
Abstract
Reactive oxygen species (ROS) and antioxidant ingredients are a series of crucial signaling molecules in oxidative stress response. Under some pathological conditions such as traumatic brain injury, ischemia/reperfusion, and hypoxia in tumor, the relative excessive accumulation of ROS could break cellular homeostasis, resulting in oxidative stress and mitochondrial dysfunction. Meanwhile, autophagy is also induced. In this process, oxidative stress could promote the formation of autophagy. Autophagy, in turn, may contribute to reduce oxidative damages by engulfing and degradating oxidized substance. This short review summarizes these interactions between ROS and autophagy in related pathological conditions referred to as above with a focus on discussing internal regulatory mechanisms. The tight interactions between ROS and autophagy reflected in two aspects: the induction of autophagy by oxidative stress and the reduction of ROS by autophagy. The internal regulatory mechanisms of autophagy by ROS can be summarized as transcriptional and post-transcriptional regulation, which includes various molecular signal pathways such as ROS–FOXO3–LC3/BNIP3–autophagy, ROS–NRF2–P62–autophagy, ROS–HIF1–BNIP3/NIX–autophagy, and ROS–TIGAR–autophagy. Autophagy also may regulate ROS levels through several pathways such as chaperone-mediated autophagy pathway, mitophagy pathway, and P62 delivery pathway, which might provide a further theoretical basis for the pathogenesis of the related diseases and still need further research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Reactive oxygen species (ROS), which mainly encompass a range of oxygen-containing highly reactive species such as oxygen anions, free radicals, and hydrogen peroxide (H2O2), are generally shortlived, small, and highly reactive molecules (Rahal et al. 2014). There are two major sources for ROS production in cells: mitochondria, which generate ROS as a by-product of respiration, and the NADPH oxidase (NOX), which actively produces superoxide across the membranes of neutrophils and phagosomes (Scherz-Shouval and Elazar 2011). In addition, ROS such as superoxide and peroxynitrite are also linked to both the generation and propagation of the inflammatory response (Chakrabarti et al. 2014). Mitochondria are essential organelles for eukaryotic cells as an important site for the production of lipids, nucleic acids, and of amino acid precursors. ROS can oxidize and damage these products to induce mitochondrial dysfunction, which could be considered as a condition called oxidative stress (Scherz-Shouval and Elazar 2007). Cells have developed various enzymatic and non-enzymatic antioxidizing agents such as glutathione, superoxide dismutase (SOD), and catalase to prevent ROS and its adverse effects on mitochondria, however, when there is occurrence of environmental stressors such as nutrient starvation, traumatic injury, ischemia/hypoxia, and hypoxia in tumor, there exists an imbalance in the homeostasis reflecting in the inadequate antioxidant capability of cells and the excessive production of ROS. ROS accumulation results in oxidative damage, which leads to mitochondrial dysfunction and cell injury (Chakrabarti et al. 2014; Fandy et al. 2014). Meanwhile, under these conditions autophagy, which is characterized by the presence of autophagosome that engulfs cytosolic aged organelles and of autolysosome that degradates these organelles such as damaged mitochondria, is also induced (Zhang et al. 2009; Li et al. 2014). Recent studies have shown that ROS could initiate autophagosome formation and autophagic degradation acting as cellular signaling molecules (Chen et al. 2009). And autophagy, in contrast, serves to reduce oxidative damage and ROS levels through removal of protein aggregates and damaged organelles such as mitochondria (Ureshino et al. 2014).
The aim of this review is to summarize recent advances of these interactions between ROS and autophagy under different stress conditions, then further to interpret and discuss the potential molecular regulatory mechanisms in it under these conditions.
Interactions Between ROS and Autophagy in Related Pathological Conditions
As mentioned above, ROS can involve in the process of autophagy and, at the same time, are also regulated by autophagy. The consequences of these interactions between ROS and autophagy could manifest under various pathological conditions such as traumatic brain injury (TBI), ischemia/reperfusion (I/R), starvation, and tumor.
