
Abstract
Naringinase which was extracted from the fermented broth of Cryptococcus albidus was purified about 42-folds with yield 0.7% by sulfate fractionation and chromatography on Toyopearl HW-60, Fractogel DEAE-650-s, and Sepharose 6B columns. Molecular weight of protein determined by gel filtration and SDS-PAGE was 50 kDa. Naringinase of C. albidus includes high content of the dicarbonic and hydrophobic amino acids. Enzyme contains also carbohydrate component, represented by mannose, galactose, rhamnose, ribose, arabinose, xylose, and glucose. The enzyme was optimally active at pH 5.0 and 60 °C. Naringinase was found to exhibit specificity towards p-nitrophenyl-α-L-rhamnose, p-nitrophenyl-β-D-glucose, naringin, and neohesperidin. Its K m towards naringin was 0.77 mM and the V max was 36 U/mg. Naringinase was inhibited by high concentrations of reaction product—L-rhamnose. Enzyme revealed stability to 20% ethanol and 500 mM glucose in the reaction mixture that makes it possible to forecast its practical use in the food industry in the production of juices and wines.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Industrial enzymology is an important area of modern biotechnology; it promotes intensification of food and chemical and pharmaceutical industries. Current research efforts are aimed at finding high-performance strains of microorganisms—producers of enzymes with different specificity. Due to their properties to carry out the biotransformation of various glycosides, glycosidases are commercially attractive for use in various industrial processes. Naringinase (E.C.3.2.1.40) is an enzyme complex expressing α-L-rhamnosidase (E.C.3.2.1.40) and β-D-glucosidase (E.C.3.2.1.23), which cleave terminal α-L-rhamnose and β-D-glucose from glycosides, glycopeptides, glycolipids, and flavonoides. The ability to cleave naringin makes naringinase extremely attractive for use in the food industry, in particular for the debittering of fruit juices [1, 2]. The flavonoide naringin consists of sugar components (α-L-rhamnose and β-D-glucose) and an aglycone part. α-L-Rhamnosidase acts on sugar complex naringin (4,5,7-trihydroxy-flavonone 7-rhamnoglucoside) releasing prunin, while β-D-glucosidase acts on prunin to produce a tasteless compound naringenin (4,5,7-trihydroxy-flavonone) in a two-step reaction. Naringenin has a great potential, especially in the food and pharmaceutical industries, due to its recognized antioxidant, anti-inflammatory, anti-ulcer, and hypocholesterolemic effects. Naringenin has also shown anti-mutagenic and neuroprotective activities, while prunin has antiviral activity. Often, the glycosidic residue is related to activity of glycoconjugate; in some cases, deglycosylation improves the biological activity and bioavailability. Such improvement may be related not only to pharmacodynamics but also to the overall molecule structure.
Naringinase improves taste of fruit juices acting on naringin, which has a bitter taste, producing naringenin, which is tasteless. This conversion does not affect product stability, organoleptic characteristics, and other inherent properties of antioxidants compounds, at the same time increasing of the product [3]. Naringinase activity in combination with arabinosidase is considered suitable for aroma enhancement in wine making [2, 4]. The α-L-rhamnosidase expressed by naringinase can be used in the biotransformation of steroids, preparation of rhamnose and antibiotics, and production of prunin and ginsenosides.
Naringinase has been studied for a few decades, but its specificity and structural and functional properties have not been investigated as extensively as it was done for many others enzymes. Many microorganisms synthesizing naringinase had been reported. Fungi Aspergillus niger, Rhizopus nigricans, Phomopsis citri, and Penicillium decumbens produce naringinase, which has been purified and characterized [5,6,7]. Bacterial α-L-rhamnosidase production has been studied in the plant pathogen Corticium rolfsii, the human intestinal Bacteroides, Sphingomonas sp., and Bacillus methylotrophicus, Clostridium stercorarium, Lactobacillus sp., Pseudomonas sp., Pediococcus acidilactici, and Staphylococcus xylosus [8,9,10,11]. Despite the contribution of microbial α-L-rhamnosidases to the improvement of wine flavor and debitterness of citrus juice, there is still lack of information about the enzyme activity in yeasts. So far, there have been only a few reports on the production of α-L-rhamnosidase in yeasts [12]. Low levels of α-L-rhamnosidase activity were found in Saccharomyces cerevisiae Tokaj 7, Hansenula anomala, and Debaryomyces polymorphus. Recently, this enzyme production by Clavispora lusitaniae, Candida tropicalis, Cryptococcus laurentii, and Pichia angusta was described [13,14,15,16]. Yet, many of such producers have different drawbacks, which make their enzymes of interest unacceptable for the use in industry due to their technical or economical disadvantages.
Earlier [17] through screening yeasts of different genus, we found Cryptococcus albidus to be the best producer of the naringinase under the conditions tested. Here, we report results of the purification and characterization of extracellular naringinase produced by C. albidus.
