
Abstract
Bone remodeling is a vital physiological process of healthy bone tissue in humans. Imbalances in this vital process lead to pathological conditions, including periodontal diseases. In this study, we characterized the effects of micromolar levels of NaF on the proliferation and osteogenic differentiation of MC3T3-E1 osteoblastic cells. NaF significantly enhanced the proliferation, alkaline phosphatase (ALP) activity, and mineralization of MC3T3-E1 cells. Quantitative real-time PCR analysis revealed that the expression of mRNAs encoding runt-related transcription factor 2 (Runx2), Osterix, Osteopontin and Osteocalcin was up-regulated in NaF-treated MC3T3-E1 cells compared with untreated controls. Western blot analysis demonstrated that Runx2 and Osterix were inhibited by Runx2 siRNA but were re-activated by treatment with NaF. Furthermore, in vivo evidence indicated that NaF protects against Porphyromonas gingivalis-induced periodontal inflammation and alveolar bone loss in a P. gingivalis-challenged experimental periodontitis animal model. These data suggest that NaF promotes the osteoblastic differentiation of MC3T3-E1 cells through the Runx2/Osterix pathway and may be effective for the treatment of bone-related disorders.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Massive bone defects and severe periodontitis cannot heal spontaneously and require some form of biological augmentation for reconstruction. Delays in treating aggressive periodontitis can result in the loss of soft connective tissues and bone that surround the teeth and is a major cause of tooth loss in adults [1]. The identification of factors that regulate osteogenic differentiation is an area of intensive investigation with the potential to identify novel targets that could enhance bone formation and repair bone diseases. Novel factors exhibiting two critical properties would prove to be assets towards a therapeutic anti-inflammatory benefit, yet preserving the bone. First, they should enhance the expression of osteoblast differentiation markers. A significant induction of alkaline phosphatase (ALP) activity leads to the osteogenic differentiation of osteoblasts. Second, they should have an anti-inflammatory potential. In the inflammation process, inflammatory and immune cells circulate in the blood and sequentially infiltrate infected sites via extravasation. Polymorphonuclear leukocytes (PMNs) are the first responders that migrate to an infected area.
Fluoride (e.g., NaF) is considered a multifunctional growth factor that has diversified actions and effects on the differentiation of osteoblasts. NaF is unique among inorganic ions in that it stimulates bone formation in vivo [2] and in vitro [3, 4]. In osteoblasts, NaF causes proliferation and differentiation, inhibits phosphotyrosil acid phosphatase, and modulates the activity of growth factors. Fluorine is an essential trace element for all mammalian species, but an excess of fluorine is toxic both to animals and to cells in culture [5]. We previously reported the effects of micromolar levels of NaF, an active compound that selectively inhibits RANKL-induced osteoclastogenesis by reducing the induction of NFATc1 [6]. As such, NaF is able to efficiently interfere with the suppressed expression of cathepsin K and matrix metalloprotease 9 (MMP9). Those effects were extended with in vivo studies showing that NaF markedly inhibited Porphyromonas gingivalis-induced osteoclastogenesis in an experimental rat periodontitis model [6]. NaF has also demonstrated a therapeutic benefit in other rat models of chronic inflammation, including experimental diabetes [7]. While being a potent inducer in the healing of destructive periodontal tissue, we demonstrate in this study that NaF increases osteoblast differentiation via its effects on the transcription factor Runx2.
