
Abstract
γ-Aminobutyrate (GABA) is an important bioactive compound synthesized through decarboxylation of L-glutamate by the glutamate decarboxylase (GAD). In this study, stabilized variants of the GAD from Lactobacillus brevis were constructed by consensus mutagenesis. Using Consensus Finder (http://cbs-kazlab.oit.umn.edu/), eight positions with the most prevalent amino acid (over 60% threshold) among the homologous family members were identified. Subsequently, these eight residues were individually mutated to match the consensus sequence using site-directed mutagenesis. Compared to the wild-type, T383K variant displayed the largest shift in thermostability among the single variants, with a 3.0 °C increase in semi-inactivation temperature (T5015), a 1.7-fold improvement of half-life (t1/2) at 55 °C, and a 1.2-fold improvement of t1/2 at 37 °C, respectively, while its catalytic efficiency (kcat/Km) was reduced. To obtain the mutant with improvement in both thermostability and catalytic activity, we performed a site-saturation mutation at T383. Notably, mutants T383V and T383G exhibited an increasement in thermostability and kcat/Km than that of wild-type. This study not only emphasizes the value of consensus mutagenesis for improving the thermostability of GAD but also sheds a powerful guidance to study the thermal stability of other enzymes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
γ-Aminobutyric acid (GABA) is a four-carbon non-protein amino acid, which is widely distributed in plants, animals, algae, fungi, and bacteria [1]. GABA has several important physiological functions, such as induction of hypotensive effects, regulating neurological disorders, and resisting cardiovascular diseases and convulsions [2,3,4,5]. In addition, GABA is an important intermediate for the synthesis of nylon-4 [6] and 2-pyrrolidone [7, 8]. As a consequence, GABA has the potential to be utilized extensively in the food, pharmaceutical and chemical industries [9]. The development of efficient approaches for production of GABA becomes an important issue to meet its increasing commercial demand.
The current methods for GABA production include chemical synthesis and biosynthesis. In contrast to the energy-intensity and environmental unfriendship of chemical synthesis method, biosynthesis has been considered as a more promising approach in pharmaceutical and food industry due to the simple reaction procedure, high catalytic efficiency, mild reaction condition, and environmental compatibility. Glutamate decarboxylase (GAD, EC 4.1.1.15) is a key enzyme in GABA biosynthesis. In the presence of co-enzyme pyridoxal-5′-phosphate (PLP), GAD can specifically catalyze the irreversible α-decarboxylation of L-glutamate to GABA. During the last few years, a large number of GADs from various organisms have been cloned and characterized [10, 11]. In addition, the thermostabilities of Bacillus megaterium GadB, Escherichia coli GadB, Lactobacillus fermentum GadB, and Lactobacillus brevisis GadA were also determined by incubating the enzymes at various temperatures and measuring their residual activities at different time intervals [8, 12,13,14,15,16]. Remarkably, in some cases, a common problem of these reported GADs is the limited stability [17]. For example, the half-life (t1/2) of the purified GAD from L. brevis was 50 min at 45 °C, while the GadB form E. coli displayed lower thermostability, with only 24.2 min at 45 °C and 1.8 min at 50 °C in t1/2, respectively [8, 14, 15]. Therefore, there is of great interest to improve the thermostability of GAD.
Effective methods for improving intrinsic enzyme stability include directed evolution and rational design. Rationally predicted stabilizing mutations can be found with less experimental screening effort and have thus been developed to address specific advantages. Recently, consensus mutagenesis based on sequence information to mutate specific amino acid in homologous genes has been applied to enhance the thermostability of proteins [18, 19]. It has been proposed that one-third variants of the β-lactamase from Enterobacter cloacae presented an increasement in thermostability, and the variant NA04.17 that contained eight consensus mutations increased the temperature transition midpoint (Tm) by 9.1 °C [20]. The mutant αW25Y of penicillin G acylase from E. coli and variant G283P of endoglucanase Cel8A from Clostridium thermocellum demonstrated a higher thermostability, with almost 3-fold increase in t1/2 at 50 °C and 14-fold increase in t1/2 at 85 °C, respectively [21, 22]. In addition, the double mutant H210N/I77L of the R-type selective amine transferase of Aspergillus terreus increased by 6.1-fold and 6.6 °C in the t1/2 at 40 °C and T5010, respectively [23]. The above examples show that the consensus mutagenesis is a feasible strategy to improve protein stability. Remarkably, the amino acid with higher frequency at the special position in homologous proteins is more valuable in improving protein stability [24].