Materials and Methods
Strain, Medium, and Growth Conditions
C. albidus 1001, from the collection of living cultures of the D.K. Zabolotny IMV NAS of Ukraine, was grown in medium containing (g/l) 1.0 of rhamnose, 5.0 of peptone, 3.0 of yeast extract, and 3.0 of malt extract. The value of the liquid medium pH was adjusted to 6, and culture was incubated at 28 °C in an orbital shaker at 220 rpm for 4 days.
Enzyme Activity Assays
The activity of naringinase was determinate using Davis method [18] with minor modification. The assay mixture contained 0.2 ml of 0.1% naringin (Sigma, USA) solution in 0.1 M phosphate-citrate buffer (PCB) pH 5.2 and 0.2 ml enzyme solution. After incubation at 37 °C for 30 min, the reaction was stopped by addition of 5 ml diethylene glycol (90%) and 0.1 ml of 4 N NaOH. The residual naringin was measured at 420 nm. One unit of naringinase activity was defined as the amount of enzyme that releases 1 μmol of naringin per min at 37 °C under conditions used.
α-L-Rhamnosidase and β-D-glucosidase activities were determined using as a substrate p-nitrophenyl-α-L-rhamnopyranoside (p-NPR) and p-nitrophenyl-β-D-glucopyranoside(p-NPG), respectively [19]. To determine the activity, 0.1 ml of the enzyme solution was mixed with 0.2 ml 0.1 M PCB pH 5.2 and 0.1 ml 0.01 M substrate solution in PCB. Reaction mixture was incubated for 10 min at 37 °C. The reaction was stopped by adding 2 ml of 1 M sodium bicarbonate. The amount of released nitrophenol as a result of hydrolysis was determined colorimetrically by the absorption at 400 nm. One unit of enzyme activity was defined as the amount of enzyme that releases 1 μmol of p-nitrophenol per minute at 37 °C in 0.1 M PCB and pH 5.2.
Enzyme Purification
Culture filtrate of C. albidus grown on rhamnose for 4 days was mixed with dry ammonium sulfate salt up to 30% saturation under pH control. Mixture was incubated for 2 h at 4 °C followed by centrifugation at 5000g, 30 min, and 4 °C. Precipitate was discarded, and the supernatant was mixed with ammonium sulfate to achieve final concentration of 90% saturation. The mixture was incubated for 6 h at 4 °C followed by centrifugation under the same conditions. Precipitate was removed, dissolved in 3 M ammonium sulfate (threefold of volume). 0.01 M of sodium azide was added for conservation. To obtain highly purified enzyme preparations, gel filtration and ion exchange chromatography techniques have been used. Precipitate resulted from the fractionating with ammonium sulfate was dialyzed against 10 mM phosphate buffer (pH 6.0). The dialyzed enzyme solution was applied to an Toyopearl HW-60 column (2.5 × 90 cm), equilibrated with 0.01 M phosphate buffer and pH 6.0. Fractions exhibiting naringinase activity were collected and concentrated by evaporating under vacuum. The resultant preparation was applied to Fractogel DEAE-650-s (“Merck”, Germany) column (3 × 35 cm), equilibrated with 0.01 M Tris-HCl buffer and pH 7.5. Elution was performed by the NaCl linear gradient (0–1 M of 250 ml each) at 24 ml/h rate. Collected fractions were screened for protein content (A280) and naringinase activity. Active fractions were combined, dialyzed against distilled water, concentrated by evaporating under vacuum, and was applied on the column (1.3 × 50 cm) with Sepharose 6B. The active fractions were combined and dialyzed against 0.1 M PCB and pH 5.2, and the dialysate was used as the purified naringinase throughout this study.
Molecular Mass Determination
Determining the enzymes molecular mass in the native system was performed by the gel filtration on the column (1.3 × 50 cm) with Sepharose 6B. Elution was performed by the 0.01 M phosphate buffer (pH 6.0) with 0.1 M NaCl. Standard curve for molecular mass calculations was plotted using high-molecular protein markers (“Pharmacia,” Sweden): ribonuclease (13.7 kDa), proteinase K (25 kDa), ovalbumin (43 kDa), and bovine serum albumin (67 kDa). Sodium dodecyl sulfate (SDS)-denatured proteins were separated by electrophoresis (Phast-System; Pharmacia, Uppsala, Sweden) on commercially available polyacrylamide gels (Pharmacia, Uppsala, Sweden) and were stained with a 0.4% silver nitrate solution (Silver Stain kit; Pharmacia, Sweden). The protocols for the separation methods and gels are described by the manufacturer’s instructions (Phast-System; Pharmacia, Uppsala, Sweden). For molecular mass estimation, the following markers were used: bovine serum albumin (67.0 kDa), ovalbumin (43.0 kDa), carboanhydrase (30.0 kDa), and trypsin inhibitor (20.0 kDa).
