
Abstract
Long-term excessive fluoride intake is linked to skeletal disease. Skeletal health is influenced by the balance between bone formation and resorption of which osteoblast function is critical. The objectives of this study were to determine the effect of fluoride treatment on osteoblast proliferation, apoptosis and caspase-3 and caspase-9 mRNA expression in vitro. Neonatal rat osteoblasts were cultured in the presence of varying concentrations (0.5–30 mg/l) of sodium fluoride and effects of treatments were determined. Treatment with sodium fluoride inhibited osteoblast proliferation in a dose-dependent fashion and effects were maximal after 120 h incubation. A significant increase in osteoblast apoptosis was observed (after 24 and 72-h treatment) in response to the lowest dose of sodium fluoride (0.5 mg/l) and osteoblast apoptosis was further increased in response to higher doses. Increased-osteoblast caspase-3 and caspase-9 mRNA was also observed in response to sodium fluoride treatment (5 mg/l) for 72 h. Results indicate that negative effects of excess fluoride on skeletal health may be mediated in part by inhibition of osteoblast survival.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fluorine is a gaseous element, which reacts with almost all kinds of metal elements to generate different fluorides widely found in nature. Fluoride (F) is most well-known for its prophylactic use over several decades to prevent dental caries via topical application or supplementation of drinking water (Yeung 2008). However, pathological effects of F intake have been reported for various organs including the brain (Wang et al. 2004a, b; Ge et al. 2005a, b; Wu et al. 2006), reproductive tissues (Zhang et al. 2006a, b, c; Wan et al. 2006a, b), thyroid gland (Ge et al. 2005c; Susheela et al. 2005), liver (Shashi 2003; Zhan et al. 2006b), kidney (Usuda et al. 1998; Guan et al. 2000; Zhan et al. 2006a), and pancreas (Matsuo et al. 2000). Furthermore, long-term excessive F intake disrupts the balance of bone deposition and remodeling activities and is linked to skeletal disease, the most prominent of which is fluorosis (Krishnamachari 1986). Fluorosis is a chronic disease caused by ingesting excessive amounts of F, mostly from water, and less often from food (Wang et al. 2002, 2003) and air (Liang et al. 1994; Wang et al. 1992, 1994). Affected organs in affected individuals include the teeth, which is referred to as dental fluorosis (Aoba and Fejerskov 2002) and bone, referred to as skeletal fluorosis (Krishnamachari 1986).
Skeletal fluorosis is characterized by extreme bone deformity with accompanying osteosclerosis, osteoporosis, osteomalacia and immobilization of joints of the axial skeleton (Krishnamachari 1986). The balance between bone deposition and resorption is tightly controlled by osteoblastic and osteoclastic activities of resident bone cells, the osteoblasts and osteoclasts, respectively (Goltzman 2002). Osteoblasts (OB) are the primary cells contributing to bone formation. Osteoblasts actively secrete type I collagen and other bone matrix components and release growth factors, enzymes and minerals into the bone matrix (Goltzman 2002). Hence, effects of excessive F intake on skeletal health could be manifest by effects on OB survival.
In the present studies, cultures of OB were established from neonatal rat calvarias. The objective of described experiments was to investigate the effects of treatment with varying concentrations of F on OB proliferation and apoptosis in vitro. To further elucidate potential mechanisms contributing to F-induced OB apoptosis, effects on treatments on caspase-3 and caspase-9 mRNA expression were also determined.
Materials and methods
Animals
Ten 24-h-old female neonatal Wistar rats were provided by the Experimental Animal Center of Shanxi Medical University (Taiyuan, China). Animals were handled in accordance with the principles of laboratory animal care and all experimental procedures were approved by the Research Commission for the Care and Use of Experimental Animal Center of Shanxi Medical University of China.