Osteoblast differentiation is a key process in bone formation that is controlled by a complex signaling network with interactions between many proteins and signaling molecules, but the mechanisms underlying those interactions are poorly understood [8]. Active mature osteoblasts on the bone surface can be distinguished by their morphological properties as well as by their temporal expression of several non-collagenous proteins such as Osteocalcin and Osteopontin, which act as markers of mature osteoblasts [9]. Osteocalcin increases bone formation and enhances the appearance of active osteoblasts and bone healing around hydroxyapatite/collagen composites. In bone, Osteopontin is secreted by osteoblastic cells and is deposited into the extracellular matrix. In osteogenesis, Osteopontin messenger RNA (mRNA) is expressed during early stages of matrix formation [10]. The transcription factor Runx2 is the master regulator of this process with many partners that act downstream and/or upstream. Runx2 plays a pivotal role in the regulation of osteogenic differentiation. Osterix, a zinc finger-containing transcription factor, is required for osteoblastic differentiation and acts as a downstream factor of Runx2 [11–13]. Given that work on the physical interaction between NaF and Runx2 has been limited [14] and that only protein kinase (PKR)-like ER kinase (PERK) has been investigated to date as a Runx2-target gene affected by NaF [15], we revisited this issue and characterized the interaction between NaF and Runx2 during the induction of osteoblast differentiation in cultures of MC3T3-E1 osteoblastic cells.
The objectives of this study were (1) to determine the effect of micromolar levels of NaF on the differentiation of osteoblastic cells and (2) to investigate whether treatment with NaF suppressed inflammation and promoted bone healing in a P. gingivalis-challenged experimental rat periodontitis model. Our findings suggest that micromolar levels of NaF function as an osteogenesis-enhancing molecule, activating osteoblastic differentiation in addition to its functions on proliferation. Moreover, if delivered into massive bony defects, NaF might enhance bone regeneration and promote the healing of those defects.
Materials and Methods
Cell Culture
MC3T3-E1 Subclone 14 osteoblastic cells (CRL-2594) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). They were cultured in sterile cell culture plates containing alpha minimal essential medium (α-MEM) with 10% heat-inactivated fetal bovine serum (FBS) (Biowest, Riverside, MO, USA), 2 mM l-glutamine, and 10 U/ml penicillin/streptomycin (Gibco, Grand Island, NY, USA), in a humidified 95% air/5% CO2 incubator at 37 °C. When the cells reached sub-confluence, they were harvested and sub-cultured.
Cell Proliferation Assay
The cell proliferation assay was performed as previously described [16]. In brief, MC3T3-E1 cells were harvested from 80% confluent monolayer cultures by a brief trypsinization with 0.1% trypsin and 0.1% EDTA. The cells were then seeded at a density of 1 × 104 cells per well in 96-well tissue culture plates and were cultured for 24 h in the regular medium with 10% FBS. The medium was replaced with serum-free medium for 24 h, and cells were then treated with medium supplemented with NaF at final concentrations of 50 or 500 μM. The concentrations have been adapted from our previous studies [6, 7, 16, 17]. Cell proliferation was assessed using the MTS assay at 24 and 48 h after treatment. CellTiter 96® AQueous One Solution Reagent (Promega, Madison, WI, USA) was added directly to each well, incubated for 2 h, and the absorbance at 490 nm was measured using a microplate reader.
ALP Assay
5 × 104 MC3T3-E1 cells per milliliter in α-MEM was seeded in 6-well plates overnight. The medium in each well was removed the next day and replaced with osteoblast differentiation-inducing medium (OIM) (MK430, Takara Bio Inc., Tokyo, Japan) and then incubated at 37 °C for 14 days in a 5% CO2 incubator. Day 0 is the date the OIM medium was added. ALP activity was determined using an ALP assay kit (ab83369, Abcam, Cambridge, MA, USA) following the manufacturer’s instructions. In brief, untreated control and NaF-treated MC3T3-E1 cells were homogenized in assay buffer after washing with cold PBS. For each sample, the supernatants were added in triplicate in 96-well plates and each well was brought to a total volume of 80 μl with assay buffer, followed by the addition of 20 μl stop solution for a background control. Standard curves were generated as detailed in the manufacturer’s manual. The ALP activity of each sample was calculated based on a comparison between the standard curve and the sample curve and is reported as units per milliliter (U/ml).