Analytical Methods
Protein concentration was determined according to Lowry et al. [20], using bovine serum albumin as standard. Neutral sugars were measured according to Dubois et al. [21]. Identification of neutral monosaccharides was carried out after hydrolysis of the preparation with 2 M HCl (100 C for 5 h). Monosaccharides were analyzed in the form of polyol acetates [22] and were performed on an Agilent 6890N/5973N chromatograph-mass spectrometer (USA) equipped with a DB-225mS column (30 m × 0.25 mm × 0.25 μm); the carrier gas was helium at a flow rate of 1 ml/min; the temperatures of evaporator and interface were 250 and 280 °C (isothermal regime). Amino acid and hexosamine content was determined by the amino acid analyzer “Hitachi KLA-5” (Japan).
Optimal pH and Temperature and Stability of the Naringinase
The optimum pH for the activities of naringinase was determined by incubating the enzyme preparation with naringine in 0.1 M сitrate, PCB, and 0.01 M Tris-HCl buffers at the pH range from 2 to 9. The pH stability was evaluated by preincubation of the enzymes in PCB over a pH range from 4 to 6 at 37 °C. Activities were measured at 24 h using the standard protocol. The optimum temperature of naringinase was determined by incubating the assay mixture for 30 min at temperature ranging from 5 to 80 °C. Thermal stability was measured by preincubation of the enzymes at the optimum pH at different temperatures (30, 37, 50, 60, and 70 °C) with the exposition time of 90 min.
Thermal inactivation of naringinase was studied at 65 °C and pH 5.2 (0.1 M PCB). The enzyme samples (3 U/ml) in 3 ml of the appropriate buffer were kept at given temperature for 1.5–3 h; the aliquots in 0.1 ml were collected in definite intervals (10–30 min) for measurement of residual activity. Bovine serum albumin (BSA) and L-rhamnose at concentration 0.5% were used to stabilize enzyme preparations.
Enzyme treatment with glutaraldehyde was carried out as follows: 10 μl of 25% glutaraldehyde solution was added to 1 ml of the purified enzyme solution (8 U/ml) and the mixture was incubated at room temperature for 60 min. The remaining reagent was removed by gel filtration on Sepharose 6B. Thermal inactivation was carried out as described above.
Kinetic Constants
Kinetic experiments were carried out at 37 °C at the optimal pH. The maximum rate (V max) and Michaelis constant (K m) were determined according to Lineweaver-Burk from curves defining dependence of the enzyme reaction rate on the substrate level (from 0.1 to 8 mM). Inhibition studies were performed using L-rhamnose at concentrations ranging from 1 to 10 mM.
Substrate Specificity
Neohesperidin and naringin (0.5–1 mM), the two flavonoids, and synthetic p-nitrophenyl substrates (0.4 mM) were used as substrates to find the naringinase substrate specificity.
Effect of Various Compounds on the Enzyme Activities
Reactions were carried out for 60 min at 37 °C and pH 5.2 (0.1 M PCB) in the presence or absence of the compounds examined.
Photooxidating was carried out upon various values of pH (3.0–6.0) and temperature (20–50 °C). As a source of light, a filament lamp (glow lamp) (200 wt) with the red color filter was placed 15 cm over the solution surface. 5 × 10−6 M methylene blue was used as a photosensibilisator. Photoactivation was carried out in a protected from light and in an open (illuminated) thermostat. Aliquots were taken at regular intervals and their enzymatic activity was measured. Control samples contained the same amount of photosensibilisator as experimental but unlike of latter kept in the darkness.
Enzymatic Hydrolysis of Grapefruit Juice
Grapefruit juice samples were obtained from fresh grapefruits. The juice samples were extracted and filtrating to remove the seeds and pomaces. Stock solution of naringin was prepared at a concentration of 1 mg/ml by dissolving in 50% aqueous ethanol. Enzyme solutions were prepared by dissolving naringinase in 0.1 M PCB (pH 5.2) at a concentration of 2 mg/ml (7 U). 0.1 ml of an enzyme solution was added to 2 ml of the juice sample. The reaction was performed at 40 and 50 °C for 60 min. After reaction, 1 ml juice or hydrolyzed sample was pipetted and vigorously mixed with 1 ml anhydrous ethanol. The mixture was then centrifuged at 10000 rpm for 10 min. The collected supernatant was filtered and further analyzed by HPLC. Standard curve was plotted using naringin.
HPLC Analysis
An Agilent 1200 HPLC coupled with a Zorbax SB C18 reverse phase column (2.5 × 150 mm, 3.5 μm) and diode matrix detector was used for analysis. The mobile phase was acetonitrile-water (25:75, v/v) programmed at a flow rate of 0.3 ml/min, the injection volume was 3 μL, and the column was at ambient temperature. Naringin in samples was detected at 280 nm and identified by comparison of retention times with standard.