In previous work of our research group, a high-yield GABA strain Lactobacillus brevis CGMCC No.1306 has been isolated from unpasteurized milk sample [25]. With the continuous efforts, the GAD1407 gene from the strain No.1306 has been cloned and expressed in E. coli BL21(DE3) [26]. Moreover, the crystal structure GAD (PDB ID: 5GP4) has been determined by X-ray crystallography, which provided a theoretical basis for unraveling the elusive structure-function relationship of this functional protein [27]. In the current study, the Consensus Finder (http://cbs-kazlab.oit.umn.edu) was adopted to predict stabilizing substitutions in GAD, and the most common residue in homologs was introduced to match the consensus by site-directed mutagenesis. Subsequently, the stabilizing effects of these mutations on catalytic activity and thermostability were experimentally identified. In addition, the thermostability mechanism of these variants was assessed by molecular dynamics (MD) simulation.
Materials and Methods
Materials and Reagents
Wild-type GAD gene incorporated in the pET28a (+) expression vector was used as the template for site-directed mutagenesis. Primers synthesis and DNA sequencing were performed by General Biosystems Co., Ltd. (Anhui, China). The PrimeSTAR® Max DNA polymerase, used for the polymerase chain reaction (PCR), was purchased from Takara Biotechnology (Dalian, China). Modified Bradford protein assay kit and Ni-NTA Sefinose™ Resin were obtained from Sangon Biotech Co., Ltd. (Shanghai, China). The SYPRO orange dye used for differential scanning fluorescence was purchased from Invitrogen (Carlsbad, CA, USA).
Design of Stable Variants
The GAD sequence with FASTA form was harvested in the NCBI gene database via searching GAD gene number (GenBank: ADG02973). Subsequently, the sequence of GAD in FASTA form was input to the Consensus Finder (http://cbs-kazlab.oit.umn.edu) to find similar sequences. All sequences were aligned (E-value maximum of 10−3) by ClustalX, and the overexpressed sequences were deleted using CD-Hit Suite (CD-Hit redundancy outstripped 0.9) [28]. The consensus generated sequences were obtained by aligning homologous sequences (Table S1). The putative mutation sites (D45, Q156, I159, A163, D179, H181, T383, and T408) were selected for site-directed mutation, at which the conservation threshold was above 60%. WebLogo online sever (http://weblogo.berkeley.edu/logo.cgi) was used to visualize the amino acid distributed as a “logo”.
Site-Directed Mutagenesis of GAD
Generally, the fifty-microliter (50-μL) reaction mixtures contained 22 μL of sterile water, 1 μL forward and reverse primers (Table S2), 1 μL (50–100 ng) of template DNA (pET28a (+)-GAD plasmid), and 25 μL of the 2× PrimeSTAR® Max DNA polymerase were subjected to PCR thermocycling program: a cycle at 98 °C for 5 min; 35 cycles of 98 °C for 20 s, 55 °C for 15 s, and 72 °C for 2 min; and a cycle at 72 °C for 10 min. After amplification, the PCR products were digested with DpnI at 37 °C for 2 h, and then transferred into E. coli DH5α by heat shock transformation. The transformants were selected on a solid Luria-Bertani (LB) plate containing 50 μg/mL kanamycin at 37 °C for 12 h. Subsequently, the recombinant plasmids, confirmed by DNA sequencing, were transformed into E. coli BL21 (DE3) cells.
Site-Saturation Mutation
The primers utilized to produce the site-saturation mutations at position 383 were designed by DNAMAN (Table S3), and the plasmid of wild-type was used as the template. The PCR reaction and cycling program of site-saturation mutagenesis were then performed as described previously. The PCR products were digested with DpnI at 37 °C for 2 h, and then transferred to E. coli BL21 (DE3) by heat shock transformation.
High-Throughput Colorimetric Screening Assay of GAD Residual Activity
Individual colonies were picked into 96-deep-well plates and cultured containing 1 mL of LB medium supplemented with 50 μg/mL kanamycin in each well, and then incubated for 8 h at 37 °C with shaking at 180 rpm. An aliquot of 100 μL of the culture was moved into a new 96-deep-well plate containing 900 μL of LB medium supplemented with 50 μg/mL kanamycin and grown at 37 °C. When OD600 reached 0.6–0.8, the culture was induced by IPTG (final concentration 0.8 mM) in each well. Subsequently, plates were incubated for 10 h at 25 °C with shaking at 180 rpm. The plates were centrifuged at 6000 rpm for 15 min at 4 °C and the supernatants were abandoned. The pellets were resuspended with 200 μL of 50 mM PBS buffer (pH 7.8) and kept at − 80 °C. After repeated freezing and thawing for three times, the pellets were resuspended in 200 μL of PBS buffer containing 1 mg/mL of lysozyme, and incubated at 37 °C for 30 min. After centrifugation at 6000 rpm for 15 min, the supernatants were collected and used for the GAD activity assay.