Alizarin Red S Staining and Quantification
After 14 days of culture in OIM, the cells in 6-well plates were washed twice with Hanks balanced salt solution (HBSS) (Gibco 14025, Thermo Fisher Scientific, Waltham, MA, USA), fixed with 4% paraformaldehyde for 15 min, washed twice with deionized water, and then incubated with 1% Alizarin Red S for 3 min at 37 °C. The samples were then washed three times with deionized water to stop the reaction. The stained samples were observed using a microscope and were photographed. Calcium deposition was quantified by destaining with 10% cetylpyridinium chloride (Sigma-Aldrich, Saint Louis, MO, USA) for 15 min, and the absorbance at 562 nm was measured using a microplate reader.
Enzyme-Linked Immunosorbent Assay
After osteogenic induction for 14 days, Osteocalcin in the supernatants was directly measured using an ELISA kit (USCN Life Science, Wu Han, China) according to the manufacturer’s instructions and a microplate reader at 450 nm. The assays were performed in triplicate.
Quantitative RT-PCR Analysis
At day 14 after incubation with OIM, total RNAs were isolated using an RNeasy Mini Kit (Qiagen, Hilden, Germany). First-strand complementary DNAs (cDNAs) were synthesized from 1 μg of each total RNA using SuperScript VILO Master Mix (Life Technologies, Carlsbad, CA, USA). Two microliters of each cDNA was subjected to real-time RT-PCR using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA) for Runx2 (Mm00501584_m1), Osterix (Mm04209856_m1), Osteopontin (Mm00436767_m1), and Osteocalcin (Mm03413826_m1) and Pre-Developed TaqMan Assay Reagents (Applied Biosystems) for β-actin (Mm02619580_g1) as an internal control. Two-step PCR cycling was carried out as follows: 50 °C 2 min for 1 cycle, 95 °C 10 min for 1 cycle, and 95 °C 15 s and 60 °C 1 min for 40 cycles. Two independent measurements were averaged, and relative mRNA expression levels were calculated as a ratio to β-actin expression of each sample.
Western Blotting
Cells cultured for 14 days were lysed in RIPA lysis buffer (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Protein concentrations were determined using a BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA). SDS-PAGE was calibrated with molecular weight markers (Bio-Rad, Hercules, CA, USA) for 15 min, and the samples were rinsed with Tris-buffered saline with 0.1% Tween 20 (TBS-T). Antibodies to Runx2 (1:500; Abcam), Osterix (1:500; Abcam), and β-actin (1:100; Cell Signaling Technology, Inc., Danvers, MA, USA) were used as primary antibodies. Anti-mouse and anti-rabbit secondary antibodies (Cell Signaling Technology) were each used at a dilution of 1:2000. Bound antibodies were visualized by chemiluminescence using an ECL Plus Western Blotting Detection System (Amersham, Uppsala, Sweden), and images were analyzed using a Luminescent Image Analyzer (LAS-3000; Fuji Film Inc., Kashiwa, Japan).
Short Interference RNA
The duplexes of each short interference RNA (siRNA) targeting Runx2 and negative controls (non-silencing siRNA or scramble siRNA) were synthesized by Qiagen. For siRNA transfection, MC3T3-E1 cells were seeded at 5 × 104 cells in 35-mm dishes. After 24 h, the siRNAs were transfected into the cells using RNAiMAX (Invitrogen, Carlsbad, CA, USA). The cells were incubated for 48 h and were then subjected to western blot analyses.
Preparation of Bacteria and the Model for Experimental Periodontitis
Cultures of P. gingivalis bacteria and a P. gingivalis-challenged experimental rat periodontitis model were established as previously described [6]. In brief, P. gingivalis ATCC 33277 was grown in a brain heart infusion broth supplemented with 5 mg/ml yeast extract, 5 μg/ml hemin, and 0.2 μg/ml vitamin K1. Bacterial cells were grown under anaerobic conditions (85% N2, 10% H2,s and 5% CO2) at 37 °C for 24 h. Eighteen 5-week-old male Sprague Dawley rats (CLEA Japan, Inc., Tokyo, Japan) were given sulfamethoxazole (1 mg/ml) and trimethoprim (200 μg/ml) in their drinking water for 4 days to reduce any original oral microorganisms, followed by a 3-day antibiotic-free period before starting the oral challenges with bacteria. Rats were divided into the following three groups of six rats each. Group A received only 5% carboxymethyl cellulose (CMC) (control group). Group B was orally challenged with P. gingivalis ATCC 33277 (P. g. group). Each rat infected with P. gingivalis received 0.5 ml (1.0 × 108 cells/ml) of the bacterial suspension in 5% CMC by oral gavage at 8, 10, and 12 days. Group C was treated with 500 μM fluoride in their drinking water (P. g. and NaF group) after the three P. gingivalis treatments. All rats were sacrificed 30 days after the final infection, and horizontal alveolar bone loss was measured using a morphometric method. The experimental procedures of this study were reviewed and approved by the Committee of Ethics on Animal Experiments of Kanagawa Dental University.