Results
Purification
Enzyme preparation demonstrating naringinase activity was purified from the C. albidus liquid culture grown on L-rhamnose as the sole carbon source. The steps of the purification procedure are presented in Table 1. The specific naringinase activity of the fraction was found to be 12.5 U/mg, with approximately 42-fold increase in specific activity and a level of recovery of 0.7% for naringinase. The purified enzyme was confirmed to be homogeneous by SDS-PAGE and gel filtration on Sepharose 6B column.
Molecular Mass and Characterization of C. albidus Naringinase
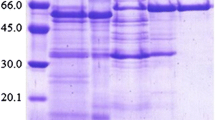
It is known that molecular masses of microbial naringinases can range widely. Fungi normally produce glycosidases of monomeric structure, with the molecular mass of about 70–100 kDa. In our study, molecular mass of 50 kDA for C. albidus naringinase was determined in the native system by gel filtration on Sepharose 6В. The SDS-PAGE of this fraction on an 8 to 25% polyacrylamide gradient gel showed a single protein band after being stained with silver nitrate (Fig. 1). Electrophoretic investigations also revealed enzyme with a molecular mass of 50 kDa.

Sodium dodecyl sulfate polyacrylamide gel electrophoresis of the purified naringinase C. albidus. Line 1. Molecular mass standards including bovine serum albumin (67.0 kDa), ovalbumin (43.0 kDa), carboanhydrase (30.0 kDa), and trypsin inhibitor (20.0 kDa). Lane 2. The purified enzyme
The naringinase tested contains the predominant amino acids (Table 2): asparaginic acid (14.84%), glutamic acid (17.5%), glycine (15.48%), alanine (8.99%), serine (5.65%), leucine (6.26%), and threonine (5.28%). It is shown that carbohydrate component (5%) is also present in the naringinase preparation. Monosaccharide composition was represented by mannose, galactose, rhamnose, ribose, arabinose, xylose, and glucose, in ratio 13:3:1:4:22:5:3, respectively.
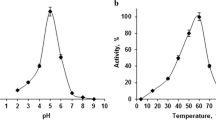
Optimum pH for the hydrolysis of naringine by C. albidus naringinase was 5.0 at 37 °С (Fig. 2), optimum temperature—60 °С (Fig. 3). Stability of enzyme was unchanged in the pH range from 3.5 to 6.0 at 20 °С during 48 h (Fig. 4). Being incubated at 60 °С during 180 min naringinase retained 100% of the activity. Also, high level of stability was shown for this enzyme if stored as 3 M solution of ammonium sulfate. Specifically, during 2 years of storage at 4 °С, no reduction of activity was determined. However, more than 50% of activity is usually being lost during lyophilization.

Effects of pH on naringinase activity (37 °C)

Effects of temperature on the activity of naringinase (pH 5.2)

Effect of pH on stability of the naringinase at 20 °C
The experiments with purified preparations showed that the introduction of 0.5% neutral protein such as BSA to the reaction mixture stabilized naringinase in thermal denaturation conditions increasing the enzyme half-life by 50% (Fig. 5). It was also shown that thermal inactivation of C. albidus naringinase was accelerated in the presence of 0.5% rhamnose (Fig. 5). Addition of 0.25% glutaraldehyde to the enzyme preparation facilitated formation of the stable enzyme conformation, which remained stable at 65 °C (Fig. 5).

Thermal stability of C. albidus naringinase, pH 5.2; t = 65 °C: control (white circle); activity of naringinase in the presence of rhamnose 0.5% (black square); activity of naringinase in the presence of BSA 0.5% (white square); activity of naringinase in the presence of glutarealdehyde 0.25% (black triangle)
Substrate Specificity
According to kinetic analysis data, obtained enzyme preparation showed high affinity to its natural substrates—naringin and neohesperidin, which are characterized by the presence of L-rhamnose bound to glycon via α-1,2 bond (Table 3). K m values for the hydrolysis by naringinase were 0.77 mM for naringin and 3.3 mM for neohesperidin. Naringinase also demonstrated narrow spectrum of activity towards synthetic substrates (Table 3).
Studies of the time of naringin hydrolysis at various temperatures showed (Fig. 6) that C. albidus naringinase completely hydrolyzed 2 mM naringin during 2–3 h at 60 °C. The time of hydrolysis increased to 4 h when the temperature lowered to 50 °C.

