoxic effects of DOX. Modulation of this pathway with BDNF or IGF-1 could provide potential therapeutic approaches against the detrimental impacts of DOX on the heart (Fig. 2).

The mTOR pathways involved in DOX-induced cardiotoxicity. AMPK AMP-activated protein kinase, BDNF brain-derived neurotrophic factor, eNOS nitric oxide synthase, IGF-1 insulin-like growth factor 1, p38 MAPKs p38 mitogen-activated protein kinases, mTOR mechanistic target of rapamycin, NF-κB nuclear factor-kappa B, PI3K phosphoinositide 3-kinase, TFEB transcription factor EB, TSC2 tuberous sclerosis complex 2, ULK1 Unc-51-like kinase 1
Moreover, numerous studies have reported that DOX treatment leads to a decrease in the phosphorylation and activation of AMPK in heart tissue [39,40,41,42,43,44,45]. This inhibition of AMPK, while potentially protective in the short term, ultimately contributes to the detrimental effects of chronic DOX-induced cardiotoxicity through increased energy deficits and activation of the mTOR pathway [39,40,41,42,43,44,45]. The Unc-51-like kinase 1 (ULK1) is a serine/threonine protein kinase that plays a central role in the initiation of autophagy by the phosphorylation of protein Beclin1 [46]. During DOX treatment, activated mTORC1 suppresses the ULK1 complex and prevents the initiation of autophagy [47]. However, other studies have revealed that DOX treatment leads to an increase in AMPK expression and a decrease in mTOR phosphorylation in the heart [9, 10]. This suggests that DOX may activate the AMPK signaling while inhibiting the mTOR pathway [9, 10]. Generally, targeting the AMPK/mTOR pathway may contribute to the development of strategies to protect the heart during DOX chemotherapy (Fig. 2).
The transcription factor E2F1 is a key regulator of cell cycle progression and proliferation [48]. DOX has been shown that activate E2F1 in cardiomyocytes [49]. Activated E2F1 inhibited autophagy in cardiomyocytes by inducing the mTORC1 activity [49]. Therefore, targeting the E2F1/mTORC1, by mTOR inhibitors, may represent a promising strategy to mitigate DOX-induced cardiotoxicity (Fig. 2).
Protein p53 is a transcription factor that plays a key role in regulating apoptosis [50]. The p53 has been shown to induce AMPK expression during stress [50]. Activated AMPK phosphorylates and activates the TSC2, a critical negative dysregulated the mTOR pathway [1]. The inhibition of the mTOR pathway by p53 can serve as a mechanism to limit cell growth, especially in response to cellular stress [50]. Zhu et al. have reported that the p53-mediated inhibition of mTOR signaling can contribute to the development of DOX-induced cardiotoxicity [51]. The downregulation of the p53/mTOR pathway may mitigate the cardiotoxic effects of DOX [51] (Fig. 2).
The transcription factor EB (TFEB) is a main regulator of autophagy and lysosomal biogenesis [52]. TFEB is normally phosphorylated and sequestered in the cytoplasm by mTORC1 [52]. During mTORC1 inhibition, TFEB is dephosphorylated and translocated to the nucleus, where it activates the transcription of genes involved in autophagy and lysosomal function [52]. Research papers have shown that DOX dysregulated autophagy and lysosomal function in cardiomyocytes by impairing the mTORC1/TFEB signaling pathway [47, 53].
TFEB can also lead to the inactivation of the transcription factor the nuclear factor-kappa B (NF-κB), a key regulator of inflammatory responses [54]. It has been reported that DOX reduced nuclear localization and expression of TFEB by activating the mTOR pathway [54]. The inhibition of nuclear TFEB by DOX led to the activation of the NF-κB pathway, which resulted in the excessive release of pro-inflammatory cytokines and cardiac dysfunction [54]. Generally, the mTOR/TFEB signaling pathway is a complex axis that regulates autophagy, lysosomal function, and inflammatory responses in cardiomyocytes. Modulating this pathway can explore a potent strategy to reducing DOX-induced cardiotoxicity (Fig. 2).