Materials and chemicals
Dulbecco’s Modified Eagle Medium (DMEM) and trypsin were obtained from Gibco (Grand Island, NY, USA). Fetal bovine serum (FBS) was purchased from Hangzhou Sijiqing Biological Engineering Material Company (Hangzhou, China). HEPES buffer was obtained from Hyclone (Logan, UT USA). NaF and MTT were purchased from Sigma (St. Louis, MO USA). The apoptosis detection kit containing Annexin-V-FITC and propidium iodide (PI) was from Becton Dickinson (San Jose, CA USA). Trizol Reagent was purchased from Invitrogen (Carlsbad, CA USA). The One-Step SYBR® RT-PCR Kit and real time PCR primers were obtained from the Takara Biotechnology Company (Dalian, China).
OB cell isolation and culture
Osteoblasts were obtained from calvarias of neonatal rats according to previously published procedures (Torricelli et al. 2003; Xue 2001) with some modifications. Approximately 1 × 106 cells/ml were seeded in 25-ml tissue culture flasks containing DMEM medium supplemented with 10% fetal calf serum (FCS), 100 IU/ml penicillin, and 100 μg/ml streptomycin and cultured at 37°C in air with 5% CO2. For subculture, cells at 80–90% confluence were passaged at a ratio of 1:3 after detachment in 0.125% trypsin and 1 mM EDTA. OB were identified by morphology after Giemsa and hemotoxylin and eosin (H&E) staining, alkaline phosphatase (ALP) staining and mineralized nodule (alizarin red) staining. Treatments were applied when cells were semi confluent and were added to 5% FCS-containing medium. For experiments described below, cells were seeded in 96-well plates for cell proliferation studies or in T25 flasks for studies of F-induced apoptosis and caspase-3 and caspase-9 mRNA expression.
Cell proliferation assays
To determine effects of F on cell proliferation, OB were cultured in the presence of increasing concentrations of sodium fluoride (NaF; 0, 0.5, 5, 10, 20 and 30 mg/l) for 24, 48, 72, 96 and 120 h. Effects of F treatment on numbers of viable OB (proliferation) were assessed using the MTT assay which quantifies the ability of viable cells to convert soluble MTT dye into an insoluble dark blue formazan reaction product. At the end of each treatment period, MTT stock solution was added (1 part to 10 parts medium) to cells in each well of a 96-well tissue culture plate and cells were incubated at 37°C for 4 h. Subsequently, the medium containing MTT was removed and 150 μl dimethylsulfoxide (DMSO) was added per well to dissolve the formazan crystals. The absorbance in each well (reflective of number of live cells) was measured at 570 nm with a Bio-Rad automated EIA Analyzer.
Apoptosis assays
For studies of effects of F treatment on OB apoptosis, cells were cultured in the presence of 0, 0.5, 5, 10 and 20 mg/l NaF for 24 and 72 h. After completion of treatment periods, cells were collected by trypsinization and centrifugation, washed three times with PBS (pH 7.4) and resuspended in binding buffer at a concentration of 1 × 106 cells/ml. For each sample, a total of 5 μl of Annexin V-FITC and 10 μl of 20 mg/ml PI were added and cells were incubated for 15 min in the dark before a further addition of 400 μl PBS. Quantitative analysis of OB apoptosis was performed using a FACS caliber flow cytometer (FCM, Becton Dickinson).
Real time PCR analysis of caspase-3 and caspase-9 mRNA
For studies of effects of F treatment on OB mRNA abundance for caspase-3 and caspase-9, cells were cultured in the presence of 0, 0.5, 5, 10 and 20 mg/l NaF for 24 and 72 h. Effects of F treatment on caspase-3 and caspase-9 mRNA were determined using quantitative real time RT-PCR (qRT-PCR) procedures. Primers (Table 1) for qRT-PCR were designed based on available rat sequences in Genbank for caspase-3 (NM_012922), caspase-9 (NM_031632) and β-actin (NM_031144) using Primer premier 5.0 software. The three pairs of primers were tested for their specificity by conventional reverse transcription polymerase chain reaction (RT-PCR) before being used in qRT-PCR studies and by melting curve analysis following qRT-PCR (described below).