Naringin hydrolysis by C. albidus naringinase at different temperatures: black triangle—60 °С, black square—50 °С, black circle—40 °С
Inhibition of Naringinase by Several Compounds
We found that L-rhamnose, D-glucose, D-mannose, D-fructose, and melibiose in a concentration of 1 М inhibit naringinase by 10–30% (Table 4). It is shown that the presence in the reaction medium from 0.5 M glucose naringinase activity is reduced by 20% (Fig. 7a). One of the important characteristics of quality wines is aromatic bouquet due to the presence of monoterpenes present in grapes, which must be hydrolyzed by naringinase. Since wine contains a percentage of ethanol, it would check its effect on the activity of enzymes. Naringinase of C. albidus retains to 75% of the activity in the presence of 20% ethanol in the reaction mixture (Fig. 7b).

Effects of glucose (a) and ethanol (b) concentrations on naringinase activity (20 °C, pH 5.2)
Debittering of Grapefruit Juice
The presence of flavonoids and limonoids in citrus fruits can have a significant effect on the quality of citrus juices, especially grapefruit and orange. The use of naringinase in the production of such juices allows the degradation of flavonoids such as naringin, thereby greatly improving the taste of the product.
In Fig. 8, the HPLC chromatograms show that as a result of enzymatic hydrolysis of grapefruit juice with C. albidus naringinase, the concentration of naringin is reduced by 84% at 40 °C and 100% at 60 °C after 60 min of incubation. Thus, the high activity of the enzyme towards the natural substrate is shown. These data suggest that naringinase from C. albidus can efficiently remove the bitterness of grapefruit juice by hydrolyzing naringin. Thus, we can conclude that naringinase of C. albidus has a high potential for use in the food industry.

HPLC chromatogram of naringin in grapefruit juice sample before hydrolysis by the naringinase (1) and after hydrolysis at 40 °C (2) and 50 °C (3). Concentration of naringin: 1—215.54 μg/ml; 2—35.45 μg/ml; 3—< 5 μg/ml
Effect of Different Chemicals on Naringinase Activity
Naringinase was stable in the presence of almost all metal ions used for the study. Ag+ significantly reduced activity of naringinase (on 72.5%). Activity of enzyme was inhibited in the presence of Mg2+ and Al3+ in a concentration of 10 mМ (Table 4). Nevertheless, we suppose that their inhibitory effect was nonspecific. The observed inhibitory effect of sulfite may indicate the presence of catalytically active SH groups.
We studied specific inhibitors and activators effect on the rate of naringinase activity. Thus, enzyme was inhibited by 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide (Table 4). Analysis of the effect of chelating agents (EDTA and о-phenanthroline) and sodium azide on enzyme activity shows that metal containing groups cannot be involved in catalysis of the naringinase. This result is in a good agreement with our previous studies of cations and anions effect. Dithiothreitol and β-mercaptoethanol at a concentration of 10 mM at 37 °C practically had not decreased the activity of naringinase (Table 4). However, we observed enzyme inactivation in their presence at 60 °C for 89 and 58.5%, respectively, compared to control. It was also shown that C. albidus naringinase is inactivated in the presence of methylene blue (Fig. 9). The observed enzyme photoinactivation is accelerated with the increase of pH and temperature. Being a photosensitizer, methylene blue promotes heterocycle of imidazol breakage and enzyme inactivation.

Photoinactivation of C. albidus naringinase in the methylene blue presence
Discussion
Naringinase catalyzes hydrolytic reaction with the releases of rhamnose and glucose from the substrate, and, thus, is the key enzyme complex in the modification and decomposition of rhamnose-related compounds. To date, only a small group of microorganisms has been shown to produce naringinase. This is the first time when the ability to produce naringinase has been shown for C. albidus. Naringinase activity of C. albidus may be presented either by complex of two enzymes, α-L-rhamnosidase and β-glucosidase [23], or by one enzyme with two active sites, as in A. niger naringinase [1]. The naringinase of C. albidus was purified 42 times with the yield of 0.7%. The enzyme possesses both α-L-rhamnosidase and β-glucosidase activities, but preparation did not contain any glycosidase or proteolytic activities. Based on the data about the correlation of α-L-rhamnosidase and β-glucosidase activity obtained in the process of separation and purification naringinase of C. albidus, we can speculate that this activity is presented by a single protein with two active sites for α-L-rhamnosidase and β-glucosidase activities. Data of the SDS-PAGE electrophoresis and gel filtration on Sepharose 6B support this conclusion. Further kinetic studies should definitively resolve the nature of naringinase C. albidus. The molecular weight of C. albidus naringinase was 50 kDa, which is slightly lower than yeast monomeric α-L-rhamnosidase of P. angusta (90 kDa) [16] and heterodimer naringinase of C. tropicalis (73 and 78 kDa) [15], but it is similar to the α-L-rhamnosidase of P. citrinum (51 kDa) [24] and Bacillus amyloliquefaciens [25]. Many reports have shown molecular weights of naringinase to range from 70 to 240 kDa [2, 3]. Among these enzymes were found monomeric α-L-rhamnosidase [26], homodimer—Aspergillus niger [6] and heterodimer forms—C. tropicalis [15], and Aspergillus aculeatus [7].