The p38 mitogen-activated protein kinases (MAPKs) is a stress-activated protein kinase that is triggered by various stimuli, including oxidative stress and inflammation [55]. The p38 MAPK family consists of four isoforms (α, β, γ, and δ) that are involved in multiple cellular functions including differentiation, cell proliferation, inflammation, and apoptosis [55]. DOX treatment has been shown to induce the p38 MAPK pathway in cardiomyocytes [56]. The possible protective effects of the p38γ and p38δ knock-out on the hearts of mice treated with an acute dose of the DOX have been studied [56]. The genetic deletion of the p38δ protein in female mice was associated with reduced fibrosis, decreased mTOR activation, increased autophagy, and improved cardiac output, ultimately leading to improved survival in the DOX-treated group [56]. Therefore, targeting p38δ MAPK/mTOR pathway could be a potential strategy in DOX chemotherapy (Fig. 2).
Sestrins are a family of stress-responsive proteins that act as cellular sensors of energy status [57]. A study has shown that sestrins (sestrin 1 and sestrin 2) neutralized the negative effects of DOX on the heart by suppressing mTORC1 activity and enhancing the autophagic pathway [58]. The inhibition of mTORC1 by sestrins leads to the activation of the ULK1 complex, which then initiates the formation of autophagosomes and the autophagy process [57]. Therefore, the sestrins/mTOR signaling may play a crucial role in the context of DOX-induced cardiotoxicity [58] (Fig. 2).
KLF15 transcription factor plays an important role in maintaining cardiac metabolic and functional homeostasis [59]. It induces the endothelial nitric oxide synthase (eNOS) expression, leading to the upregulation of eNOS expression [60]. The eNOS is responsible for the production of nitric oxide (NO), which has been shown to activate the mTORC1 by stimulating the AKT pathway [60]. It has been revealed that the expression of KLF15 and eNOS were reduced in DOX-treated cardiomyocytes [61]. The decreased eNOS level then resulted in the inactivation of the mTORC1 complex [61]. Therefore, targeting the KLF15/eNOS/mTORC1 axis could potentially be a therapeutic approach to mitigate the harmful cardiac effects of DOX during cancer treatment (Fig. 2). Overall, to address the dual role of mTOR, a potential solution could be the use of combination therapies that target both the anticancer and cardioprotective effects of mTOR modulation. Using mTOR inhibitors, such as rapamycin, in combination with DOX can enhance the anticancer effects by further suppressing mTOR activity in cancer cells. This combination therapy could potentially reduce the cardiotoxic side effects of DOX by preventing the dysregulation of the mTOR pathway in the heart. Moreover, exploring strategies that modulate the upstream regulators (e.g., AMPK, PI3K/AKT) or downstream effectors of mTOR could provide a more comprehensive approach to managing the dual role of mTOR. Identifying and developing cardioprotective agents that can specifically target the mTOR pathway in the heart could help prevent DOX-induced cardiotoxicity without compromising the anticancer effects of DOX. Generally, by understanding the dual role of mTOR in the anticancer effects and cardiotoxicity of DOX, researchers can explore combination therapies and targeted approaches to maximize the therapeutic benefits of DOX while minimizing its detrimental effects on the heart.
Although the study identified several pathways, future investigation for a deeper understanding of the role of these pathways in the anticancer and cardiotoxic effects of DOX is needed. Additionally, it is crucial to develop targeted inhibitors or activators for these pathways. Current inhibitors, such as rapamycin, have limitations, highlighting the necessity for new and more precise agents. Furthermore, the findings need validation in clinical research to ensure that mTOR-targeting treatments are safe and effective for humans.
Abbreviations
- AMPK:
-
AMP-activated protein kinase
- BDNF:
-
brain-derived neurotrophic factor
- DOX:
-
doxorubicin
- eNOS:
-
nitric oxide synthase
- IGF-1:
-
insulin-like growth factor 1
- MAPKs:
-
p38 mitogen-activated protein kinases
- mTOR:
-
mechanistic target of rapamycin
- NF-κB:
-
nuclear factor-kappa B
- NO:
-
nitric oxide
- PI3K:
-
phosphoinositide 3-kinase
- PIP3:
-
phosphatidylinositol-3,4,5-trisphosphate
- PRISMA:
-
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- PROSPERO:
-
Prospective Register of Systematic Reviews
- TFEB:
-
transcription factor EB
- TrkB:
-
tropomyosin receptor kinase B
- TSC2:
-
tuberous sclerosis complex 2
- ULK1:
-
unc-51-like kinase 1
References
Yarmohammadi, F., Hesari, M., & Shackebaei, D. (2023). The role of mTOR in doxorubicin-altered cardiac metabolism: A promising therapeutic target of natural compounds. Cardiovascular Toxicology. https://doi.org/10.1007/s12012-023-09820-7.