Histological Analysis
The tissues were fixed in 4% paraformaldehyde at 4 °C for 48 h and were then decalcified in 10% EDTA (pH 7.0) for 4 weeks. Paraffin-embedded tissue sections were prepared at a thickness of 4 μm. The sections were stained with hematoxylin and eosin (H-E) or Azan, and histopathological observations were performed using light microscopy.
Immunohistochemistry
Immunohistochemical staining was performed on 4% formaldehyde-fixed, paraffin-embedded rat maxilla specimens. Sections were initially immersed in citrate buffer pH 6.0 (Abcam) at 97 °C for 12 min, and subsequent steps were performed according to the manufacturer’s instructions. Mouse monoclonal anti-Runx2 antibody (1:200; Abcam) and rabbit polyclonal anti-PMN antibody (1:100; Accurate, Westbury, NY, USA) were used as primary antibodies to detect immunoreactivity in rat tissues. After overnight incubation with 3% hydrogen peroxide–methanol at 4 °C to block endogenous peroxidase activity, the specimens were rinsed with PBS and were incubated at room temperature for 1 h with the appropriate secondary antibody Histofine Simple Stain Rat MAX PO (Multi) (Nichirei Biosciences, Tokyo, Japan). After rinsing with PBS, all specimens were color-developed with diaminobenzidine (DAB) solution (DAKO, Carpinteria, CA, USA) and were counterstained with hematoxylin.
Statistical Analysis
Statistical analyses were performed using one-way ANOVA and Student’s t test. A P value of less than 0.05 is considered statistically significant.
Results
Effects of NaF on MC3T3-E1 Cell Proliferation
Optimal NaF concentrations for MC3T3-E1 cell proliferation were determined using the MTS assay. MC3T3-E1 cells (1 × 104cells/well) in α-MEM medium were seeded in 6-well plates and were treated with or without various concentrations of NaF for 24 and for 48 h. The results of the MTS assay indicated that NaF actually stimulated the proliferation of MC3T3-E1 cells at concentrations of 50 or 500 μM at 24 and at 48 h (Fig. 1a). No significant morphological changes occurred at 500 μM NaF that could be observed under a light microscope (data not shown).

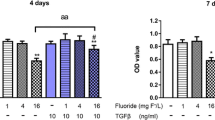
a Effect of NaF on MC3T3-E1 cell proliferation. Cell growth was assessed using the MTS assay at 24 and 48 h after treatment with 50 or 500 μM NaF. Exposure of MC3T3-E1 cells to 50 or 500 μM NaF for 24 or 48 h resulted in a significant increase in the number of living cells compared with non-treated cells (P < 0.05). b Effect of NaF on ALP activity of MC3T3-E1 cells. MC3T3-E1 cells were cultured with OIM and various concentrations of NaF for 7 days. Data are expressed as units per milliliter. c NaF promotes osteogenesis in MC3T3-E1 cells. Osteocalcin levels in the supernatant were measured after a 14-day induction as described in the “Materials and Methods” section. Asterisk denotes significant difference between OIM and NaF-treated cells (P < 0.05). d Effect of NaF on mineralization in MC3T3-E1 cells. Quantitation of mineralization using Alizarin Red staining as described in the “Materials and Methods” section. Values are expressed as means ± SD (n = 3). *, significant difference between OIM and NaF-treated cells (P < 0.05)
NaF Enhances ALP Activity, Extracellular Matrix Mineralization, and Osteocalcin Production in MC3T3-E1 Cells
Colorimetric analysis demonstrated that the addition of NaF increased ALP activity in MC3T3-E1 cells cultured in the OIM medium in a concentration-dependent manner (Fig. 1b). Fifty or 500 μM NaF increased ALP activity in a concentration-dependent manner compared to the untreated control (P < 0.05). The addition of 50 or 500 μM NaF increased Osteocalcin protein secretion at day 14 (P < 0.05 (Fig. 1c). On day 14, the formation of dense and intensive Alizarin red-positive mineralized nodules was greater in the NaF-treated culture than in the untreated culture (Fig. 1d, left). The total calcium content in the day 14 culture was higher in the NaF-treated culture than in the untreated control (Fig. 1d, right). These results indicate that NaF increased ALP activity and mineralization during the early stages of osteoblast differentiation in MC3T3-E1 cells.