Purified naringinase was resistant to changes in reaction conditions. Enzyme was highly stable in the pH range of 3.5–6.0 and at 20–60 °C temperature range. Thermal stability of naringinase from C. albidus is similar to α-rhamnosidase from thermophilic bacteria [27] and micromycete A. kawachii [28], and significantly exceeds the thermal stability of the yeast rhamnosidase [15, 16]. The activity and stability of C. albidus naringinase in acidic and neutral pH at higher temperatures is an important property of the enzyme if considered for further practical use. Typically, yeast and bacterial rhamnosidases are more active in neutral and alkaline medium, which limits their use in processing of juices and wines. Unlike them, naringinase of C. albidus can be used in wider pH range, from low to high, during the processing of citrus fruits. Stability of the enzyme can be explained by high level of nonpolar hydrophobic amino acids (42%) in the protein molecule. High percentage of acidic amino acids (32%) in the preparation of C. albidus naringinase is also a feature of the fungal glycosidases. Many rhamnosidases have isoelectric point in acidic range: the α-L-rhamnosidase from P. augusta has isoelectric point at 4.9, pI α-L-rhamnosidase of A. terreus—4.6 [4, 11, 16]. Significant impact on the stability of molecule naringinase from C. albidus may have the carbohydrate component. Most fungal rhamnosidases contain a high percentage of carbohydrates, for example, rhamnosidase of P. decumbens contains 50% of carbohydrate [23]; in enzymes isolated from the genus Aspergillus, the carbohydrate content amounted 15–24% [11]. Carbohydrate component (5% by weight) in naringinase C. albidus was detected with mannose and arabinose (43 and 25% of total peak area, respectively) being predominant monosaccharides.
Some approaches to naringinase stabilization were investigated. Slowdown in thermal denaturation was observed in the presence of a neutral protein and after treatment with glutaraldehyde. Biocatalysts based on cross-linked aggregates exhibit enhanced stability and better stereochemical accessibility for immobilizing agents [29]. Glutaraldehyde is the most commonly used cross-linking agent owing to its ability to form polymers acting as cross-linkers with varying length of bridges. Our results showed that formation of a rigid structure of the enzyme active site may not be crucial for the activity of naringinase also possible aggregation of molecules preventing thermal denaturation.
The high thermal stability of the enzyme can significantly increase the efficiency of hydrolysis of rhamnoglycosides. It has been shown that the rate of naringin hydrolysis at 60 °C is twofold higher than at 50 °C, and under these conditions, the enzyme does not lose activity during 3 h. In this respect, C. albidus naringinase surpasses many fungal and yeast enzymes [4, 15, 16].
Naringinase of C. albidus, like most rhamnosidases, displays activity towards alpha-1,2-linked rhamnosides and exhibits the greatest affinity to naringin, while β-glucosidase activity presents 28% of max enzyme activity. It has also been shown that naringinase of C. albidus is able to effectively hydrolyze naturally occurring flavonoid (naringin) in grapefruit juice. The V max/K m ratio corroborates greater specificity of the C. albidus enzyme for naringin than p-NP-α-L-rhamnose, as typical for most α-rhamnosidases [11]. P. augusta α-L-rhamnosidase with similar activity towards naringin has been reported [16].
End product inhibition by L-rhamnose can significantly limit the rate of bioflavonoids hydrolysis. It is known that carbohydrates are competitive inhibitors of the most rhamnosidases [1, 11]. It was shown that activity of C. albidus naringinase decreases in the presence of 1 M rhamnose and glucose, but more than 80% activity was retained with 500 mM glucose present in the reaction medium. This glucose concentration exceeds normal content of this sugar in the grape juice (21% w/v). In addition, enzyme stability in the presence of ethanol (up to 20%) may indicate high potential for use of C. albidus naringinase in the processing of beverages and wines.
Studies of the effect of different anions, cations, and specific chemicals can help to make some assumptions about the enzyme catalytically active groups. These data also allow to develop methods for chemical stabilization of proteins. Our results provide ground for some conclusions about the functionally active groups of C. albidus naringinase. Participation of carboxyl group of C-terminal amino acid and imidazole group of histidine in the catalytic action of naringinase was proposed based on the inhibition and kinetic analysis. Important role of the carboxyl group in activity of naringinase enzyme is confirmed by inhibition of the enzyme activity by 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide. Involvement of sulphydryl groups in catalysis carried out by naringinase of C. albidus was proposed based on the inhibition of the enzyme activity by sulfite and thiol reagents such as dithiothreitol and β-mercaptoethanol. Cysteine residues located, presumably, outside of the active center of the enzyme, ensuring stability of C. albidus naringinase due to thiol-disulfide exchange. Inhibition of the enzyme activity by silver ions demonstrates carboxyl, imidazole, and sulfhydryl groups involvement in the catalysis. Partial inhibition indicates the presence of other amino acid residues on the protein surface that are important for enzymatic activity. These results are in agreement with the existing concept of the glycosidase active center structure. These data are consistent with those of structural studies of the family GH78 enzymes [30]. Glu636 is predicted to donate protons to the glycosidic oxygen, and Glu895 is the likely catalytic general base, activating the nucleophilic water and indicating that the enzyme operates through an inverting mechanism [31]. Several negatively charged residues, such as Asp567, Glu572, Asp579, and Glu841, conserved in GH family 78 enzymes, interact with rhamnose, and RhaB mutants of these residues have drastically reduced enzyme activity, indicating that their or its residues are crucial for enzyme catalysis and/or substrate binding [32].