The colorimetric method for determination of GAD residual activity was performed in 96-deep-well plate as previously referenced [29]: 20 μL cell lysate and 400 μL acetate buffer (20 mM, pH 4.8) containing 100 mM glutamate, 0.01 mM PLP, and 50 μM bromocresol purple were incubated in 96-well plate at 48 °C with shaking at 400 rpm. The assay is based on the color change of the pH indicator bromocresol purple as protons are consumed during the decarboxylation of glutamate catalyzed by GAD [5].
Protein Expression and Purification
Recombinant E. coli BL21 (DE3) cells harboring pET28a (+)-GAD plasmids were incubated in 5 mL of the LB medium containing 50 μg/mL kanamycin at 37 °C overnight. The overnight culture was transferred to 200 mL of LB medium containing 50 μg/mL kanamycin and incubated at 37 °C. When OD600 reached 0.6–0.8, the culture was induced for 8 h at 25 °C by adding 0.8 mM IPTG. The cells were harvested by centrifugation at 6000 rpm for 10 min at 4 °C, and resuspended in 50 mM PBS buffer (pH 7.8). Subsequently, the cells were broken by sonication at 4 °C and the enzymes were released. The cell debris was removed by centrifugation at 8000 rpm for 40 min at 4 °C. The recombinant protein was purified by Ni-NTA affinity chromatography.
Protein concentration was measured using the modified Bradford protein assay kit, with bovine serum albumin (BSA) as the standard protein. The purity of the protein was identified by SDS-PAGE (12% separation gel and 5% stacking gels).
Enzyme Activity Assay
The enzymatic activities of wild-type and mutants at different substrate concentrations at 37 °C and 55 °C were determined as previously described [15]. Briefly, the 400 μL reaction mixtures (pH 4.8) containing 20 mM sodium acetate buffer, 100 mM L-MSG, 0.01 mM PLP, and 20 μL purified enzyme were incubated for 15 min at 37 °C and 55 °C, respectively. Then, 100 μL reaction product was mixed with 900 μL sodium bicarbonate solution (0.2 M) to terminate the reaction. Subsequently, the derivatization was carried out by addition of 500 μL of above mixture into 500 μL of 4 g/L dansyl chloride (DNS-Cl) solution, followed by incubation at 40 °C for 1 h. After derivatization, the products of the reactions catalyzed by wild-type and the mutants were subjected to ultra-performance liquid chromatography coupled with mass spectrometry (UPLC-MS) analysis using the method of Stragierowicz et al. [30] with minor modifications to confirm the production of GABA. In addition, HPLC assay was used to determine the concentration of GABA using a gradient elution procedure as described previously [15]. One unit of GAD activity (U) was defined as the amount of enzyme that produced 1 μmol of GABA per second.
Kinetic and Thermodynamic Stability
The purified enzymes of the wild-type and mutants were incubated for 15 min at 55 °C, and rapidly kept on ice for 5 min. Subsequently, 20 μL purified enzyme was mixed with 400 μL buffer B (20 mM sodium acetate buffer, 100 mM L-MSG, 0.01 mM PLP, and pH 4.8) and reacted for 15 min at 37 °C. The enzymatic activity at 4 °C after 15 min was taken as 100%.
The T5015 is defined as the temperature at which the enzyme activity is reduced to 50% of its original enzyme activity after a 15-min heat treatment. The purified enzymes of the wild-type and variants were incubated for 15 min at 30–70 °C, and cooled on ice for 5 min. The relative activity of each sample was detected as described above. The T5015 was calculated by fitting the data of relative enzyme activity at certain temperatures to a four-parameter Boltzmann equation using Origin 8.0.
The time required for the residual activity to be reduced to half (t1/2) were determined by incubating the purified enzymes of the wild-type and mutants for different times at 37 °C or 55 °C and then keeping them on ice for 5 min. The relative activities of the wild-type and mutants were detected as described above.
The differential scanning fluorimetry (DSF) method was modified based on the previously methods [31, 32]. Briefly, the mixture consisted of 1 × SYPRO orange dye, 0.15 mg/mL purified enzyme, and buffer A (150 mM NaCl, 50 mM sodium phosphate, and pH 7.5), with a total volume of 50 μL. The fluorescence intensity change of the sample was determined by StepOne Real-Time PCR System (version 2.2.2) at 28–56 °C (0.7 °C increments), with each temperature keeping for 30 s. The excitation wavelength is 490 nm, and the emission wavelength is 605 nm. The PBS buffer was served as a negative control to correct background fluorescence.