Traumatic Brain Injury (TBI)
Lai et al. (2008) established TBI mice model and observed that lipidated microtubule-associated protein light chain 3, a biochemical footprint of autophagy referred to as LC3 II, was increased at 2 and 24 h after TBI. However, after treatment with the antioxidant cysteine-donor GCEE, oxidative stress was significantly reduced and LC3 II formation was also partially inhibited, suggesting that reducing oxidative stress could reduce autophagosomal vacuole formation after acute brain injury. In addition, treatment with GCEE also improved Morris water maze performance and partially reduced histologic damage, which suggested that antioxidant intervention may lead to partial improvement in behavioral and histologic outcome.
Taken together, in the pathological condition of TBI, oxidative stress can be induced and initiate autophagy, which may contribute to the neuropathology after TBI. While the treatment with antioxidant can improve histologic damage, by inhibiting autophagosomal vacuole formation more strongly suggesting this tight relationship between ROS and autophagy.
Ischemia/Reperfusion (I/R)
The same as in TBI, the role of interactions between ROS and autophagy also reflects in I/R tissue. Hariharan et al. (2011) found that under starvation conditions autophagic flux was increased after the treatment of H2O2 in mice cardiac myocytes, which was attenuated in the presence of N-2-mercaptopropionyl glycine (MPG), an antioxidant. Hariharan et al. further established mice model of myocardial I/R and found that treatment with MPG also attenuated I/R-induced increases in oxidative stress and autophagic flux, accompanied by a decrease in the size of myocardial infarction (MI)/area at risk (AAR). This study suggested that in cardiac myocytes, H2O2 acting as one of ROS could induce autophagy under pathological conditions of starvation or I/R. ROS also played a facilitating role in myocardial injury after I/R through enhancing autophagy.
Furthermore, (Hamacher-Brady et al. 2006) found that the main role of autophagy in cardiac myocytes subjected to IR is the clearance of ROS-damaged mitochondria and proteins as part of the cell’s effort to minimize cell damage and promote survival. Chien CT’s study (2007) in the kidney after IR also supported this view.
In short, it can clearly be seen that in cardiac myocytes of I/R, ROS generate excessively and oxidative stress is induced, which may increase autophagic flux. Activated autophagy may conversely remove ROS-damaged mitochondria and proteins, which could contribute to cell survival.
Others
In tumor cells, hypoxia-induced autophagy could also reduce oxidative damage, which may promote cell survival. Hypoxia is a pathological condition of oxygen limitation or deprivation, which has been confirmed as a feature of most tumors (Wilson and Hay 2011). In response to hypoxia, HIF-1 and its target genes (BNIP3 and NIX) are activated to induce autophagy (Semenza 2010). Activation of autophagy could clear cellular components damaged by excessive production of ROS, thus supporting tumor cell survival and promoting cancer (Koritzinsky and Wouters 2013).
As in tumor, autophagy also could decrease cell injury in neurodegenerative diseases acting as an effective antioxidant pathway by clearing increased mitochondrial or cytosolic ROS (Giordano et al. 2013; Mortiboys et al. 2008; Rodríguez-Navarro et al. 2010).
In summary, the interactions between ROS and autophagy exist in a variety of pathological conditions. ROS may contribute to the cell injury through oxidative damage. At the same time, initiated autophagy by oxidative stress could clear mitochondria and proteins damaged by ROS. However, as mentioned above, the consequences of these interactions between ROS and autophagy could manifest in two different aspects: cell damage or cell survival. The current studies tend to support that the excessive production of ROS could increase cell damage, while the role of autophagy could exhibit duality. Increasing evidence supports the notion that autophagy is a double-edged sword (Chen et al. 2014; Evangelisti et al. 2015; Chen et al. 2013). The activation levels of autophagy were critical for the survival or death of cells: the physiological levels of autophagy are extremely important in maintaining cellular homeostasis through continual turnover of nonfunctional proteins and organelles, whereas insufficient or excessive levels of autophagy may promote cell death due to breaking cellular homeostasis. In addition, the induction time of autophagy also determines its role in cells. However, the specific molecular mechanisms of this double role remain unclear, which need to be further researched.