The kinetic properties, thermostability, substrate specificity, and high tolerance to glucose and ethanol which are displayed by the C. albidus naringinase allow its use in industry. Identification of the catalytically important groups present in the enzyme active center helps to predict its behavior in reaction environment and subsequently leads to the increased efficiency of enzymatic processing used in biotechnological industry.
References
Puri, M., & Banerjee, U. C. (2000). Production, purification, and characterization of the debittering enzyme naringinase. Biotechnology Advances, 18, 207–217.
Ribeiro, M. H. (2011). Naringinase: occurrence, characteristics, and applications. Applied Microbiology and Biotechnology, 90, 1883–1895. https://doi.org/10.1007/s00253-011-3176-8.
Puri, M. (2012). Updates on naringinase: structural and biotechnological aspects. Applied Microbiology and Biotechnology, 93, 49–60. https://doi.org/10.1007/s00253-011-3679-3.
Gallego, M. V., Pinaga, F., Ramon, D., & Valles, S. (2001). Purification and characterization of an α-L-rhamnosidase from Aspergillus terreus of interest in winemaking. J. Food Science, 66, 204–209.
Eliades, L. A., Rojas, N. L., Cabello, M. N., Voget, C. E., & Saparrat, M. C. (2011). α-L-Rhamnosidase and β-D-glucosidase activities in fungal strains isolated from alkaline soils and their potential in naringin hydrolysis. Journal of Basic Microbiology, 51, 659–665. https://doi.org/10.1002/jobm.201100163.
Ni, H., Chen, F., Cai, H., Xiao, A., You, Q., & Lu, Y. (2012). Characterization and preparation of Aspergillus niger naringinase for debittering citrus juice. Journal of Food Science, 77, 1–7. https://doi.org/10.1111/j.1750-3841.2011.02471.x.
Chen, Y., Ni, H., Chen, F., Cai, H., Li, L., & Su, W. (2013). Purification and characterization of a naringinase from Aspergillus aculeatus JMUdb058. Journal of Agricultural and Food Chemistry, 61, 931–938. https://doi.org/10.1021/jf303512q.
Michlmayr, H., Brandes, W., Eder, R., Schümann, C., del Hierro, A. M., & Kulbe, K. D. (2011). Characterization of two distinct glycosyl hydrolase family 78 alpha-L-rhamnosidases from Pediococcus acidilactici. Applied and Environmental Microbiology, 77, 6524–6530.
Mukund, P., Belur, P. D., & Saidutta, M. B. (2014). Production of naringinase from a new soil isolate, Bacillus methylotrophicus: isolation, optimization and scale-up studies. Preparative Biochemistry & Biotechnology, 44, 146–163. https://doi.org/10.1080/10826068.2013.797910.
Puri, M., Kaur, A., Barrow, C. J., & Singh, R. S. (2011). Citrus peel influences the production of an extracellular naringinase by Staphylococcus xylosus MAK2 in a stirred tank reactor. Biotechnology Relevant Enzymes Proteins, 89, 715–722.
Yadav, V., Yadav, P. K., Yadav, S., & Yadav, K. D. S. (2010). Alpha-L-rhamnosidase: a review. Process Biochemistry, 45, 1226–1235.
Singh, P., Sahota, P. P., & Singh, R. K. (2015). Evaluation and characterization of new α-L-rhamnosidase-producing yeast strains. The Journal of General and Applied Microbiology, 61, 149–156. https://doi.org/10.2323/jgam.61.149.
Li, L., Ni, H., Xiao, A., & Cai, H. (2010). Characterization of Cryptococcus sp. jmudeb008 and regulation of naringinase activity by glucose. Wei Sheng Wu Xue Bao, 50, 1202–1207.
Sahota, P. P., & Kaur, N. (2015). Characterization of enzyme naringinase and the production of debittered low alcoholic kinnow (Citrus raticulata blanco) beverage. International Journal of Advanced Research, 36, 1220–1233.
Saranya, D., Shahmugam, S., Kumar, S., Thayumanavan, B., & Rajasekaran, P. (2009). Purification and characterization of naringinase from Candida tropicalis. Advance Biotechnology, 9, 11–13.
