
Abstract
An alpha-galactosidase was purified from Pseudobalsamia microspora (PMG) to 1224.1-fold with a specific activity of 11,274.5 units/mg by ion-exchange chromatography and gel filtration. PMG is a monomeric protein with a molecular mass of 62 kDa as determined by SDS-PAGE and by gel filtration. Chemical modification using N-bromosuccinimide (NBS) resulted in a complete abrogation of the activity of PMG, suggesting that Trp is an amino acid essential to its activity. The activity was strongly inhibited by Hg2+, Cd2+, Cu2+, and Fe3+ ions. Three inner peptide sequences for PMG were obtained by liquid chromatography–tandem mass spectrometry (LC–MS–MS) analysis. When 4-nitrophenyl α-d-glucopyranoside (pNPGal) was used as substrate, the optimum pH and temperature of PMG were 5.0 and 55 °C, respectively. The Michaelis constant (K m) value of the alpha-galactosidase on pNPGal was 0.29 mM, and the maximal velocity (V max) was 0.97 μmol ml−1 min−1. Investigation by thin-layer chromatography (TLC) demonstrated its ability to hydrolyze raffinose and stachyose. Hence, it can be exploited in degradation of non-digestible oligosaccharides from food and feed industries.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Instruction
Pseudobalsamia microspora is a fungus first described in the USA by Diehl and Lamber (1930) from material received in the winter of 1929 from a mushroom grower in Ohio. It is a competitive infectious microbe noted in the cultivation of Agaricus bisporus and Pleurotus ostreatus, initially found in the north and west of the USA and subsequently introduced into China by strains carrying the microbe. P. microspore has a rapid growth rate and was able to completely occupy the mushroom beds, thus discouraging growth of mushrooms. This disease was unknown before 1927. It was introduced into the mushroom beds with the casing soil [1]. In recent years, since research on P. microspore is mainly focused on the prevention and treatment of diseases in the edible fungus, it is of great significance to study the active substance of P. microspore to understand and use the infectious microbe.
Alpha-galactosidases are categorized as alpha-d-galactoside galactohydrolases (EC 3.2.1.22), according to the general classification based on the catalytic activity of the enzymes [2]. They are exoglycosidases that catalyze the hydrolysis of the terminal non-reducing −1.6-linked-galactose residues from oligosaccharides.
Alpha-galactosidases are widely distributed in microorganisms, animals, and plants and have been extracted and purified from microorganisms [3] and plants [4] especially from seed tissues containing large amounts of sugars from the raffinose family.
Alpha-galactosidases have a variety of biotechnological, medical, and industrial applications. Fabry disease, a lysosomal storage disorder caused by accumulation of glycosphingolipids in body fluids and tissue lysosomes [5], is alleviated by the action of alpha-galactosidase. Besides, alpha-galactosidases are able to remove the terminal galactose residues from glycans and thus are helpful in converting blood group B to blood group O [6]. Alpha-galactosidases also hydrolyze raffinose oligosaccharides, which are widely distributed in soy products and cause flatulence. Alpha-galactosidase treatment for reducing stachyose and raffinose content in the soybean flour has been investigated. This can be utilized effectively for improving the nutritional quality of soy-based foods on a large scale, which could be one of the best alternatives for fulfilling the protein requirement in the lactose-intolerant population [7].
In the present study, we reported, for the first time, the isolation and characterization of an alpha-galactosidase from P. microspora (PMG). We also noted the effect of PMG on degradation of raffinose oligosaccharides. This work would lay the foundation for future investigations on the biological applications of PMG and provide a scientific basis and possibility for converting the damage brought about by the infectious microbe P. microspora into something of value.
Materials and Methods
Materials
P. microspora was picked from the button mushroom A. bisporus. Diethylaminoethyl (DEAE)-cellulose and Q-sepharose were obtained from Sigma Chemical Company, USA. Mono Q 4.6/100 PE, Superdex 75 HR 10/30, and AKTA Purifier were from GE Healthcare, USA. The substrates, pNPGal, locust bean gum, guar gum, melibiose, stachyose, and raffinose were purchased from Sigma Chemical Company (St. Louis, MO, USA). Bicinchoninic Acid (BCA) Protein Assay Kit was purchased from Sigma-Aldrich Company. All other chemicals used were of analytical grade unless otherwise stated.