Szwed, A., Kim, E., & Jacinto, E. (2021). Regulation and metabolic functions of mTORC1 and mTORC2. Physiological Reviews, 101(3), 1371–1426. https://doi.org/10.1152/physrev.00026.2020.
El-Tanani, M., Nsairat, H., Aljabali, A. A., Serrano-Aroca, Á., Mishra, V., Mishra, Y., & Tambuwala, M. M. (2023). Role of mammalian target of rapamycin (mTOR) signalling in oncogenesis. Life Sciences, 323, 121662. https://doi.org/10.1016/j.lfs.2023.121662.
Yarmohmmadi, F., Rahimi, N., Faghir-Ghanesefat, H., Javadian, N., Abdollahi, A., Pasalar, P., & Dehpour, A. R. (2017). Protective effects of agmatine on doxorubicin-induced chronic cardiotoxicity in rat. European Journal of Pharmacology, 796, 39–44. https://doi.org/10.1016/j.ejphar.2016.12.022.
Maayah, Z. H., Zhang, T., Forrest, M. L., Alrushaid, S., Doschak, M. R., Davies, N. M., & El-Kadi, A. O. S. (2018). DOX-Vit D, a novel doxorubicin delivery approach, inhibits human osteosarcoma cell proliferation by inducing apoptosis while inhibiting Akt and mTOR signaling pathways. Pharmaceutics, 10(3). https://doi.org/10.3390/pharmaceutics10030144
Ji, C., Yang, B., Yang, Y.-L., He, S.-H., Miao, D.-S., He, L., & Bi, Z.-G. (2010). Exogenous cell-permeable C6 ceramide sensitizes multiple cancer cell lines to doxorubicin-induced apoptosis by promoting AMPK activation and mTORC1 inhibition. Oncogene, 29(50), 6557–6568. https://doi.org/10.1038/onc.2010.379.
Xu, J., Ji, J., & Yan, X.-H. (2012). Cross-talk between AMPK and mTOR in regulating energy balance. Critical Reviews in Food Science and Nutrition, 52(5), 373–381. https://doi.org/10.1080/10408398.2010.500245.
Yao, H., Han, X., & Han, X. (2014). The cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling pathway. American Journal of Cardiovascular Drugs, 14, 433–442. https://doi.org/10.1007/s40256-014-0089-9.
Cao, Y., Shen, T., Huang, X., Lin, Y., Chen, B., Pang, J., & Li, J. (2017). Astragalus polysaccharide restores autophagic flux and improves cardiomyocyte function in doxorubicin-induced cardiotoxicity. Oncotarget, 8(3), 4837–4848. https://doi.org/10.18632/oncotarget.13596.
Lee, Y., Kwon, I., Jang, Y., Cosio-Lima, L., & Barrington, P. (2020). Endurance exercise attenuates doxorubicin-induced cardiotoxicity. Medicine and Science in Sports and Exercise, 52(1), 25–36. https://doi.org/10.1249/MSS.0000000000002094.
Timm, K. N., & Tyler, D. J. (2020). The role of AMPK activation for cardioprotection in doxorubicin-induced cardiotoxicity. Cardiovascular Drugs and Therapy, 34(2), 255–269. https://doi.org/10.1007/s10557-020-06941-x.
Wang, S., Song, P., & Zou, M.-H. (2012). Inhibition of AMP-activated protein kinase α (AMPKα) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: role of p53 and SIRT1. The Journal of Biological Chemistry, 287(11), 8001–8012. https://doi.org/10.1074/jbc.M111.315812.
Yarmohammadi, F., Hayes, A. W., & Karimi, G. (2021). Natural compounds against cytotoxic drug-induced cardiotoxicity: A review on the involvement of PI3K/Akt signaling pathway. Journal of Biochemical and Molecular Toxicology, 35(3), e22683. https://doi.org/10.1002/jbt.22683.
Lin, K., Rong, Y., Chen, D., Zhao, Z., Bo, H., Qiao, A., & Wang, J. (2020). Combination of ruthenium complex and doxorubicin synergistically inhibits cancer cell growth by down-regulating PI3K/AKT signaling pathway. Frontiers in Oncology, 10, 141. https://doi.org/10.3389/fonc.2020.00141.