Molecular Dynamics Simulation
The three-dimensional (3D) structures of wild-type and mutants were subjected to FoldX algorithm, and MD simulation was performed at constant temperature (313 K) for 10 ns using the Amber 14 force field of YASARA (version 16.4.6) software (http://www.yasara.org) [33, 34]. The 3D structures were filled with water with a density of 0.998 mg/L and inserted into a cube with edge lengths of 10 Å. The sodium ion and chloride ion were acted as counter ions to make an electrically neutral system, and the ionizable groups were protonated according to their pKa values at pH 4.8 in the medium. The van der Waals interactions were handled with a cutoff of 7.86 Å. The long-range electrostatic interactions were calculated by the particle mesh Ewald (PME) method [35]. The magnitude of a time step was 2.5 fs, and the trajectory was collected every 25 ps. Analyses of protein structures including root mean square deviation (RMSD) of backbone atom positions and root mean square fluctuation (RMSF) for individual residues were performed using YASARA. The simulation trajectories were visualized by the Visual Molecular Dynamics (VMD) software.
Results and Discussion
Selection of Sites for Consensus Mutagenesis
The potentially stable amino acid substitutions were predicted using the Consensus Finder, and the potential mutations were visualized using WebLogo. After aligning the sequences of 500 wild-type GAD homologs, eight mutants (D45Y, I159L, Q156K, A163S, D179E, H181R, T383K, and T408A) with more than 60% consistent residues were obtained and individually mutated to match the consensus. The PyMOL (http://pymol.org) was used to display the three-dimensional structure of wild-type GAD and eight single substituents (Fig. 1). According to the secondary structure of GAD, D45Y was located at the α-helix, Q156K, A163S, D179E, T383K, and T408A were located at the loop regions, and I159L and H181R were located at the β-sheet.

The positions of eight mutants in the GAD sequence. Substituted residues are represented by spheres. Substituted residue pairs are represented as WebLogo markers (the abscissa indicates, amino acid position; the symbol height, relative frequency of the corresponding amino acid at that position)
Screening of Mutants and Site-Directed Saturation Mutagenesis of T383
To evaluate the thermostability of consensus mutagenesis mutants, the relative activities of eight variants were determined after incubation at 55 °C for 15 min. As shown in Fig. 2, T383K was the most stable mutant, with a 1.5-fold improvement of residual activity than that of wild-type.

The relative activities of all consensus mutagenesis mutants. The relative activity of wild-type was taken as 100%, and the standard deviations from three independent experiments were showed by error bars
To obtain the mutant with enhanced thermostability as well as improved catalytic performance at position 383, a saturated library was established by site-saturation mutation at this site with wild-type plasmid as template. The colorimetric method for determination of GAD residual activity was performed in 96-deep-well plate as previously referenced, and based on the color change of the pH indicator bromocresol purple as protons are consumed during the decarboxylation of glutamate catalyzed by GAD (Fig. S1). Twenty mutants from saturation libraries with larger color change than wild-type were screened and sequenced. Subsequently, eight potential mutants (T383S, T383L, T383G, T383N, T383V, T383A, T383D, and T383H) were obtained, and their relative activities were determined. Finally, T383V and T383G exhibited the larger shift in thermostability than that of wild-type, with a 1.33-fold and a 1.28-fold improvement of relative activity after incubation at 55 °C for 15 min, respectively (Fig. 3).

The relative activities of wild-type and eight variants at position 383. The relative activity of wild-type was taken as 100%, and the standard deviations were calculated from at least three independent experiments and showed by error bars
SDS-PAGE and UPLC-MS Analysis
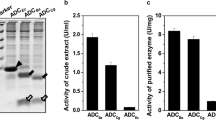
As shown in Fig. S2, the variants (T383K, T383V, T383G) had a single band and the same molecular weight with wild-type. The samples were derivatized with DNS-Cl and identified by UPLC-MS with positive electron spray ionization mode. The retention time and mass-to-charge (m/z) of hydrogenation adduct [(GABA-DNS) + H]+ were 7.9 min and 371 according to extracted ion chromatogram (EIC) by UPLC-MS. After DNS-Cl derivation, the products catalyzed by wild-type and three mutants (T383K, T383V, and T383G) have the same retention time and m/z value (Fig. 4).