Molecular Regulatory Mechanisms of Interactions
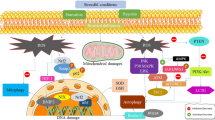
As previously described, ROS induce autophagy and that autophagy, in turn, contributes to reduce levels of ROS under various stress conditions. Studies have generally accepted that there are multiple molecular pathways involved in the regulation of these interactions between ROS and autophagy. The molecular regulatory mechanisms of autophagy by ROS in cells occur in the nucleus and cytoplasm, which can be correspondingly considered as transcriptional and post-transcriptional regulation (Fig. 1). Autophagy also regulated levels of ROS by other pathways such as chaperone-mediated autophagy (CMA) pathway, P62 delivery pathway, and mitophagy pathway.

Regulatory mechanisms of autophagy by ROS. The molecular regulatory mechanisms of autophagy by ROS mainly include transcriptional and post-transcriptional regulation. Under certain conditions such as starvation, traumatic brain injury (TBI), ischemia/reperfusion (I/R), and hypoxia in tumor, ROS could accumulate excessively and induce oxidative damages in cells. Increased ROS levels stimulate respectively the activity of P53, HIF-1, NRF2, FOXO3 in the nucleus, enhancing transcription of the corresponding genes: TIGAR/DRAM, BNIP3/NIX, p62, and LC3/BNIP3. In addition, ROS can also regulate PERK, whose downstream effectors could regulate transcription of autophagy-related genes. These genes’ protein products in the cytoplasm finally induce autophagy. In the cytoplasm, ROS also negatively regulates Atg4’s activity in the process of autophagosome to autolysosome, which can prevent Atg8-PE from deconjugating by the Atg4 protease
Regulatory Mechanisms of Autophagy by ROS
Transcriptional regulatory mechanisms of autophagy by ROS usually occur in the nucleus. Increased levels of ROS-enhanced oxidative stress response, excessive generation of ROS activates respectively HIF-1, p53, FOXO3, and NRF2. These transcription factors then induce respectively the transcription of BNIP3 and NIX, TIGAR, LC3 and BNIP3 and p62. The corresponding protein products finally induce autophagy in the cytoplasm, which can be considered as post-transcriptional regulation. In addition, the ER stress sensor PERK, whose downstream effectors then induce the expression of the autophagy genes, also increase autophagic flux (Scherz-Shouval and Elazar 2011).
ROS–TIGAR–Autophagy
p53 is a tumor suppressor protein that has a critical function in inhibiting cancer development, both TIGAR (TP53-induced glycolysis and apoptosis regulator) and DRAM (DNA damage-regulated autophagy modulator) can be transcriptionally activated by p53. TIGAR modulates the glycolytic pathway and interacts with hexokinase 2, resulting in the regulation of mitochondrial membrane potential, thus increasing NADPH production and decreasing intracellular ROS levels (Cheung et al. 2012). In response to nutrient starvation or metabolic stress, TIGAR could inhibit autophagy and it might modulate autophagy as part of its constitutive activity in the cellular antioxidant defense system with no clear effects on the mTOR pathway, and is p53 independent (Bensaad et al. 2009). DRAM also positively regulates autophagy (Crighton et al. 2006), however, a connection between ROS and DRAM has not been made.
Bensaad et al. (2009) found through Western blotting and fluorescence microscopy that nutrient starvation or metabolic stress strongly increases ROS and LC-3 II (specific autophagic marker) levels in U2OS cells, however, over-expression of TIGAR effectively inhibited these enhancements. Conversely, knockdown of TIGAR expression resulted in an increase in ROS levels, and this increase was further elevated after inhibition of the endogenous TIGAR protein by siRNA knockdown. These studies showed that TIGAR and its protein products could reduce ROS levels and inhibit autophagy in cells under nutrient starvation or metabolic stress. Bensaad et al. also found that the enhanced autophagy response to TIGAR inhibition was still retained in cells depleted of p53, indicating that TIGAR may modulate autophagy independent of p53. Ye et al. (2013) also demonstrated that silencing TIGAR by RNAi (RNA interference) in HepG2 cells down-regulated TIGAR mRNA, which led to the induction of LC-3 II and the intracellular ROS levels.