Chen, C., Lu, L., Yan, S., Yi, H., Yao, H., Wu, D., & Deng, X. (2018). Autophagy and doxorubicin resistance in cancer. Anti-Cancer Drugs, 29(1), 1–9. https://doi.org/10.1097/CAD.0000000000000572.
He, L., Wang, J., Yang, Y., Zou, P., Xia, Z., & Li, J. (2022). SIRT4 suppresses doxorubicin-induced cardiotoxicity by regulating the AKT/mTOR/autophagy pathway. Toxicology, 469, 153119. https://doi.org/10.1016/j.tox.2022.153119.
Xiao, B., Hong, L., Cai, X., Mei, S., Zhang, P., & Shao, L. (2019). The true colors of autophagy in doxorubicin-induced cardiotoxicity. Oncology Letters, 18(3), 2165–2172. https://doi.org/10.3892/ol.2019.10576.
Zhang, X., Zhou, H., & Chang, X. (2023). Involvement of mitochondrial dynamics and mitophagy in diabetic endothelial dysfunction and cardiac microvascular injury. Archives of Toxicology, 97(12), 3023–3035. https://doi.org/10.1007/s00204-023-03599-w.
Koleini, N., & Kardami, E. (2017). Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget, 8(28), 46663–46680. https://doi.org/10.18632/oncotarget.16944.
Chang, X., Liu, R., Li, R., Peng, Y., Zhu, P., & Zhou, H. (2023). Molecular mechanisms of mitochondrial quality control in ischemic cardiomyopathy. International Journal of Biological Sciences, 19(2), 426–448. https://doi.org/10.7150/ijbs.76223.
Li, Y., Yu, J., Li, R., Zhou, H., & Chang, X. (2024). New insights into the role of mitochondrial metabolic dysregulation and immune infiltration in septic cardiomyopathy by integrated bioinformatics analysis and experimental validation. Cellular & Molecular Biology Letters, 29(1), 21. https://doi.org/10.1186/s11658-024-00536-2.
Chang, X., Zhou, S., Liu, J., Wang, Y., Guan, X., Wu, Q., & Liu, R. (2024). Zishen Tongyang Huoxue decoction (TYHX) alleviates sinoatrial node cell ischemia/reperfusion injury by directing mitochondrial quality control via the VDAC1-β-tubulin signaling axis. Journal of Ethnopharmacology, 320, 117371. https://doi.org/10.1016/j.jep.2023.117371.
Yu, W., Sun, H., Zha, W., Cui, W., Xu, L., Min, Q., & Wu, J. (2017). Apigenin attenuates adriamycin-induced cardiomyocyte apoptosis via the PI3K/AKT/mTOR pathway. Evidence-Based Complementary and Alternative Medicine eCAM, 2017, 2590676. https://doi.org/10.1155/2017/2590676.
Wu, Y., Wang, J., Yu, X., Li, D., Han, X., & Fan, L. (2017). Sevoflurane ameliorates doxorubicin-induced myocardial injury by affecting the phosphorylation states of proteins in PI3K/Akt/mTOR signaling pathway. Cardiology Journal, 24(4), 409–418. https://doi.org/10.5603/CJ.a2017.0018.
Merino, H., & Singla, D. K. (2018). Secreted frizzled-related protein-2 inhibits doxorubicin-induced apoptosis mediated through the Akt-mTOR pathway in soleus muscle. Oxidative Medicine and Cellular Longevity, 2018, 6043064. https://doi.org/10.1155/2018/6043064.
Hullin, R., Métrich, M., Sarre, A., Basquin, D., Maillard, M., Regamey, J., & Martin, D. (2018). Diverging effects of enalapril or eplerenone in primary prevention against doxorubicin-induced cardiotoxicity. Cardiovascular Research, 114(2), 272–281. https://doi.org/10.1093/cvr/cvx162.
Sahu, R., Dua, T. K., Das, S., De Feo, V., & Dewanjee, S. (2019). Wheat phenolics suppress doxorubicin-induced cardiotoxicity via inhibition of oxidative stress, MAP kinase activation, NF-κB pathway, PI3K/Akt/mTOR impairment, and cardiac apoptosis. Food and Chemical Toxicology, 125, 503–519. https://doi.org/10.1016/j.fct.2019.01.034.