UPLC-MS spectrum of GABA, L-MSG, and the reaction product catalyzed by wild-type and its mutants in positive electron ionization mode. a The Base Peak Intensity (BPI) chromatograms from the derivative of GABA, L-MSG, and the reaction product catalyzed by wild-type and its mutants. The reaction mixtures were derivatized by adding DNS-Cl for 1 h at 40 °C. Absorption of the derivative was determined at 254 nm. b The ion mass spectra of the [(GABA-DNS) + H]+ ions (m/z 371) of standard GABA. c The ion mass spectra of the [(GABA-DNS) + H]+ ions (m/z 371) of the reaction product catalyzed by wild-type
Kinetic and Thermodynamic Stability of Wild-Type and Its Mutants
To evaluate the thermal inactivation profiles of the enzymes, the purified enzymes were incubated at different temperatures for different times. As shown in Table 1 and Fig. S3, the T5015 and t1/2 values of wild-type were 56.2 °C and 24.1 min (55 °C), respectively. Compared with wild-type, the T5015 values of mutants T383K, T383V, and T383G increased to 59.2 °C, 58.8 °C, and 58.1 °C, respectively. In addition, the t1/2 values of mutants T383K, T383V, and T383G at 55 °C increased by 1.73-fold, 1.67-fold, and 1.66-fold, respectively. The results show that mutants T383K, T383V, and T383G have higher thermostability than wild-type. Moreover, the t1/2 values of mutants T383K, T383V, and T383G were 137.6 h, 134.0 h, and 129.4 h at 37 °C, which correspond to increases of 23.5 h, 19.9 h, and 15.3 h over that of the wild-type (114.1 h), respectively (Table 1 and Fig. S4).
DSF is a rapid and simple method for studying protein thermostability. At unfolded states of a protein, hydrophobic portion of the protein will be exposed and specifically bound to the SYPRO orange dye [36]. The melting temperature (Tm) is regard as the temperature transition midpoint value between the initial point and the maximum point. In order to estimate the thermodynamic stability of wild-type and thermostable mutants, the Tm of proteins were determined by DSF. As shown in Table 1 and Fig. S3, the Tm values of mutants T383K, T383V, and T383G were 41.9 °C, 41.8 °C, and 41.0 °C, which increased by 1.3 °C, 1.2 °C, and 0.4 °C than that of the wild-type (40.6 °C). The data indicates the consistence of the thermodynamic stability and kinetic stability.
Enzyme Activity Assay
To compare the catalytic performance of wild-type and its variants, the kinetic constants at 37 °C and 55 °C were investigated by determining the initial reaction rates at different substrate concentrations. As shown in Tables 2 and 3, T383K showed lower catalytic efficiency (kcat/Km) than that of the wild-type at 37 °C and 55 °C, indicating a decrease in catalytic efficiency. In contrast, T383V and T383G presented higher kcat/Km values than that of wild-type, with 1.5-fold and 1.3-fold increase at 37 °C, and 1.8-fold and 1.5-fold increase at 55 °C, respectively. It is indicated that the catalytic performance and the thermostability of T383V and T383G were simultaneously increased.
The GABA-producing capacity of wild-type and its variants at 37 °C and 55 °C was determined to estimate the enzymatic activity. As shown in Fig. S5, it was observed that mutants T383V and T383G displayed higher GABA produced than that of wild-type, while mutant T383K possessed lower GABA produced, indicating that variants T383V and T383G exhibited relatively stronger enzymatic activity than wild-type. In addition, the GABA produced of wild-type and its variants at 55 °C was obviously lower than those at 37 °C; this behavior probably resulted from enzyme inactivation at high temperature.
MD Simulation Analysis
MD simulation is an effective method to understand the molecular mechanism of protein stability [37]. The RMSD, as a function of time, was used to represent the flexibility of the overall structure of the protein, and high RMSD values show there are significant structural changes during the simulation. After a rapid increase during the initial 1 ns, the wild-type structure reached stability at about 8.5 ns (Fig. 5a), while the most stable mutant T383K structure reached stability at about 2.5 ns. In addition, the average RMSD value of the wild-type GAD over the whole MD simulation was higher than the variant T383K, indicating that the wild-type was more flexible than variant. The RMSF is commonly used to reflect the flexibilities of individual residues, and a high RMSF value corresponds to higher flexibility of a given residue [38]. As can be seen in Fig. 5b, the RMSF value for the residue T383 in the wild-type protein was higher than that of T383K at 313 K.