These studies all highlighted that TIGAR gene and its protein could decrease levels of ROS and inhibit autophagy under nutrient starvation or metabolic stress. However, this regulatory function by TIGAR may be p53 independent even though it is a p53 target gene.
ROS–HIF1–BNIP3/NIX–Autophagy
Hypoxia-inducible factor (HIF) is a major factor in the cell survival response to hypoxia, which could induce the transcription of BNIP3 and NIX genes (Mahalingaiah and Singh 2014). Their protein products compete with beclin-1 for the binding of BCL2, thereby releasing beclin-1 and allowing it to induce autophagy. By triggering mitochondrial selective autophagy, HIF-1 could reduce ROS production (Semenza 2011).
Zhang et al. (2008) found that levels of BNIP3 and LC3-II were markedly increased in Wild type (WT) mouse embryo fibroblasts (MEFs) subjected to 1 % O2 as compared with HIF-1α knockout (KO) MEFs. The exposure of HIF-1α-KO MEFs to 1 % O2 for 48 h resulted in a marked increase in ROS levels, on the contrary, ROS levels decreased in response to hypoxia in WT MEFs. The same results were found in BNIP3-KO MEFs in contrast to WT MEFs. This study showed that in MEFs exposed in prolonged hypoxia, autophagy could be induced through HIF-1-dependent expression of BNIP3, which is necessary to prevent increased levels of ROS.
Bellot et al. (2009) also found that siRNA-mediated ablation of either BNIP3 or BNIP3L had little effect on autophagy, however, the combined silencing of these two HIF target genes could suppress hypoxia-mediated autophagy. The ectopic expression of both BNIP3 and BNIP3L in normoxia activates autophagy. They further proposed that the atypical BH3 domains of hypoxia-induced BNIP3/BNIP3L have been designed to induce autophagy by disrupting the Bcl-2–Beclin1 complex without inducing cell death.
These studies demonstrated that HIF-1 could induce the transcription of BNIP3, BNIP3L, or NIX in response to hypoxia; the protein products could induce autophagy by releasing beclin-1 from the Bcl-2–Beclin1 complex. HIF-1 could also decrease levels of ROS to cell survival by autophagy, which may selectively clear mitochondria in cells.
ROS–NRF2–P62–Autophagy
Oxidative stress could also activate NRF2, which belongs to the basic leucine zipper (bZIP) family of transcription factors. In response to an oxidative stress such as H2O2, NRF2 specifically binds to the antioxidant-responsive element (ARE motif) located in the p62 promoter to promote the expression of p62 mRNA. P62, in turn, positively regulates the transcription of NRF2 (Puissant et al. 2012). p62 is also a substrate for lysosomal proteases. Hence, stimuli such as hypoxia and amino acid deprivation have been shown to induce autophagy, as well as p62 degradation and, subsequently, decrease p62 intracellular levels (Larsen et al. 2010).
Rubio et al. (2014) have investigated that the formation of p62-associated Ub aggregates in normal and cancer cells was stimulated after hypericin-mediated photodynamic therapy (Hyp-PDT), a procedure known to incite levels of ROS. Autophagy ultimately removed this formation through a mechanism partially regulated by p38MAPK. Genetic or pharmacological p38MAPK inhibition impaired NRF2 activation and reduced p62 levels, thus increasing oxidative stress. This study indicated that abundant ROS generation could activate NRF2 and induce transcription of p62; autophagy could inhibit p62-associated Ub aggregates through p38MAPK passway, which mainly negatively regulated NRF2 activation and reduced p62 levels.