NaF Up-Regulates Bone-Related mRNA Expression in Osteoblastic Culture
The expression of bone matrix-related mRNAs, including Runx2, Osterix, Osteopontin, and Osteocalcin, in osteoblastic cells cultured in the OIM medium, was enhanced by the addition of 50 or 500 μM NaF compared to the untreated control at day 14. The expression levels of bone matrix mRNAs in MC3T3-E1 cells treated with NaF were markedly higher than in untreated controls at day 7 (data not shown). Throughout the culture period, mRNA expression levels in cells treated with 500 μM NaF reached and stabilized at levels comparable to those seen in cells cultured in OIM at day 14. Runx2 mRNA expression after treatment with 50 or 500 μM NaF was accelerated and boosted in contrast to the untreated cells (Fig. 2a). Osterix expression following treatment with 50 or 500 μM NaF increased to a level greater than in cells not treated with NaF at day 14 (Fig. 2a). Likewise, Osteopontin and Osteocalcin mRNA expression levels were markedly enhanced by the addition of 50 or 500 μM NaF compared to untreated cells, showing a similar profile to expression to Runx2 (Fig. 2a).

a Up-regulation of bone-related mRNA expression in osteoblastic culture. The expression of Runx2, Osterix, Osteopontin (OPN), and Osteocalcin (OCN) mRNAs was analyzed by quantitative RT-PCR in MC3T3-E1 cells in OIM media with or without 50 or 500 μM NaF at day 14. The relative mRNA expression levels were calculated as a ratio to β-actin expression of each sample. Values represent means ± SE (bars) from three independent experiments. *P < 0.05, as determined using the t test. b Runx2 is required for NaF-induced osteogenesis. MC3T3-E1 cells were transfected with a scramble siRNA or with Runx2 siRNA. At 48 h post-transfection, proteins were solubilized and subjected to western blot analysis for Runx2, Osterix, and β-actin. One representative sample from at least three independent experiments with similar results is shown. Values represent means ± SE (bars) from three independent experiments. *P < 0.05, as determined using the t test
Involvement of Runx2 in NaF-Induced Osteogenesis
To determine whether Runx2 plays an important role in NaF-induced osteogenesis in MC3T3-E1 cells, we used a specific siRNA targeting Runx2 to examine the effects of the siRNA on Runx2 and Osterix expression in MC3T3-E1 cells in α-MEM medium. As shown in Fig. 2b, NaF induced Runx2 and Osterix protein expression in the control samples. Transfection with Runx2 siRNA for 48 h significantly knocked down the NaF-stimulated Runx2 and Osterix protein expression and had no effect on the housekeeping actin expression. These results suggest that Runx2/Osterix plays an important role in the NaF-induced osteogenesis in MC3T3-E1 cells.
Histological Examination of P. gingivalis-Challenged Experimental Rat Periodontitis Tissues
There was little inflammation in the absence of P. gingivalis, while treatment with P. gingivalis induced a moderate level of inflammation as observed by microscopy at ×100 magnification (Fig. 3, upper panels). Rats in the P. g.- and NaF-treated group showed a reduction in bone loss compared to the P. gingivalis-only group (Fig. 3, lower panels).