MD simulation analysis of wild-type and T383K for 10 ns at 313 K using YASARA. a The RMSD values at 313 K during a 10 ns simulation for the wild-type and mutant T383K. b The RMSF values at 313 K during a 10 ns simulation for the wild-type and mutant T383K
Intramolecular interactions, such as hydrogen bonds, hydrophobic interactions, and disulfide bond played decisive roles in improving the thermostability of proteins [14, 23]. In order to further probe the molecular mechanism of stability, the 3D structures of wild-type and T383K were compared. It is clear that residues Thr383/Lys383 and Arg388 constitute the hydrogen bonds (Fig. 6). The O atom of Thr383 forms two hydrogen bonds with the NH1 atom of Arg388 (1.8 Å and 3.0 Å) in the wild-type. Particularly, the O atom of Lys383 not only forms two hydrogen bonds with the NH1 atom of Arg388 (1.8 Å and 3.5 Å) but also results in one additional hydrogen bonds with the NH2 atom of Arg388 (2.3 Å) in the mutant T383K. This indicated that the improvement in thermostability of the mutant T383K might be attributed to the additional hydrogen bonds.

Modeling analysis of hydrogen bonds at position 383 in wild-type GAD and variant T383K. a wild-type GAD, b variant T383K. The hydrogen bonds formed between the O atom of Thr383/Lys383 and the NH1 atom of Arg388 are displayed by yellow, and the hydrogen bond formed between the O atom of Lys383 and the NH2 atom of Arg388 is displayed by red
Conclusion
Three mutants of GAD with higher thermostability were obtained by consensus mutagenesis and site-saturation mutation. Compared to the T5015 and the t1/2 values of the wild-type GAD, the T5015 values of T383K, T383V, and T383G increased by 3.0 °C, 2.6 °C, and 1.9 °C, respectively; the t1/2 values at 55 °C of T383K, T383V, and T383G all improved 1.7-fold; the t1/2 values at 37 °C of T383K, T383V, and T383G improved 1.2-fold, 1.2-fold, and 1.1-fold, respectively. As measured by DSF, the Tm values of T383K, T383V, and T383G were greater than that of the wild-type GAD by 1.3 °C, 1.2 °C, and 0.4 °C respectively. In addition, the kcat/Km values of T383V and T383G at 37 °C and 55 °C are higher than that of wild-type GAD, indicating that variants T383V and T383G exhibited superior performance in the thermostability and catalytic efficiency. Furthermore, the thermostability of variant T383K and other reported GADs derived from different origins were compared. As shown in Table 4, the t1/2 values of T383K were 137.6 h at 37 °C and 41.8 min at 55 °C, respectively, which are higher than that of the GADs from Bacillus megaterium, E. coli, and Lactobacillus fermentum. In summary, the study demonstrates that consensus mutagenesis can not only be regarded as an effective strategy to enhance the thermostability of GAD, but may also be applied to other enzymes for improvement in the stability.
References
Shelp, B. J., Bozzo, G. G., Trobacher, C. P., Zarei, A., Deyman, K. L., & Brikis, C. J. (2012). Hypothesis/review: contribution of putrescine to 4-aminobutyrate (GABA) production in response to abiotic stress. Plant Science, 193-194, 130–135.
Kaila, K., Ruusuvuori, E., Seja, P., Voipio, J., & Puskarjov, M. (2014). GABA actions and ionic plasticity in epilepsy. Current Opinion in Neurobiology, 26, 34–41.
Yin, Y. Q., Cheng, C., & Fang, W. M. (2018). Effects of the inhibitor of glutamate decarboxylase on the development and GABA accumulation in germinating fava beans under hypoxia-NaCl stress. RSC Advances, 8, 20456–20461.
Bednar, D., Beerens, K., Sebestova, E., Bendl, J., Khare, S., Chaloupkova, R., Prokop, Z., Brezovsky, J., Baker, D., & Damborsky, J. (2015). FireProt: energy- and evolution-based computational design of thermostable multiple-point mutants. PLoS Computational Biology, 11(11), e1004556.
Lyu, C. J., Zhao, W. R., Hu, S., Huang, J., Lu, T., Jin, Z. H., Mei, L. H., & Yao, S. J. (2017). A physiology-priented engineering strategy to improve gamma-aminobutyrate production in Lactobacillus brevis. Journal of Agricultural and Food Chemistry, 65, 858–866.
Park, S. J., Kim, E. Y., Noh, W., Oh, Y. H., Kim, H. Y., Song, B. K., Cho, K. M., Hong, S. H., Lee, S. H., & Jegal, J. (2013). Synthesis of nylon 4 from gamma-aminobutyrate (GABA) produced by recombinant Escherichia coli. Bioprocess and Biosystems Engineering, 36, 885–892.