Yanai, T., & Sato, M. (2000). Purification and characterization of an alpha-L-rhamnosidase from Pichia angusta X349. Bioscience, Biotechnology, and Biochemistry, 64, 2179–2185. https://doi.org/10.1271/bbb.64.2179.
Rzaieva, O. M., Varbanets, L. D., & Nagorna, S. S. (2010). Screening of producers of α-L-rhamnosidase among yeasts. Microbiology Zhurnal, 72, 11–17.
Davis, D. W. (1947). Determination of flavonones in citrus juice. Analytical Biochemistry, 19, 476–478.
Chaplin, M. E., & Kennedy, J. E. (Eds.). (1986). Carbohydrate analysis: a practical approach. Washington: Oxford IRL Press.
Lowry, O. H., Rosebrough, N. J., Farr, A. L., & Randall, R. J. (1951). Protein measurement with the Folin phenol reagent. The Journal of Biological Chemistry, 193, 265–275.
Dubois, M., Gilles, K. A., & Hamilton, J. K. (1956). Colorimetric method for determination of sugars and related substances. Analytical Chemistry, 28, 350–356.
Albersheim, P., Nevins, D. J., English, P. D., & Karr, A. (1967). A method for the analysis of sugars in plant cell-wall polysaccharides by gas-liquid chromatography. Carbohydrate Research, 5, 340–345. https://doi.org/10.1016/S0008-6215(00)80510-8.
Young, N. M., Johnston, R. A. Z., & Richards, J. C. (1989). Purification of α-L-rhamnosidase from Penicillium decumbens and characterization of two glycopeptides components. Carbohydrate Research, 191, 53–62.
Yadav, S., Yadav, V., Yadav, S., & Yadav, K. D. S. (2012). Purification, characterisation and application of α-L-rhamnosidase from Penicillium citrinum MTCC-8897. International Journal of Food Science and Technology, 47, 290–298.
Zhu, Y., Jia, H., Xi, M., Xu, L., Wu, S., & Li, X. (2017). Purification and characterization of a naringinase from a newly isolated strain of Bacillus amyloliquefaciens 11568 suitable for the transformation of flavonoids. Journal of Food Chemistry, 214, 39–46.
Ichinose, H., Fujimoto, Z., & Kaneko, S. (2013). Characterization of an α-L-rhamnosidase from Streptomyces avermitilis. Bioscience, Biotechnology, and Biochemistry, 77, 213–216.
Birgisson, H., Wheat, J. O., Hreggvidsson, G. O., Kristjansson, J. K., & Mattiason, B. (2004). Immobilization of a recombinant E. coli producing a thermostable rhamnosidase: creation of a bioreactor for hydrolysis of naringin. Enzyme and Microbial Technology, 40, 1181–1187.
Koseki, T., Mese, Y., Nishibori, N., Masaki, K., Fuhii, T., Handa, T., Yamane, Y., Shiono, Y., Murayama, T., & Iefuji, H. (2008). Characterization of an α-L-rhamnosidase from Aspergillus kawachii. Applied Microbiology and Biotechnology, 80, 1007–1013.
Ribeiro, M. H., & Rabaca, M. (2011). Cross-linked enzyme aggregates of naringinase: novel biocatalysts for naringin hydrolysis. Enzyme Research, 2011, ID851272. https://doi.org/10.4061/2011/851272.
Fujimoto, Z., Jackson, A., Michikawa, M., Maehara, T., Momma, M., Henrissat, B., Gilbert, H., & Kaneko, S. (2013). The structure of a Streptomyces avermitilis α-L-rhamnosidase reveals a novel carbohydrate-binding module CBM67 within the six-domain arrangement. The Journal of Biological Chemistry, 288, 12376–12385. https://doi.org/10.1074/jbc.M113.460097.
O'Neill, E. C., Stevenson, C. E. M., Paterson, M. J., Rejzek, M., Chauvin, A.-L., Lawson, D. M., & Field, R. A. (2015). Crystal structure of a novel two domain GH78 family α-rhamnosidase from Klebsiella oxytoca with rhamnose bound. Proteins, 83, 1742–1749. https://doi.org/10.1002/prot.24807.
Cui, Z., Maruyama, Y., Mikami, B., Hashimoto, W., & Murata, K. (2007). Crystal structure of glycoside hydrolase family 78 α-L-rhamnosidase from Bacillus sp. GL1. Journal of Molecular Biology, 374, 384–398. https://doi.org/10.1016/j.jmb.2007.09.003.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Borzova, N., Gudzenko, O. & Varbanets, L. Purification and Characterization of a Naringinase from Cryptococcus albidus . Appl Biochem Biotechnol 184, 953–969 (2018). https://doi.org/10.1007/s12010-017-2593-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-017-2593-2