Nie, L., Liu, M., Chen, J., Wu, Q., Li, Y., Yi, J., & Yang, J. (2021). Hydrogen sulfide ameliorates doxorubicin‑induced myocardial fibrosis in rats via the PI3K/AKT/mTOR pathway. Molecular Medicine Reports, 23(4), 1–11. https://doi.org/10.3892/mmr.2021.11938.
Qin, Y., Lv, C., Zhang, X., Ruan, W., Xu, X., Chen, C., & Guo, X. (2022). Protective effect of qiliqiangxin against doxorubicin-induced cardiomyopathy by suppressing excessive autophagy and apoptosis. Cardiovascular Therapeutics, 2022, 9926635. https://doi.org/10.1155/2022/9926635.
Hu, Y., Jiang, H., Xu, Y., Chen, G., Fan, R., Zhou, Y., & Qiu, Z. (2023). Stomatin-like protein 2 deficiency exacerbates adverse cardiac remodeling. Cell Death Discovery, 9(1), 63. https://doi.org/10.1038/s41420-023-01350-z.
Singla, D. K. (2015). Akt-mTOR pathway inhibits apoptosis and fibrosis in doxorubicin-induced cardiotoxicity following embryonic stem cell transplantation. Cell Transplantation, 24(6), 1031–1042. https://doi.org/10.3727/096368914X679200.
Zhang, X., Hu, C., Kong, C.-Y., Song, P., Wu, H.-M., Xu, S.-C., & Tang, Q.-Z. (2020). FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death and Differentiation, 27(2), 540–555. https://doi.org/10.1038/s41418-019-0372-z.
Yu, W., Qin, X., Zhang, Y., Qiu, P., Wang, L., Zha, W., & Ren, J. (2020). Curcumin suppresses doxorubicin-induced cardiomyocyte pyroptosis via a PI3K/Akt/mTOR-dependent manner. Cardiovascular Diagnosis and Therapy, 10(4), 752. https://doi.org/10.21037/cdt-19-707.
Zhang, J., Wang, M., Ding, W., Zhao, M., Ye, J., Xu, Y., & Liu, J. (2020). Resolvin E1 protects against doxorubicin-induced cardiotoxicity by inhibiting oxidative stress, autophagy and apoptosis by targeting AKT/mTOR signaling. Biochemical Pharmacology, 180, 114188. https://doi.org/10.1016/j.bcp.2020.114188.
Feng, N., Huke, S., Zhu, G., Tocchetti, C. G., Shi, S., Aiba, T., & Mankowski, J. L. (2015). Constitutive BDNF/TrkB signaling is required for normal cardiac contraction and relaxation. Proceedings of the National Academy of Sciences, 112(6), 1880–1885. https://doi.org/10.1073/pnas.1417949112.
Hang, P., Zhao, J., Sun, L., Li, M., Han, Y., Du, Z., & Li, Y. (2017). Brain-derived neurotrophic factor attenuates doxorubicin-induced cardiac dysfunction through activating Akt signalling in rats. Journal of Cellular and Molecular Medicine, 21(4), 685–696. https://doi.org/10.1111/jcmm.13012.
Sakai, H., Asami, M., Naito, H., Kitora, S., Suzuki, Y., Miyauchi, Y., & Ikarashi, N. (2021). Exogenous insulin‐like growth factor 1 attenuates cisplatin‐induced muscle atrophy in mice. Journal of Cachexia, Sarcopenia and Muscle, 12(6), 1570–1581. https://doi.org/10.1002/jcsm.12760.
Shati, A. A. (2020). Doxorubicin-induces NFAT/Fas/FasL cardiac apoptosis in rats through activation of calcineurin and P38 MAPK and inhibition of mTOR signalling pathways. Clinical and Experimental Pharmacology & Physiology, 47(4), 660–676. https://doi.org/10.1111/1440-1681.13225.
Gratia, S., Kay, L., Potenza, L., Seffouh, A., Novel-Chaté, V., Schnebelen, C., & Tokarska-Schlattner, M. (2012). Inhibition of AMPK signalling by doxorubicin: at the crossroads of the cardiac responses to energetic, oxidative, and genotoxic stress. Cardiovascular Research, 95(3), 290–299. https://doi.org/10.1093/cvr/cvs134.