Enzyme Activity Assay and Protein Determination
The activity of alpha-galactosidase was routinely assayed as described by Malhotra and Dey (1967) using 4-nitrophenyl α-d-glucopyranoside (pNPGal) as substrate with slight modifications. Fifty microliters of 10 mM pNPGal were incubated with diluted enzyme (50 μl) at 50 °C for 10 min, and the reaction was suspended by adding 400 μl of 0.5 M Na2CO3, and in the control, the reaction was terminated by adding 400 μl of 0.5 M Na2CO3 before incubation, and the released p-nitrophenol was determined spectrophotometrically at 405 nm. One unit (U) of alpha-galactosidase activity was defined as the amount of enzyme liberating 1.0 μmol of p-nitrophenol per minute under the assay conditions described above.
Protein concentration was determined as described by Brown et al. [8] with Bicinchoninic Acid (BCA) Protein Assay Kit purchased from Sigma-Aldrich Company, using bovine serum albumin (BSA) as a standard. In the standard assay, 20 parts of the BCA working reagent (prepared by mixing 50 parts of reagent A with 1 part of reagent B) were mixed with 1 part of a protein sample. The sample was either a blank, a BSA protein standard, or an unknown sample. The blank consisted of buffer with no protein. The BSA protein standard consisted of a known concentration of bovine serum albumin, and the unknown sample was the solution to be assayed. BCA assays were routinely performed at 37 °C for 30 min. The absorbance at 562 nm was recorded, and the protein concentration was determined with reference to a standard curve.
Purification of PMG
P. microspora (50 g) was smashed in distilled water (10 ml g−1) using a pounding machine. The homogenate was centrifuged at 1000 rpm for 10 min after overnight extraction at 4 °C. After adjusting the supernatant concentration to 10 mM with 0.2 M phosphate buffer solution (pH 6.4), it was applied on a 2.5 cm × 20 cm column of DEAE-cellulose previously equilibrated and subsequently eluted with 10 mM phosphate buffer solution (pH 6.4) to remove unadsorbed materials (fraction D1). The column was eluted successively with 0.05, 0.15, 0.3, and 1 M NaCl in the sample buffer to yield fractions D2, D3, D4, and D5. After dialysis against distilled water, fraction D4 was collected and was loaded on a column of Q-sepharose (0.5 cm × 10 cm) which had previously been equilibrated with 10 mM phosphate buffer (pH 6.4). Unbound material was eluted with the same buffer (fraction Q1), and then, adsorbed materials were eluted with a linear 0–1 M NaCl gradient in the starting buffer. The active fraction Q3 was dialyzed and freeze-dried. The dried sample was dissolved in 10 mM NaAc-HAc buffer (pH 5.0), and the active fractions from Q-sepharose were respectively loaded on a Mono Q 4.6/100 PE column after centrifugation at 12,000 rpm for 10 min. This process was carried out with the help of an AKTA Purifier (GE Healthcare, USA). The active fraction Mono Q2 was collected, dialyzed, lyophilized, and dissolved in 10 mM NaAc-HAc buffer (pH 5.0) and further separated on a Superdex G-75 HR10/30 column by fast protein liquid chromatography to yield PMG.
Determination of Molecular Mass and Amino Acid Sequence
The purified alpha-galactosidase was subjected to SDS-PAGE as described by Laemmli and Favre [9]. A 12 % resolving gel and a 5 % stacking gel were used for molecular mass determination. The purified protein bands were excised and dispatched to Hui Jun Biotechnology Company (Guangzhou, China) for identification of inner amino acid sequences by liquid chromatography–tandem mass spectrometry (LC–MS–MS). The electrophoretic mobility of the protein was compared with those of protein markers for molecular mass determination.