Hematoxylin and eosin (H-E) and Azan staining of P. gingivalis-challenged experimental rat periodontitis tissues. Maxillae were fixed, decalcified, and stained for H-E and Azan staining. There was little inflammation in the absence of P. gingivalis, while treatment with P. gingivalis induced a moderate level of inflammation observed by microscopy at ×100 magnification. Rats in the P. g.- and NaF-treated group showed a reduction in bone loss compared to the P. gingivalis group. Scale bars = 100 μm
Effects of NaF in a P. gingivalis-Challenged Experimental Periodontitis Animal Model
To investigate the effects of NaF in the P. gingivalis-challenged experimental periodontitis model, rats challenged with P. gingivalis were given NaF (500 μM) in their drinking water. After P. gingivalis infection for 4 weeks, the number of PMNs was increased 3.9-fold in rats (P < 0.05) (Fig. 4a). Treatment with NaF significantly reduced the PMN infiltration in the P. gingivalis and NaF group (P < 0.05). Immunohistochemical analysis showed an increased expression of Runx2 in the P. gingivalis and NaF rats, while Runx2 expression was decreased in P. gingivalis-treated tissues (Fig. 4b).

a NaF decreases the number of PMNs of P. gingivalis-challenged rats. The number of PMNs infiltrating the gingival epithelium was measured. Each value is the mean of all rats in the group ± SEM. Asterisk denotes significant difference between P. gingivalis and control rats (P < 0.05). Plus symbol, significant difference between P. gingivalis and P. gingivalis and NaF rats (P < 0.05). Scale bars = 100 μm. b Treatment with NaF induced Runx2 expression in P. gingivalis-challenged rats. Immunohistochemical analysis showed an increased expression of Runx2 in the P. gingivalis and NaF rats, while Runx2 expression was decreased in P. gingivalis-treated tissues. Scale bars = 20 μm
Discussion
The osteogenic differentiation of osteoblasts is characterized by cell proliferation, differentiation, and mineralization. An increase in total protein content was achieved by treatment with a micromolar level of NaF in the osteogenic differentiation media. This is in agreement with our previous reports demonstrating that NaF affected the proliferation of epithelial cells [16, 17]. Our results showed that NaF promotes the proliferation of osteoblasts in a dose-dependent manner, suggesting that NaF can induce the osteogenic differentiation of osteoblasts at a micromolar level.
The functional role of osteogenic-enhancing factors rests on at least two distinct elements, osteoinduction, and the modulation of osteoblastic differentiation leading to bone matrix synthesis and mineralization. This study was an effort to establish the molecular mechanism of the NaF-induced bone gain and to provide insights into the potential contribution of NaF to the regulation of osteoblast differentiation and bone formation. In this study, we examined the stimulatory effect of NaF on the osteogenic differentiation of MC3T3-E1 cells in the presence of OIM medium containing ascorbic acid 2-phosphate, b-glycerophosphate, and dexamethasone, compounds known to favor the expression of the osteoblastic phenotype in several bone cell systems [18]. MC3T3-E1 cells and human mesenchymal stem cells are commonly used in studies of osteogenic differentiation, and they have a similar mineralization process. The addition of NaF to MC3T3-E1 cells resulted in the marked enhancement of ALP activity and matrix mineralization in a dose-dependent manner. NaF accelerated and enhanced the expression of representative bone matrix proteins such as Runx2, Osterix, Osteopontin, and Osteocalcin in the osteoblastic culture. Up-regulation of those markers was already observed during the initial phase of the culture period (day 7; data not shown) and remained consistent through day 14. In addition, NaF enhanced Osteocalcin production in MC3T3-E1 cells. Our findings clearly demonstrate that NaF invokes osteoinduction and progressively modulates osteoblastic differentiation, leading to bone matrix synthesis and mineralization through the enhanced expression of Runx2 and Osterix.