Riley et al. (2010) also showed that the increase in Ub conjugates in Atg7(−/−) liver and brain was completely suppressed by simultaneous knockout of either p62 or NRF2, proposing that the accumulation of poly-Ub chains in autophagy-deficient circumstances is an indirect consequence of activation of NRF2.
These studies described that when ROS generate excessively, NRF2 could be activated and then regulate the transcription and expression of p62 gene, and thus actively participate in protein degradation in the process of autophagy.
ROS–FOXO3–LC3/BNIP3–Autophagy
Oxidative stress not only activates NRF2 but the transcription factor forkhead box O3 (FOXO3). NRF2 induces transcription of p62, whereas FOXO3 stimulates the transcription of LC3 and BNIP3, which involve in autophagy-lysosome systems (Mahalingaiah and Singh 2014). Currently more researches on this mechanism tend to focus on some diseases in skeletal muscle, such as muscle atrophy (Sandri 2013). Aucello et al. (2009) found that increased ROS in muscle cells may trigger FOXO3 signaling pathways. In response to accumulation of oxidative stress, FOXO3 may activate both the ubiquitin-proteasome pathway and the transcription of autophagy-related genes such as those encoding LC3 and BNIP3, inducing the formation of autophagy.
ROS–Atg4–Autophagy
The current studies have also been focused on the redox regulation of Atg4 almost occurred in the cytoplasm. In the cytoplasm, the elongation of the autophagosomal membrane is mediated by two ubiquitin-like conjugation systems: the conjugation of Atg12 to Atg5 and, downstream of it, the Atg8 conjugation to phosphoethanolamine (PE). Atg4 is responsible for the priming of Atg8 by its cleaving C terminus, which exposes a glycine residue. Then Atg8-PE undergoes deconjugation by the Atg4 protease, a step regulated by ROS that allows recycling of this protein. ROS could inhibit ATG4 cysteine protease activity, thereby supporting autophagosome formation (Gurusamy and Das 2009).
Scherz-Shouval et al. (2007) found that Atg4 in CHO cells is attenuated in response to starvation in a redox-dependent manner and Cys81 is a target for the redox regulation of HsAtg4A through cell culture, transfection, and immunofluorescence techniques. This oxidative signal leads to inactivation of Atg4 at the site of autophagosome formation, thereby promoting lipidation of Atg8, an essential step in the process of autophagy.
Li et al. (2012) found that the growth inhibitory effect of N-Benzoyl-O-d-phenylalanyl-d-phenylalaninol (BBP) on human breast carcinoma MCF-7 cells was associated with the induction of autophagy, which was demonstrated by the development of acidic vesicular organelles, cleavage of LC3, and upregulation of Atg4 in BBP-treated MCF-7 cells. LC3 is an Atg8 homolog that is essential for autophagosome formation. Since the application of Atg4 siRNA totally blocked the cleavage of LC3, they demonstrated a central role of Atg4 in BBP-induced autophagy. BBP also increased the levels of ROS. These results suggest that BBP produces its growth inhibitory effect through induction of the autophagy in MCF-7 cells, which is modulated by Atg4 upregulation involving ROS production. In short, ROS could promote Atg8 combining to PE by adjusting Atg4 activity in the cytoplasm and finally complete the regulation mechanisms of autophagy.
Apart from the above five pathways, researchers have found that hypoxia also induces the ER stress sensor PERK, whose downstream effectors could induce the expression of the autophagy genes LC3 and ATG5 (Mahalingaiah and Singh 2014).
The molecular regulatory signaling passways that participate in the regulation of autophagy by ROS mainly include transcriptional and post-transcriptional progresses. In the nucleus, the HIF-1 transcription factor, p53, FOXO3, and NRF2 are sequentially activated in response to generation of ROS, and they respectively stimulate the transcription of BNIP3 and NIX, TIGAR, LC3 and BNIP3 and p62. The ER stress sensor PERK is also induced, whose downstream effectors induce the expression of the autophagy genes. Then the protein products in the cytoplasm finally induce autophagy. ROS may also affect the formation of autophagic membrane by adjusting Atg4 activity.