The RT-PCR assays were performed on the Mx3000P™ QRT-PCR system (Stratagene, USA) and using the One-Step SYBR® QRT-PCR kit (20 μl reaction mixture). Relative quantification of mRNA abundance for caspase-3 and caspase-9 was performed using the comparative ΔΔCT method expression,with the house-keeping gene β-actin used as a calibrator (Pfaffl 2001). Thermal cycling conditions were as follows: an initial reverse transcription step of 5 min at 42°C and 40 cycles at 95°C for 10 s, 58°C for 20 s, and 72°C for 6 s. The reaction was then subjected to a melting protocol from 55 to 95°C with a 0.2°C increment and 1 s holding at each increment to verify specificity of the amplified products.
Statistical analysis
Experimental data were expressed as mean ± SEM of three independent experiments, with three replicates of each treatment/experiment. Comparisons among treatments mean for cell proliferation and apoptosis data were performed using Kruskal–Wallis two-way ANOVA. Effects of treatments on caspase-3 and caspase-9 mRNAs were determined by analysis of variance (ANOVA) for the dose-response experiments and using 2-tailed Student’s t tests. Values of P < 0.05 were considered statistically significant.
Results
Establishment of OB cultures
After 24 h in culture, cells adhered to the flasks and showed classical morphological characteristics of OB including spindle, triangle or polyangular shape as revealed by H&E staining (Fig. 1a). Oval nuclei and obvious cytoplasm were observed following Giemsa stain (Fig. 1b). Cytochemical staining for ALP in OB revealed black deposits in cytoplasm (Fig. 1c). The OB overlapped each other after reaching confluence with a single layer, at the center of which calcification occurred, forming sporadic white spots visible with the naked eye. Round red mineralized nodules were visualized by Alizarin red staining, further confirming that deposition of calcium had occurred (Fig. 1d).

Morphological and histochemical characterization of osteoblasts (OB) cultured from neonatal rat calvaria. a Hemotoxylin and eosin staining of OB cultured in vitro. Note spindle,triangle or polyangular appearance (×200). b Giemsa staining of cultured OB. Note cells with enlarged nuclei and obvious cytoplasm and cell protrusions (×200). c Alkaline phosphatase staining of cultured OB. Note black deposits in cytoplasm (×200). d Alazarin red staining of cultured OB. Note mineralized nodule stained by alizarin red with red calcified extracellular matrix (×100)
Effects of NaF treatment on OB proliferation
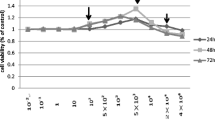
MTT assays were performed to determine if F treatment influences OB proliferation during 24–120 h of culture. As depicted in Fig. 2, proliferation of cells cultured in the absence of NaF (control) was evident during the 120 h of in vitro culture. Numbers of viable OB were slightly increased (relative to control) following 72 h exposure to 0.5 and 5 mg/l concentrations of NaF, but the increase was not statistically significant (P > 0.05). However, higher concentrations of NaF (20 and 30 mg/l) suppressed OB proliferation through 72 h of culture (P < 0.01). NaF at 10.0 mg/l did not influence OB proliferation after 72 h of exposure (P > 0.05). In contrast, cell proliferation was reduced following culture of OB for 96 and 120 h in the presence of all concentrations of NaF tested (P < 0.05; Fig. 2). Results indicate that extended culture in the presence of NaF dramatically inhibits OB cell proliferation.