Endogenous Runx2 is expressed in pre-osteoblasts, immature osteoblasts, early mature osteoblasts, and pre-odontoblasts and serves as a master regulator in osteoblastic differentiation and bone formation [19]. Runx2 and Osterix are two major transcription factors that play an essential role in the formation of adult bones and the expression of osteoblast genes [20, 21]. Runx2 is an upstream controller of Osterix [22, 23], a zinc finger-containing transcription factor. A number of bone matrix protein genes, such as Osteopontin and Osteocalcin, are regulated by Runx2 [22]. The conditional deletion of Osterix postnatally severely disrupts the maturation, morphology, and function of osteocytes [24]. The manipulation of Runx2 in various mesenchymal cell culture models strongly impacts the expression of osteoblast marker genes and related phenotypes such as ALP activity [25, 26]. We found that NaF increases Osteopontin and Osteocalcin mRNA levels and significantly up-regulates the levels of Runx2 and Osterix mRNA/protein, thereby up-regulating downstream gene expression and restoring ALP activity. ALP is thought to be a defined marker for osteoblastogenesis [27, 28] and is a key regulator that promotes the mineralization of the bone matrix [27, 29]. As expected, NaF also strengthened mineralized nodule formation, a marker for a later stage of osteogenic differentiation [30]. These data suggest that the bone-protective effects of NaF are mediated through the regulation of Runx2 and Osterix expression.
The maintenance of anti-inflammatory effects by NaF is of particular importance, since non-hematopoietic cells, such as fibroblasts, also contribute to the inflammatory process in periodontitis and facilitate bone erosion. In the present study, NaF was found to be effective in attenuating P. gingivalis-induced alveolar bone loss in vivo and facilitating bone formation in vitro, as evidenced through its restoration of calcium deposition and its regulation of bone differentiation and mature-associated proteins in osteoblasts. These results indicate that NaF exerts protective effects against bone diseases.
The major limitation of our study is that we tested only a few specific markers of bone remodeling, but it is likely that other proteins could be involved in the mechanism of bone differentiation, including markers of wound healing. Therefore, further research is needed to better clarify the effects of NaF on hard and soft tissues.
In conclusion, this study demonstrated that NaF stimulates the osteogenesis of osteoblasts at least in part, by increasing the expression levels of Runx2 and Osterix. More importantly, NaF protects against the P. gingivalis-induced inhibition of osteoblast differentiation in an experimental rat periodontitis model. These results suggest that NaF may be useful for treating bone-related disorders. Notably, our findings not only enrich the understanding of NaF regulation of bone development and osteogenic differentiation but also enable new opportunities for bone tissue engineering, and especially the prevention and treatment of periodontitis and other bone metabolism-related diseases.
References
Pihlstrom BL, Michalowicz BS, Johnson NW (2005) Periodontal diseases. Lancet 366:1809–1820
Harrison JE, McNeill KG, Sturtridge WC et al (1981) Three-year changes in bone mineral mass of osteoporotic patients based on neutron activation analysis of the central third of the skeleton. J Clin Endocrinol Metab 52:751–758
Farley JR, Wergedal JE, Baylink DJ (1983) Fluoride directly stimulates proliferation and alkaline phosphatase activity of bone forming cells. Science 222:330–332
Khokher MA, Dandona P (1990) Fluoride stimulates [3H]thymidine incorporation and alkaline phosphatase production by human osteoblasts. Metabolism 39:1118–1121
Guan ZZ, Xiao KQ, Zeng XY et al (2000) Changed cellular membrane lipid composition and lipid peroxidation of kidney in rats with chronic fluorosis. Arch Toxicol 74:602–608
Bhawal UK, Lee HJ, Arikawa K et al (2015) Micromolar sodium fluoride mediates anti-osteoclastogenesis in Porphyromonas gingivalis-induced alveolar bone loss. Int J Oral Sci 7:242–249