Regulation Mechanisms of ROS by Autophagy
It is now widely accepted that autophagy is crucial for the removal of damaged mitochondria. Removal of oxidized proteins by the ubiquitin/proteasome system has been considered as the main responsible mechanism.
Chaperone-Mediated Autophagy (CMA) Pathway
Selective targeting of proteins to lysosomes for their degradation is possible via CMA. The CMA pathway, the main major intracellular proteolytic system, was suggested to selectively degradate proteins (Kaushik and Cuervo 2006).
CMA is the type of autophagy wherein a particular pool of soluble cytosolic proteins is selectively targeted to lysosomes for degradation. The substrate proteins are recognized by a chaperone–co-chaperone complex, then this complex delivers them to the lysosomal membrane. On the lysosomal membrane these substrate proteins bind to a receptor protein, the lysosomal-associated membrane protein type 2A (LAMP-2A). Along with unfolding, these substrate proteins are translocated across the lysosomal membrane through a lysosomal-resident chaperone, then they are degraded in the lysosome (Massey et al. 2004). The process of unfolding that typically associates with oxidative damage may expose hidden CMA-targeting motifs, which may facilitate their recognition by the cytosolic chaperone complex (Kiffin and Christian 2004).
Mitophagy Pathway
Selective forms of autophagy are now well appreciated that exist for degradation of specific organelles such as mitophagy. ROS generated by damaged mitochondria might induce mitophagy, which in turn eliminates the damaged organelles to decrease the levels of ROS. It was originally proposed that loss of mitochondrial membrane potential (DC) serves as a cue for mitophagy (Kim et al. 2007).
The ROS scavenger catalase also undergoes selective protein degradation through autophagy. Although the mechanism in this selectivity is still unclear, it occurs in response to caspase inhibition (Yu et al. 2006), as well as TrkA activation (Oh et al. 2008).
P62 Delivery Pathway
Recent studies have also showed that in response to oxidative stress, NRF2 transcription factor could induce p62 expression, which has been implicated in the delivery of oxidized proteins to autophagosomes for degradation so that it can decrease the oxidative injury (Jain et al. 2010; Komatsu et al. 2010).
In short, intracellular oxidative stress can increase ROS production significantly, while the number of ROS may play an important role in regulating the formation of autophagy via various signaling pathways including ROS–FOXO3–LC3/BNIP3–autophagy, ROS–NRF2–P62–autophagy, ROS–HIF1–BNIP3/NIX–autophagy, ROS–TIGAR–autophagy, and the ER stress sensor PERK, as well as the regulation mainly by inhibiting Atg4 activity. Meanwhile, autophagy also could reduce levels of ROS in cells under certain conditions by other pathways such as CMA pathway, P62 delivery pathway, and mitophagy pathway.
Concluding Remarks
In this review, we discuss and summarize the interactions between ROS and autophagy in related pathological conditions such as TBI, I/R, starvation, and tumor. ROS may aggravate cell injury through oxidative stress, however, autophagy could conversely clear mitochondria and proteins damaged by ROS to decrease cell injury and promote cell survival. These interactions need various molecular regulatory signaling passways to participate in. The process mainly includes transcriptional and post-transcriptional regulation of autophagy by ROS. In the nucleus, the HIF-1 transcription factor, p53, FOXO3, and NRF2 are sequentially activated in response to the generation of ROS, and they respectively stimulate the transcription of BNIP3 and NIX, TIGAR, LC3 and BNIP3 and p62. The ER stress sensor PERK is also induced, whose downstream effectors induce the expression of the autophagy genes. Then the protein products finally induce autophagy. In the cytoplasm, ROS may also affect the formation of autophagic membrane by adjusting Atg4 activity. Autophagy, in turn, also could reduce ROS levels by other pathways such as CMA, mitophagy, and P62 delivery pathway. In short, the interactions between ROS and autophagy could manifest in different conditions and diseases. And the internal molecular regulatory mechanisms are also various and still need further research.