Effects of sodium fluoride treatment on osteoblast proliferation (as determined by the MTT assay)
Effects of NaF treatment on OB cell apoptosis:
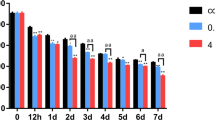
Incidence of apoptosis was determined by Annexin V-propidium iodide staining following 24 and 72 h culture in the presence of increasing concentrations of NaF. Annexin V-FITC binding is utilized to quantify phosphatidylserine residues redistributed from the inner to the outer leaflet of the cell membrane, which is an early event in apoptosis. Cell membrane integrity is lost with progression of apoptosis. Hence, binding of the membrane impermeable PI to DNA is used as an index of cells in later stages of apoptosis/dead cells (Wang et al. 2008). NaF treatment significantly increased the numbers of OB in early stages of apoptosis (Annexin V+/PI−) and late stages of apoptosis/dead cells (Annexin V+/PI+) following 24 h (Fig. 3a–e) and 72 h of treatment (Fig. 4a–e). Following 24 h of treatment, the percent of Annexin V positive cells and percent of PI positive cells were significantly increased in response to as little as 0.5 mg/l NaF. The percentage of cells in early stages of apoptosis were increased by 4.42 (P < 0.05), 7.23, 13.15 and 9.72-fold (P < 0.01) in response to culture in the presence of 0.5, 5, 10 and 20 mg/l NaF, respectively, and the percentage of late apoptotic/dead OB were increased by 12.4 (P < 0.05), 13.5, 18.2 and 15.05-fold (P < 0.01), respectively, in response to the same treatments (Fig. 3f). Moreover, following 72 h exposure of OB to 0.5, 5, 10 and 20 mg/l NaF, the percentage of cells in early stages of apoptosis were increased by 5.94, 8.28, 8.74 and 11.46-fold (P < 0.01), respectively, and the percentage of late apoptotic/dead cells were increased by 4.87, 5.02, 8.51 and 22.57-fold, respectively, for the same treatments (P < 0.01) relative to control cells cultured in the absence of NaF (Fig. 4f). Results indicate that NaF treatment can induce OB apoptosis in a dose-dependent fashion.

Effect of 24-h treatment with sodium fluoride (NaF) on apoptosis of cultured osteoblasts. a–e Two-dimension scatter plots depicting distribution of cells positively stained for Annexin V, propidium iodide (PI) and their combination following 24-h treatment with a 0 mg/l NaF, b 0.5 mg/l NaF, c 5 mg/l NaF, d 10 mg/l (NaF) or e 20 mg/l NaF. Cells in lower left quadrant of each picture correspond to normal cells (Annexin V −/PI −). Cells in right lower quadrant correspond to early apoptotic cells (Annexin V + /PI −). Cells in right upper quadrant correspond to late apoptotic/dead cells (Annexin V + /PI +). f Quantification of effects of treatment with various concentrations of NaF on percent apoptotic osteoblasts staining positive for Annexin V and for PI. Values depicted in (f) represent the mean ± SE (** P < 0.01 relative to untreated control; * P < 0.05 relative to untreated control)

Effect of 72-h treatment with sodium fluoride (NaF) on apoptosis of cultured osteoblasts. a–e Two-dimension scatter plots depicting distribution of cells positively stained for Annexin V, propidium iodide (PI) and their combination following 24-h treatment with a 0 mg/l NaF, b 0.5 mg/l NaF, c 5 mg/l NaF, d 10 mg/l (NaF) or e 20 mg/l NaF. Cells in lower left quadrant of each picture correspond to normal cells (Annexin V−/PI−). Cells in right lower quadrant correspond to early apoptotic cells (Annexin V+/PI−). Cells in right upper quadrant correspond to late apoptotic/dead cells (Annexin V+/PI+). f Quantification of effects of treatment with various concentrations of NaF on percent apoptotic osteoblasts staining positive for Annexin V and for PI. Values depicted in (f) represent the mean ± SE (** P < 0.01 relative to untreated control)
Effect of NaF treatment on OB cell caspase-3 and caspase-9 mRNA abundance
To further investigate the potential mechanisms involved in OB cell apoptosis induced by F, qRT-PCR analysis of caspase-3 and caspase-9 mRNA abundance following 24 and 72 h incubation in the presence of increasing concentrations of NaF (0, 0.5. 5 and 20 mg/l) was conducted. No significant effects of NaF on caspase-3 and caspase-9 mRNA were observed following 24 h of treatment (Fig. 5a, b). However, treatment with 5 mg/l NaF significantly increased caspase-3 and caspase-9 mRNA abundance (P < 0.05) by over twofold following 72 h of treatment (Fig. 5c, d).