Nazari Soltan Ahmad, S., Sanajou, D., Kalantary-Charvadeh, A., Hosseini, V., Roshangar, L., Khojastehfard, M., & Mesgari-Abbasi, M. (2020). β-LAPachone ameliorates doxorubicin-induced cardiotoxicity via regulating autophagy and Nrf2 signalling pathways in mice. Basic & Clinical Pharmacology & Toxicology, 126(4), 364–373. https://doi.org/10.1111/bcpt.13340.
Wang, Y., Zhu, S., Liu, H., Wei, W., Tu, Y., Chen, C., & Xu, Z. (2019). Thyroxine alleviates energy failure, prevents myocardial cell apoptosis, and protects against doxorubicin-induced cardiac injury and cardiac dysfunction via the LKB1/AMPK/mTOR axis in mice. Disease Markers, 2019, 7420196. https://doi.org/10.1155/2019/7420196.
Li, X., Wang, X., Wang, B., Chi, W., Li, Z., Zhang, M., & Liu, Y. (2022). Dihydromyricetin protects against doxorubicin-induced cardiotoxicity through activation of AMPK/mTOR pathway. Phytomedicine: International Journal of Phytotherapy and Phytopharmacology, 99, 154027 https://doi.org/10.1016/j.phymed.2022.154027.
Sun, X., Zhou, L., Han, Y., Yang, Q., Li, X., Xin, B., & Guo, C. (2023). Scutellarin attenuates doxorubicin-induced cardiotoxicity by inhibiting myocardial fibrosis, apoptosis and autophagy in rats. Chemistry & Biodiversity, 20(1), e202200450. https://doi.org/10.1002/cbdv.202200450.
Ma, T., Yang, L., Zhang, B., Lv, X., Gong, F., & Yang, W. (2023). Hydrogen inhalation enhances autophagy via the AMPK/mTOR pathway, thereby attenuating doxorubicin-induced cardiac injury. International Immunopharmacology, 119(37), 110071. https://doi.org/10.1016/j.intimp.2023.110071.
Zhang, S., Wei, X., Zhang, H., Wu, Y., Jing, J., Huang, R., & Li, Y. (2023). Doxorubicin downregulates autophagy to promote apoptosis-induced dilated cardiomyopathy via regulating the AMPK/mTOR pathway. Biomedicine & Pharmacotherapy, 162, 114691. https://doi.org/10.1016/j.biopha.2023.114691.
Zhang, L., Ouyang, L., Guo, Y., Zhang, J., & Liu, B. (2018). UNC-51-like Kinase 1: From an autophagic initiator to multifunctional drug target: miniperspective. Journal of Medicinal Chemistry, 61(15), 6491–6500. https://doi.org/10.1021/acs.jmedchem.7b01684.
Wang, X., Li, C., Wang, Q., Li, W., Guo, D., Zhang, X., & Wang, Y. (2019). Tanshinone IIA restores dynamic balance of autophagosome/autolysosome in doxorubicin-induced cardiotoxicity via targeting Beclin1/LAMP1. Cancers, 11(7), 910. https://doi.org/10.3390/cancers11070910.
Denechaud, P.-D., Fajas, L., & Giralt, A. (2017). E2F1, a novel regulator of metabolism. Frontiers in Endocrinology, 8, 311. https://doi.org/10.3389/fendo.2017.00311.
Gu, J., Fan, Y.-Q., Zhang, H.-L., Pan, J.-A., Yu, J.-Y., Zhang, J.-F., & Wang, C.-Q. (2018). Resveratrol suppresses doxorubicin-induced cardiotoxicity by disrupting E2F1 mediated autophagy inhibition and apoptosis promotion. Biochemical Pharmacology, 150, 202–213. https://doi.org/10.1016/j.bcp.2018.02.025.
Beyfuss, K., & Hood, D. A. (2018). A systematic review of p53 regulation of oxidative stress in skeletal muscle. Redox Report, 23(1), 100–117. https://doi.org/10.1080/13510002.2017.1416773.
Zhu, W., Soonpaa, M. H., Chen, H., Shen, W., Payne, R. M., Liechty, E. A., & Field, L. J. (2009). Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation, 119(1), 99–106. https://doi.org/10.1161/CIRCULATIONAHA.108.799700.