References
Aucello M, Dobrowolny G, Musarò A (2009) Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy 5(4):527–529
Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29(10):2570–2581
Bensaad K, Cheung EC, Vousden KH (2009) Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 28(19):3015–3026
Chakrabarti S, Jahandideh F, Wu J (2014) Food-derived bioactive peptides on inflammation and oxidative stress. Biomed Res Int 2014:608979
Chen Y, Azad MB, Gibson SB (2009) Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 16(7):1040–1052
Chen G, Zhang W, Li YP, Ren JG, Xu N, Liu H, Wang FQ, Sun ZJ, Jia J, Zhao YF (2013) Hypoxia-induced autophagy in endothelial cells: a double-edged sword in the progression of infantile haemangioma? Cardiovasc Res 98(3):437–448
Chen W, Sun Y, Liu K, Sun X (2014) Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res 9(12):1210–1216
Cheung EC, Ludwig RL, Vousden KH (2012) Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc Natl Acad Sci U S A 109(50):20491–20496
Chien CT, Shyue SK, Lai MK (2007) Bcl-xL augmentation potentially reduces ischemia/reperfusion induced proximal and distal tubular apoptosis and autophagy. Transplantation 84(9):1183–1190
Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM (2006) DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126(1):121–134
Evangelisti C, Evangelisti C, Chiarini F, Lonetti A, Buontempo F, Neri LM, McCubrey JA, Martelli AM (2015) Autophagy in acute leukemias: a double-edged sword with important therapeutic implications. Biochim Biophys Acta 1853:14–26
Fandy TE, Jiemjit A, Thakar M, Rhoden P, Suarez L, Gore SD (2014) Decitabine induces delayed reactive oxygen species (ROS) accumulation in leukemia cells and induces the expression of ROS generating enzymes. Clin Cancer Res 20(5):1249–1258
Giordano S, Darley-Usmar V, Zhang J (2013) Autophagy as an essential cellular antioxidant pathway in neurodegenerative disease. Redox Biol 2:82–90
Gurusamy N, Das DK (2009) Autophagy, redox signaling, and ventricular remodeling. Antioxid Redox Signal 11(8):1975–1988
Hamacher-Brady A, Brady NR, Gottlieb RA (2006) Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 281(40):29776–29787
Hariharan N, Zhai P, Sadoshima J (2011) Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal 14(11):2179–2190
Jain A, Lamark T, Sjøttem E, Larsen KB, Awuh JA, Øvervatn A, McMahon M, Hayes JD, Johansen T (2010) p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem 285(29):22576–22591
Kaushik S, Cuervo AM (2006) Autophagy as a cell-repair mechanism: activation of chaperone-mediated autophagy during oxidative stress. Mol Aspects Med 27(5–6):444–454
Kiffin R, Christian C (2004) Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 15(11):4829–4840
Kim I et al (2007) Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462:245–253
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12(3):213–223
Koritzinsky M, Wouters BG (2013) The roles of reactive oxygen species and autophagy in mediating the tolerance of tumor cells to cycling hypoxia. Semin Radiat Oncol 23(4):252–261
Lai Y, Hickey RW, Chen Y, Bayir H, Sullivan ML, Chu CT, Kochanek PM, Dixon CE, Jenkins LW, Graham SH, Watkins SC, Clark RS (2008) Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J Cereb Blood Flow Metab 28(3):540–550
Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G (2010) A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 6(6):784–793
Li Y, Luo Q, Yuan L, Miao C, Mu X, Xiao W, Li J, Sun T, Ma E (2012) JNK-dependent Atg4 upregulation mediates asperphenamate derivative BBP-induced autophagy in MCF-7 cells. Toxicol Appl Pharmacol 263(1):21–31
Li Lulu, Zhang Qiang, Tan Jin, Yunyun Fang Xu, An Baoyuan Chen (2014) Autophagy and hippocampal neuronal injury. Sleep Breath 18(2):243–249
Mahalingaiah PK, Singh KP (2014) Chronic oxidative stress increases growth and tumorigenic potential of mcf-7 breast cancer cells. PLoS ONE 9(1):e87371