Quantitative real time PCR analysis of the effect of 24 and 72 h sodium fluoride (NaF) treatment on osteoblast (OB) caspase-3 and caspase-9 mRNA expression in vitro. a Effect of 24-h treatment with increasing doses of NaF on OB caspase-3 mRNA abundance. b Effect of 24-h treatment with increasing doses of NaF on OB caspase-9 mRNA abundance. c Effect of 72-h treatment with increasing doses of NaF on OB caspase-3 mRNA abundance. d Effect of 72-h treatment with increasing doses of NaF on OB caspase-9 mRNA abundance. Relative mRNA abundance was calculated using ΔΔCT method. Abundance of caspase-3 and caspase-9 mRNAs was normalized relative to that of β-actin mRNA. Data are shown as mean ± SE. Doses of NaF without a common superscript for each timepoint/mRNA analyzed are different at P < 0.05
Discussion
Skeletal health is dictated by the balance between bone deposition and resorption. Chronic excessive F intake has been linked to skeletal disease, such as fluorosis (Krishnamachari 1986), but the effects of F directly on bone forming cells (osteoblasts) that promote skeletal disease are not well understood. In the present studies, the effects of NaF treatment on proliferation and apoptosis of neonatal rat OB and on caspase-3 and caspase-9 mRNA expression were investigated. Collectively, results demonstrate a pronounced negative effect of F on OB survival, and suggest that such effects could potentially contribute to the abnormal bone metabolism, characteristic of F-induced skeletal diseases such as fluorosis.
Potent inhibitory effects of F treatment on OB proliferation were observed in the present studies. Culture of OB in the presence of low concentrations of NaF (0.5 and 5 mg/l) for 72 h resulted in a subtle, but nonsignificant increase in OB proliferation. Culture in the presence of higher concentrations (20 and 30 mg/l) dramatically reduced OB proliferation. Proliferation was reduced at all other timepoints examined in response to all concentrations of NaF tested. Dose-dependent effects of NaF treatment on proliferation of caprine OB have been reported (Qu et al. 2008), where low doses of NaF stimulated proliferation and higher doses inhibited proliferation after 48 h of culture, and such divergent effects of NaF on caprine OB proliferation were observed through 96 h of culture. However, in the present studies, all doses of NaF tested inhibited OB proliferation following 96 h of culture. Treatment with NaF increased chicken embryonic OB proliferation in vitro and increased bone formation in embryonic calvaria cultures at concentrations that stimulated bone formation in vivo (Farley et al. 1983) and no inhibitory effects of NaF treatment on chick embryonic OB proliferation were observed. The duration of exposure of chicken embryonic OB to NaF was much shorter than that in described studies, suggesting that negative effects of NaF on OB proliferation are not acute, but rather require more prolonged NaF exposure. However, culture of human OB for up to 4 months in the presence of NaF did not influence OB proliferation (Chavassieux et al. 1993). Hence, reported effects of NaF treatment on OB proliferation are clearly variable.