Vega-Rubin-de-Celis, S., Peña-Llopis, S., Konda, M., & Brugarolas, J. (2017). Multistep regulation of TFEB by MTORC1. Autophagy, 13(3), 464–472. https://doi.org/10.1080/15548627.2016.1271514.
Che, Y., Wang, Z., Yuan, Y., Zhou, H., Wu, H., Wang, S., & Tang, Q. (2022). By restoring autophagic flux and improving mitochondrial function, corosolic acid protects against Dox-induced cardiotoxicity. Cell Biology and Toxicology, 38(3), 451–467. https://doi.org/10.1007/s10565-021-09619-8.
Wang, X., Wang, Q., Li, W., Zhang, Q., Jiang, Y., Guo, D., & Wang, Y. (2020). TFEB-NF-κB inflammatory signaling axis: a novel therapeutic pathway of Dihydrotanshinone I in doxorubicin-induced cardiotoxicity. Journal of Experimental & Clinical Cancer Research: CR, 39(1), 93. https://doi.org/10.1186/s13046-020-01595-x.
Han, J., Wu, J., & Silke, J. (2020). An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F1000Research, 9. https://doi.org/10.12688/f1000research.22092.1
George, S. A., Kiss, A., Obaid, S. N., Venegas, A., Talapatra, T., Wei, C., & Efimov, I. R. (2020). P38δ genetic ablation protects female mice from anthracycline cardiotoxicity. American Journal of Physiology—Heart and Circulatory Physiology, 318(5), 775–786. https://doi.org/10.1152/AJPHEART.00415.2020.
Cordani, M., Sánchez-Álvarez, M., Strippoli, R., Bazhin, A. V., & Donadelli, M. (2019). Sestrins at the interface of ROS control and autophagy regulation in health and disease. Oxidative Medicine and Cellular Longevity, 2019(1), 1283075. https://doi.org/10.1155/2019/1283075.
Li, R., Huang, Y., Semple, I., Kim, M., Zhang, Z., & Lee, J. H. (2019). Cardioprotective roles of sestrin 1 and sestrin 2 against doxorubicin cardiotoxicity. American Journal of Physiology—Heart and Circulatory Physiology, 317(1), H39–H48. https://doi.org/10.1152/ajpheart.00008.2019.
Zhao, Y., Song, W., Wang, L., Rane, M. J., Han, F., & Cai, L. (2019). Multiple roles of KLF15 in the heart: underlying mechanisms and therapeutic implications. Journal of Molecular and Cellular Cardiology, 129, 193–196. https://doi.org/10.1016/j.yjmcc.2019.01.024.
Luo, J.-Y., Cheng, C. K., Gou, L., He, L., Zhao, L., Zhang, Y., & Chen, A. F. (2023). Induction of KLF2 by exercise activates eNOS to improve vasodilatation in diabetic mice. Diabetes, 72(9), 1330–1342. https://doi.org/10.2337/db23-0070.
Tedesco, L., Rossi, F., Ragni, M., Ruocco, C., Brunetti, D., Carruba, M. O., & Nisoli, E. (2020). A special amino-acid formula tailored to boosting cell respiration prevents mitochondrial dysfunction and oxidative stress caused by doxorubicin in mouse cardiomyocytes. Nutrients, 12(2), 282. https://doi.org/10.3390/nu12020282.
Acknowledgements
The authors are grateful to the Kermanshah University of Medical Sciences Office of Vice Chancellor for Research, Kermanshah, Iran.
Funding
This work was supported by the Kermanshah University of Medical Sciences Office of Vice Chancellor for Research, Kermanshah, Iran (Code number 4020316).
Author information
Authors and Affiliations
Contributions
D.S.H.: conceptualization, reviewing, and editing. S.G.: data curation, investigation, and reviewing. Z.V.: visualization and investigation. S.S.: investigation and editing. M.H.: supervision, reviewing, and editing. F.Y.: conceptualization, methodology, visualization, writing—reviewing, and editing.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shackebaei, D., Hesari, M., Gorgani, S. et al. The Role of mTOR in the Doxorubicin-Induced Cardiotoxicity: A Systematic Review. Cell Biochem Biophys (2024). https://doi.org/10.1007/s12013-024-01475-7
Accepted:
Published:
DOI: https://doi.org/10.1007/s12013-024-01475-7