Lammens, T. M., Franssen, M. C. R., Scott, E. L., & Sanders, J. P. M. (2010). Synthesis of biobased N-methylpyrrolidone by one-pot cyclization and methylation of γ-aminobutyric acid. Green Chemistry, 12, 1430–1436.
Lammens, T. M., De Biase, D., Franssen, M. C. R., Scott, E. L., & Sanders, J. P. M. (2009). The application of glutamic acid α-decarboxylase for the valorization of glutamic acid. Green Chemistry, 11, 1562–1567.
Wu, C., & Sun, D. (2014). GABA receptors in brain development, function, and injury. Metabolic Brain Disease, 30, 367–379.
Zhuang, Y. L., Ren, G. J., He, C. M., Li, X. Y., Meng, Q. M., Zhu, C. F., Wang, R. C., & Zhang, J. R. (2010). Cloning and characterization of a maize cDNA encoding glutamate decarboxylase. Plant Molecular Biology Reporter, 28, 620–626.
Ji, J., Zheng, L. Y., Yue, J. Y., Yao, X. M., Chang, E. M., Xie, T. T., Deng, N., Chen, L. Z., Huang, Y. W., Jiang, Z. P., & Shi, S. Q. (2017). Identification of two CiGADs from Caragana intermedia and their transcriptional responses to abiotic stresses and exogenous abscisic acid. PeerJ, 5, e3439.
Liu, Q. D., Cheng, H. J., Ma, X. Q., Xu, N., Liu, J., & Ma, Y. H. (2016). Expression, characterization and mutagenesis of a novel glutamate decarboxylase from Bacillus megaterium. Biotechnology Letters, 38(7), 1107–1113.
Huang, Y., Su, L., & Wu, J. (2016). Pyridoxine supplementation improves the activity of recombinant glutamate decarboxylase and the enzymatic production of gama-aminobutyric acid. PLoS One, 11, e0157466.
Fan, L. Q., Li, M. W., Qiu, Y. J., Chen, Q. M., Jiang, S. J., Shang, Y. J., & Zhao, L. M. (2018). Increasing thermal stability of glutamate decarboxylase from Escherichia coli, by site-directed saturation mutagenesis and its application in GABA production. Journal of Biotechnology, 278(20), 1–9.
Huang, J., Mei, L. H., Sheng, Q., Yao, S. J., & Lin, D. Q. (2007). Purification and characterization of glutamate decarboxylase of Lactobacillus brevis CGMCC 1306 isolated from fresh milk. Chinese Journal of Chemical Engineering, 15(2), 157–161.
Lin, Q., Li, D. N., & Qin, H. Z. (2017). Molecular cloning, expression, and immobilization of glutamate decarboxylase from Lactobacillus fermentum YS2. Electronic Journal of Biotechnology, 27, 8–13.
Ueno, Y., Hayakawa, K., Takahashi, S., & Oda, K. (1997). Purification and characterization of glutamate decarboxylase from Lactobacillus brevis IFO 12005. Bioscience Biotechnology and Biochemistry, 61(7), 1168–1171.
Jones, B. J., Lim, H. Y., Huang, J., & Kazlauskas, R. J. (2017). Comparison of five protein engineering strategies to stabilize an α/β-hydrolase. Biochemistry, 56, 6521–6532.
Saab-Rincón, G., Alwaseem, H., Guzmán-Luna, V., Olvera, L., & Fasan, R. (2018). Stabilization of the reductase domain in the catalytically self-sufficient cytochrome P450BM3 via consensus-guided mutagenesis. ChemBioChem, 11(6), 622–632.
Amin, N., Liu, A. D., Ramer, S., Aehle, W., Meijer, D., Metin, M., Wong, S., Gualfetti, P., & Schellenberger, V. (2004). Construction of stabilized proteins by combinatorial consensus mutagenesis. Protein Engineering, Design & Selection: PEDS, 17(11), 787–793.
Polizzi, K. M., Chaparro-Riggers, J. F., Vazquez-Figueroa, E., & Bommarius, A. S. (2006). Structure-guided consensus approach to create a more thermostable penicillin G acylase. Biotechnology Journal, 1, 531–536.
Abraham, M. J., Murtola, T., Schulz, R., Páll, S., Smith, J. C., Hess, B., & Lindahl, E. (2015). GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1-2, 19–25.