The numbers of OB in bone (survival) are influenced by the equilibrium between rates of cell proliferation and cell death. Thus, effects of various NaF treatments (for 24 and 72 h) on OB apoptosis were also determined in the present studies. Results demonstrate that cultured neonatal rat OB are highly sensitive and respond to concentrations of NaF as low as 0.5 mg/ml (within 24 h) with a significant increase in apoptosis. Up to 10% of cells treated with the maximal dose of NaF (20 mg/l) were found to be in the early stages of apoptosis following 72 h of exposure in vitro. Acute effects of F exposure on bone-derived cell apoptosis have been reported previously. Apoptosis was detected within 8 h of F treatment of UMR 106 osteosarcoma cells (Hirano and Ando 1997) and NaF-induced apoptosis of caprine OB was recently reported (Qu et al. 2008), but effects of NaF on OB apoptosis were only examined following 48 h of treatment in the latter study. Even more pronounced effects of NaF on OB cell apoptosis were found following 72 h of exposure in the present studies. Furthermore, long-term exposure of female rats to NaF through the drinking water prior to mating resulted in a decrease in OB numbers in calvaria of offspring and increased incidence of apoptosis (Zhong et al. 2005). Collectively, results of above described studies support a potential role for increased OB apoptosis as a potential causative factor in the abnormal bone metabolism characteristic of F-induced skeletal disease.
One of the molecular hallmarks of apoptosis is the activation of caspases (Thornberry and Lazebnik 1998). The caspases are a subfamily of cysteine proteases that play an active regulatory role in the apoptotic cascade. Based on their order of activation, the caspases are classified into two families: the initiator and the effector caspases (Shi 2002). Caspase-9 is the key initiator caspase for the intrinsic pathway to cell death. Upon cleavage and activation from its proform, caspase-9 cleaves and activates caspase-3, the key effector caspase. Activation of caspase-3 leads to cytoskeletal breakdown and nuclear demise (Shi 2002). Hence, to gain initial insight into potential mechanisms involved in NaF-induced OB apoptosis, effects of NaF treatment for 24 and 72 h on OB cell mRNA for caspase-3 and caspase-9 were determined. Results demonstrate a stimulatory, dose dependent effect of NaF treatment on caspase-3 and caspase-9 mRNA abundance following 72 h of treatment. To our knowledge, effects of F treatment on caspase-3 and caspase-9 mRNA abundance have not been reported previously.
Elucidation of the mechanisms involved in F-induced OB apoptosis will require further investigation. Based on above results, a potential role for caspase-9 and the intrinsic, mitochondrial pathway (Dragovich et al. 1998) in NaF-induced apoptosis seems plausible. Oxidative stress induced by F treatment in vitro (Xu et al. 2008) and in vivo (Shivarajashankara et al. 2001) may trigger cytochrome c release and activation of the intrinsic pathway of apoptosis. However, further experiments would be required to confirm a functional role for caspase-9 in NaF-induced OB apoptosis, including measurement of caspase-9 activity and mitochondrial cytochrome c release (Dragovich et al. 1998). In addition, NaF-induced apoptosis of human gingival fibroblasts is mediated by both the mitochondrial (intrinsic) and death receptor (extrinsic) apoptotic pathways (Lee et al. 2008). Hence the involvement of the extrinsic pathway in NaF-induced OB apoptosis also merits further investigation.
In summary, results of described in vitro studies demonstrate a pronounced negative effect of NaF treatment on indices of OB survival, including decreased proliferation, increased apoptosis and increased caspase-3 and caspase-9 mRNA. Results support a potential role for reduced OB cell survival in the abnormal bone metabolism associated with skeletal disease induced by chronic, excessive F exposure. Further study will be required to elucidate the intracellular and extracellular mechanisms associated with NaF-induced OB apoptosis and potential therapeutic targets.