Lee HJ, Arikawa K (2015) Influence of low level sodium fluoride on expression of IGF-1 and IGF-2 protein in experimental type 2 diabetes with periodontitis model. J Hard Tissue Biol 24:319–324
Liu TM, Lee EH (2013) Transcriptional regulatory cascades in Runx2-dependent bone development. Tissue Eng Part B Rev 19:254–263
Eijken M, Koedam M, van Driel M et al (2006) The essential role of glucocorticoids for proper human osteoblast differentiation and matrix mineralization. Mol Cell Endocrinol 248:87–93
Sodek J, Ganss B, McKee MD (2000) Osteopontin. Critical Rev Oral Biol Med 11:279–303
Komori T, Yagi H, Nomura S et al (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89:755–764
Fakhry M, Hamade E, Badran B et al (2013) Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J Stem Cells 5:136–148
Nishio Y, Dong Y, Paris M et al (2006) Runx2-mediated regulation of the zinc finger Osterix/Sp7 gene. Gene 372:62–67
Duan XQ, Zhao ZT, Zhang XY et al (2014) Fluoride affects calcium homeostasis and osteogenic transcription factor expressions through L-type calcium channels in osteoblast cell line. Biol Trace Elem Res 162:219–226
Lü P, Li X, Ruan L et al (2014) Effect of siRNA PERK on fluoride-induced osteoblastic differentiation in OS732 cells. Biol Trace Elem Res 159:434–439
Arakawa Y, Bhawal UK, Ikoma T et al (2009) Low concentration fluoride stimulates cell motility of epithelial cells in vitro. Biomed Res 30:271–277
He D, Bhawal UK, Hamada N et al (2013) Low level fluoride stimulates epithelial- mesenchymal interaction in oral mucosa. J Hard Tissue Biol 22:59–66
Coelho MJ, Fernandes MH (2000) Human bone cell cultures in biocompatibility testing. Part II: effect of ascorbic acid, b-glycerophosphate and dexamethasone on osteoblastic differentiation. Biomaterials 21:1095–1102
Komori T (2010) Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 339:189–195
Wysokinski D, Pawlowska E, Blasiak J (2015) RUNX2: a master bone growth regulator that may be involved in the DNA damage response. DNA Cell Biol 34:305–315
Sinha KM, Zhou X (2013) Genetic and molecular control of osterix in skeletal formation. J Cell Biochem 114:975–984
Komori T (2005) Regulation of skeletal development by the Runx family of transcription factors. J Cell Biochem 95:445–453
Nakashima K, Zhou X, Kunkel G et al (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108:17–29
Zhou X, Zhang Z, Feng JQ et al (2010) Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc Natl Acad Sci U S A 107:12919–12924
Banerjee C, McCabe LR, Choi JY et al (1997) Runt homology domain proteins in osteoblast differentiation: AML3/CBFA1 is a major component of a bone-specific complex. J Cell Biochem 66:1–8
Ducy P, Zhang R, Geoffroy V et al (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89:747–754
Golub E, Boesze-Battaglia K (2007) The role of alkaline phosphatase in mineralization. Curr Opin Orthop 18:444–448
Mikami Y, Asano M, Honda MJ et al (2010) Bone morphogenetic protein 2 and dexamethasone synergistically increase alkaline phosphatase levels through JAK/STAT signaling in C3H10T1/2 cells. J Cell Physiol 223:123–133
Harmey D, Hessle L, Narisawa S et al (2004) Concerted regulation of inorganic pyrophosphate and osteopontin by Akp2, Enpp1, and Ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol 164:1199–1209
Orimo H, Shimada T (2008) The role of tissue-nonspecific alkaline phosphatase in the phosphate-induced activation of alkaline phosphatase and mineralization in SaOS-2 human osteoblast-like cells. Mol Cell Biochem 315:51–60
Acknowledgements
The authors wish to express their gratitude to Ujjal K. Bhawal for his help and contribution to this study. This research was partially supported by a Grant-in-Aid for Scientific Research of Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Lee, M., Arikawa, K. & Nagahama, F. Micromolar Levels of Sodium Fluoride Promote Osteoblast Differentiation Through Runx2 Signaling. Biol Trace Elem Res 178, 283–291 (2017). https://doi.org/10.1007/s12011-017-0930-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12011-017-0930-5