Xie, D. F., Yang, J. X., Lv, C. J., Mei, J. Q., Wang, H. P., Hu, S., Zhao, W. R., Cao, J. R., Tu, J. L., Huang, J., & Mei, L. H. (2019). Construction of stabilized (R)-selective amine transaminase from Aspergillus terreus by consensus mutagenesis. Journal of Biotechnology, 293, 8–16.
Steipe, B., Schiller, B., Plückthun, A., & Steinbacher, S. (1994). Sequence statistics reliably predict stabilizing mutations in a protein domain. Journal of Molecular Biology, 240, 188–192.
Huang, J., Mei, L.-H., & Xia, J. (2007). Application of artificial neural network coupling particle swarm optimization algorithm to biocatalytic production of GABA. Biotechnology and Bioengineering, 96(5), 924–931.
Fan, E., Huang, J., Hu, S., Mei, L. H., & Yu, K. (2011). Cloning, sequencing and expression of a glutamate decarboxylase gene from the GABA-producing strain Lactobacillus brevis CGMCC 1306. Annals of Microbiology, 62, 689–698.
Huang, J., Fang, H., Gai, Z. C., Mei, J. Q., Li, J. N., Hu, S., Lv, C. J., Zhao, W. R., & Mei, L. H. (2018). Lactobacillus brevis CGMCC 1306 glutamate decarboxylase: Crystal structure and functional analysis. Biochemical and Biophysical Research Communications, 503, 1703–1709.
Huang, Y., Niu, B. F., Gao, Y., Fu, L. M., & Li, W. Z. (2010). CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics, 26(5), 680–682.
Yu, K., Hu, S., Huang, J., & Mei, L. H. (2011). A high-throughput colorimetric assay to measure the activity of glutamate decarboxylase. Enzyme and Microbial Technology, 49(3), 272–276.
Stragierowicz, J., Darago, A., Brzeznicki, S., & Kilanowicz, A. (2017). Optimization of ultra-performance liquid chromatography (UPLC) with fluorescence detector (FLD) method for the quantitative determination of selected neurotransmitters in rat brain. Medycyna Pracy, 68(5), 583–591.
Niesen, F. H., Berglund, H., & Vedadi, M. (2007). The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature Protocols, 2(9), 2212–2221.
Kim, S. J., Lee, J. A., Joo, J. C., Yoo, Y. J., Kim, Y. H., & Song, B. K. (2010). The development of a thermostable CiP (Coprinus cinereus peroxidase) through in silico design. Biotechnology Progress, 26(4), 1038–1046.
Ishak, S. N. H., Aris, S. N. A., Halim, K. B. A., Ali, M. S. M., Leow, T. C., Kamarudin, N. H. A., Masomian, M., & Rahman, R. N. Z. R. (2017). Molecular dynamic simulation of space and earth-grown crystal structures of thermostable T1 lipase Geobacillus zalihae revealed a better structure. Molecules, 22(10), 1574.
Guerois, R., Nielsen, J. E., & Serrano, L. (2002). Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. Journal of Molecular Biology, 320, 369–387.
Darden, T., York, D., & Pedersen, L. (1993). Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98, 10089–10092.
Zhu, W. L., Hu, S., Lv, C. J., Zhao, W. R., Wang, H. P., Mei, J. Q., Mei, L. H., & Huang, J. (2019). A single mutation increases the thermostability and activity of Aspergillus terreus amine transaminase. Molecules, 24(7), 1194.
Purmonen, M., Valjakka, J., Takkinen, K., Laitinen, T., & Rouvinen, J. (2007). Molecular dynamics studies on the thermostability of family 11 xylanases. Protein Engineering, Design & Selection: PEDS, 20(11), 551–559.
Dong, Y. W., Liao, M. L., Meng, X. L., & Somero, G. N. (2018). Structural flexibility and protein adaptation to temperature: molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proceedings of the National Academy of Sciences of the United States of America, 115, 1274–1279.
Funding
This study was funded by the National Natural Science Foundation of China (Nos. 31470793, 31670804, and 31971372) and Zhejiang Natural Science Foundation (Nos. LZ13B060002, LY16B060008, and LQ18B060002); and Innovation Fund for Graduate students of Zhejiang University of Science and Technology (2019YJSKC08).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(DOCX 1.16 mb).
Rights and permissions
About this article

Cite this article
Hua, Y., Lyu, C., Liu, C. et al. Improving the Thermostability of Glutamate Decarboxylase from Lactobacillus brevis by Consensus Mutagenesis. Appl Biochem Biotechnol 191, 1456–1469 (2020). https://doi.org/10.1007/s12010-020-03283-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-020-03283-0