Massey A, Kiffin R, Cuervo AM (2004) Pathophysiology of chaperone-mediated autophagy. Int J Biochem Cell Biol 36(12):2420–2434
Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, Wood NW, Willems PH, Smeitink JA, Cookson MR, Bandmann O (2008) Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol 64(5):555–565
Oh SH, Kim YS, Lim SC, Hou YF, Chang IY, You HJ (2008) Dihydrocapsaicin (DHC), a saturated structural analog of capsaicin, induces autophagy in human cancer cells in a catalase-regulated manner. Autophagy 4(8):1009–1019
Puissant A, Fenouille N, Auberger P (2012) When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res 2(4):397–413
Rahal A, Kumar A, Singh V, Yadav B, Tiwari R, Chakraborty S, Dhama K (2014) Oxidative stress, prooxidants, and antioxidants: the interplay. Biomed Res Int 2014:761264
Riley BE, Kaiser SE, Shaler TA, Ng AC, Hara T, Hipp MS, Lage K, Xavier RJ, Ryu KY, Taguchi K, Yamamoto M, Tanaka K, Mizushima N, Komatsu M, Kopito RR (2010) Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol 191(3):537–552
Rodríguez-Navarro JA, Rodríguez L, Casarejos MJ, Solano RM, Gómez A, Perucho J, Cuervo AM, García de Yébenes J, Mena MA (2010) Trehalose ameliorates dopaminergic and tau pathology in parkin deleted/tau overexpressing mice through autophagy activation. Neurobiol Dis 39(3):423–438
Rubio N, Verrax J, Dewaele M, Verfaillie T, Johansen T, Piette J, Agostinis P (2014) p38(MAPK)-regulated induction of p62 and NBR1 after photodynamic therapy promotes autophagic clearance of ubiquitin aggregates and reduces reactive oxygen species levels by supporting Nrf2-antioxidant signaling. Free Radic Biol Med 67:292–303
Sandri M (2013) Protein breakdown in muscle wasting: role of autophagy-lysosome and ubiquitin-proteasome. Int J Biochem Cell Biol 45(10):2121–2129
Scherz-Shouval R, Elazar Z (2007) ROS, mitochondria and the regulation of autophagy. Trends Cell Biol 17(9):422–427
Scherz-Shouval R, Elazar Z (2011) Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 36(1):30–38
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z (2007) Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 26(7):1749–1760
Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20(1):51–56
Semenza GL (2011) Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 1813(7):1263–1268
Ureshino RP, Rocha KK, Lopes GS, Trindade CB, Smaili SS (2014) Calcium signaling alterations, oxidative stress and autophagy in aging. Antioxid Redox Signal 21(1):123–137
Wilson WR, Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11(6):393–410
Ye L, Zhao X, Lu J, Qian G, Zheng JC, Ge S (2013) Knockdown of TIGAR by RNA interference induces apoptosis and autophagy in HepG2 hepatocellular carcinoma cells. Biochem Biophys Res Commun 437(2):300–306
Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M (2006) Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A 103(13):4952–4957
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283(16):10892–10903
Zhang H, Kong X, Kang J, Su J, Li Y, Zhong J, Sun L (2009) Oxidative stress induces parallel autophagy and mitochondria dysfunction in human glioma U251 cells. Toxicol Sci 110(2):376–388
Acknowledgement
This work was supported by National Natural Science Foundation of China (Grant No. 81370183), Tianjin Natural Science Foundation (Grant No. 14JCYBJC27800), and National Clinical Key Subject Construction Project of NHFPC Fund.
Conflict of interest
We confirm that all the listed authors do not have any possible conflicts of interest.
Presentation at a Conference Statement
The manuscript is an original work and has not been previously submitted or is under consideration for publication in another journal. The study complies with current ethical consideration.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, L., Tan, J., Miao, Y. et al. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell Mol Neurobiol 35, 615–621 (2015). https://doi.org/10.1007/s10571-015-0166-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-015-0166-x