References
Aoba T, Fejerskov O (2002) Dental fluorosis: chemistry and biology. Crit Rev Oral Biol Med 13:155–170
Chavassieux P, Chenu C, Valentin-Opran A, Delmas PD, Boivin G, Chapuy MC, Meunier PJ (1993) In vitro exposure to sodium fluoride does not modify activity or proliferation of human osteoblastic cells in primary cultures. J Bone Miner Res 8:37–44
Dragovich T, Rudin CM, Thompson CB (1998) Signal transduction pathways that regulate cell survival and cell death. Oncogene 17:3207–3213
Farley JR, Werdegal JE, Baylink DJ (1983) Fluoride directly stimulates proliferation and alkaline phosphatase activity in bone-forming cells. Science 222:330–332
Ge YM, Ning HM, Wang SL, Wang JD (2005a) DNA damage in thyroid gland cells of rats exposed to long-term intake of high fluoride and low iodine. Fluoride 38:318–323
Ge YM, Ning HM, Wang SL, Wang JD (2005b) Comet assay of DNA damage in brain cells of adult rats exposed to high fluoride and low iodine. Fluoride 38:209–214
Ge YM, Ning HM, Wang SL, Wang JD (2005c) Effects of high fluoride and low iodine on brain histopathology in offspring rats. Fluoride 38:127–132
Goltzman D (2002) Discoveries, drugs and skeletal disorders. Nat Rev Drug Discov 10:784–796
Guan ZZ, Xiao KQ, Zeng XY, Long YG, Cheng YH, Jiang SF, Wang YN (2000) Changed cellular membrane lipid composition and lipid peroxidation of kidney in rats with chronic fluorosis. Arch Toxicol 74:602–608
Hirano S, Ando M (1997) Fluoride mediates apoptosis in osteosarcoma UMR 106 and its cytotoxicity depends on the PH. Arch Toxicol 72:52–58
Krishnamachari KA (1986) Skeletal fluorosis in humans: a review of recent progress in the understanding of the disease. Prog Food Nutr Sci 10:279–314
Lee JH, Jung JY, Jeong YJ, Park JH, Yang KH, Choi NK, Kim SH, Kim WJ (2008) Involvement of both mitochondrial- and death receptor-dependent apoptotic pathways regulated by Bcl-2 family in sodium fluoride-induced apoptosis of the human gingival fibroblasts. Toxicology 243:340–347
Liang CK, Sun SZ, Ji RD, Cao SR (1994) A dose-response relationship between population’s total intake of fluoride and fluorosis in burning coal fluorosis areas in China. In: Proceedings of the 20th Conference of the International Society for Fluoride Research (ISFR ‘94), Beijing, pp 84–85
Matsuo S, Nakaqawa H, Kiyomiya K, Kurebe M (2000) Fluoride-induced ultrastructural changes in exocrine pancreas cells of rats: fluoride disrupts the export of zymogens from the rough endoplasmic reticulum (rER). Arch Toxicol 73:611–617
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:2002–2007
Qu WJ, Zhong DB, Wu PF, Wang FJ, Han B (2008) Sodium fluoride modulates caprine osteoblast proliferation and differentiation. J Bone Mineral Metab 26:328–334
Shashi A (2003) In vivo studies concerning toxic effects of sodium fluoride on hepatic function in rabbits. Fluoride 36(1):30–37
Shi YG (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell 9:459–470
Shivarajashankara YM, Shivashankara AR, Hanumanth Rao S, Gopalakrishna Bhat P (2001) Oxidative stress in children with endemic skeletal fluorosis. Fluoride 34:103–107
Susheela AK, Bhatnagar M, Vig K, Mondal NK (2005) Excess fluoride ingestion and thyroid hormone derangements in children living in Delhi, India. Fluoride 38:98–108
Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281:1312–1316
Torricelli P, Fini M, Giavaresi G, Giavaresi G, Borsari V, Carpi A, Nicolini A (2003) Comparative interspecies investigation on osteoblast cultures: data on cell viability and synthetic activity. Biomed Pharmacother 57(1):57–62
Usuda K, Kono K, Dote T, Nishiura K, Miyata K, Nishiura H, Shimahara M, Suqimoto K (1998) Urinary biomarkers monitoring for experimental fluoride nephrotoxicity. Arch Toxicol 72:104–109
Wan SX, Zhang JH, Wang JD (2006a) Effects of high fluoride on sperm quality and testicular histology in male rats. Fluoride 39:17–21
Wan SX, Zhang JH, Wang JD (2006b) Fluoride-induced changes in the expression of epidermal growth factor and its receptor in testicular tissues of young male rats. Fluoride 39:121–125
Wang JD, Zhan CW, Chen YF, Li JX, Hong JP, Wang WF (1992) A study of damage to hard tissue of goats due to industrial fluoride pollution. Fluoride 25:123–128
Wang JD, Hong JH, Li JX, Cai JP (1994) The effect of nutrition supplementation during the annual dry grass season on tooth wear in industry-fluorosed goats. Fluoride 27:136–140
Wang JD, Hong JH, Li JP, Guo YH, Zhang JF, Hao JH (2002) Effect of high fluoride and low protein on tooth matrix development in goats. Fluoride 35:51–55
Wang JD, Guo YH, Liang ZX, Hao JH (2003) Amino acid composition and histopathology of goat teeth in an industrial fluoride polluted area. Fluoride 36:177–184
Wang JD, Ge YM, Ning HM, Wang SL (2004a) Effects of high fluoride and low iodine on biochemical indexes of the brain and learning-memory of offspring rats. Fluoride 37:201–208
Wang JD, Ge YM, Ning HM, Wang SL (2004b) Effects of high fluoride and low iodine on oxidative stress and antioxidant defense of the brain in offspring rats. Fluoride 37:264–270
Wang SP, Wang JA, Li J, Zhou JH, Wang H (2008) Voltage-dependent potassium channels are involved in staurosporine-induced apoptosis of rat mesenchymal stem cells. Cell Biol Int 32:312–319
Wu CX, Gu XL, Ge YM, Zhang JH, Wang JD (2006) Effects of high fluoride and arsenic on brain biochemical indexes and learning-memory in rats. Fluoride 39:274–279
Xu H, Wang CH, Zhao ZT, Zhang WB, Li GS (2008) Role of oxidative stress in osteoblasts exposed to sodium fluoride. Biol Trace Elem Res 123:109–115
Xue QS (2001) Principle and theory of in vitro culture (in Chinese). Science Press Publications, Beijing, ISBN:7-03-008125-0
Yeung CA (2008) A systematic review of the efficacy and safety of fluoridation. Evid Based Dent 9:39–43
Zhan XA, Wang M, Xu ZR, Li JX (2006a) Toxic effects of fluoride on kidney function and histological structure in young pigs. Fluoride 39:22–26
Zhan XA, Wang M, Xu ZR, Li WF, Li JX (2006b) Evaluation of caspase-dependent apoptosis during fluoride-induced liver lesion in pigs. Arch Toxicol 80:74–80
Zhang JH, Liang C, Ma JJ, Niu RY, Wang JD (2006a) Effects of sodium fluoride and sulfur dioxide on sperm motility and serum testosterone in male rats. Fluoride 39:126–131
Zhang JH, Liang C, Ma JJ, Zhou BH, Wang JD (2006b) Changes in testis protein and metabolic enzyme activities in rats induced by sodium fluoride and sulfur dioxide. Fluoride 39:179–184
Zhang JH, Liang C, Ma JJ, Zhou BH, Wang JD (2006c) Effects of sodium fluoride and sulfur dioxide on oxidative stress and antioxidant defenses in rat testes. Fluoride 39:185–190
Zhong DB, Sun LT, Wu PF, Su JL, Jin JS, Han B (2005) Regulatory aspects of fluoride on proliferation, differentiation and calcification of caprine osteoblasts cultured in vitro. Chin J Vet Sci Technol 35:895–899
Acknowledgments
This research was sponsored by the China National Natural Science Foundation (Grant No. 30471303). (Grant No. 30671545), the Shanxi Province Returnee’s Science Foundation (Grant No. 2004043), and the Shanxi Province Science and Technology Bureau Program (Grant No. 2006031065).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yan, X., Feng, C., Chen, Q. et al. Effects of sodium fluoride treatment in vitro on cell proliferation, apoptosis and caspase-3 and caspase-9 mRNA expression by neonatal rat osteoblasts. Arch Toxicol 83, 451–458 (2009). https://doi.org/10.1007/s00204-008-0365-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-008-0365-z