Gel filtration on an FPLC-Superdex 75 column, which had been calibrated with molecular mass markers (GE Healthcare), was also employed to determine the molecular mass and the number of subunits by comparison with the molecular mass determined by SDS-PAGE.
Biochemical Characteristics of the Enzyme
The effect of pH on the enzyme was evaluated by measuring the activity of alpha-galactosidase using pNPGal as substrate in 0.1 M Mcilvaine’s buffer (pH 2.0–8.0) and 0.1 M NaHCO3-Na2CO3 buffer (pH 9.18–10.14). The reaction mixture was incubated for 10 min at 50 °C. The optimum temperature was determined by conducting the enzymatic hydrolysis reaction at different temperatures, ranging from 4 to 60 °C for 10 min. And the alpha-galactosidase activity was analyzed by determining A 405 as described above.
The assays of the effects of different metal ions (Na+, K+, Ca2+, Cd2+, Cu2+, Hg+, Mg2+, Mn2+, Pb2+, Zn2+, Al3+, and Fe3+ ions) and chemical modification regents (N-bromosuccinimide (NBS), dithiothreitol (DTT), diacetyl (DIC), and carbodiimide (EDC)) on PMG were performed by incubating the alpha-galactosidase with different concentrations of these metal ions and chemical modification regents for 60 min at 4 °C, and then, the residual activity was determined by comparing with the control in which the enzyme was assayed without the additions of regents.
Substrate Specificity and Kinetic Parameters
Substrate specificity was tested toward synthetic substrates such as pNPGal, oNPGal, 4-nitrophenyl-β-d-glucuronide (10 mM) in 0.1 M NaAc-HAc buffer (pH 4.6) at 50 °C for 10 min. Enzyme activity was estimated by measuring the amount of p-nitrophenol released at 405 nm as described earlier. Natural substrates including oligosaccharides such as raffinose and stachyose and some polysaccharides including guar gum and locust bean gum were assayed in a reaction mixture containing 50 μl of suitably diluted alpha-galactosidase enzyme and 50 μl of a 0.1 M substrate solution in Mcilvaine’s buffer (pH 5.0). The amount of reducing sugar produced was determined by adding 150 μl 3,5-dinitrosalicylic acid reagent as described by Miller [10]. Sucrose was used as the control for the raffinose or stachyose assay. One unit of alpha-galactosidase activity was defined as the amount of enzyme that brings about the release of 1 μmol of reducing sugar per minute under the assay conditions. The activity with melibiose as substrate was determined as described above, and the glucose released was estimated using a commercially available kit (Beijing BHKT Clinical Reagent Co., Ltd.) based on a glucose oxidase–peroxidase (GOD–POD) method as described by Dwevedi and Kayastha [11]. One unit of enzyme activity was defined as the amount of enzyme releasing 1 μmol of glucose per minute at 50 °C.
The Michaelis–Menten constant (K m) and maximal velocity (V max) of the PMG were determined based on different pNPGal concentrations (1–10 mM). The K m and V max of the enzyme were calculated based on the Lineweaver–Burk plot constructed by plotting the reciprocal of the substrate concentration on the x-axis and the reciprocal of the enzyme reaction velocity on the y-axis. All determinations were performed in triplicate at pH 4.6 and 50 °C.
Enzymatic Hydrolysis of Oligosaccharides
A mixture of 50 μl of purified enzyme, 30 μl deionized water, and 20 μl of raffinose (60 mM) in 0.1 M acetate buffer, pH 5.0, or stachyose (60 mM) in 0.1 M acetate buffer, pH 6.0, was incubated for various durations at 50 °C. The hydrolysates were then analyzed qualitatively by thin-layer chromatography (TLC). The reaction mixtures were spotted onto a silica gel plate (Merck Silica Gel 60 F254, Germany) and developed by ascending chromatography using containing propanol/acetic acid/water at 1:1.5:0.1 (v/v/v) [12] as the solvent system. The sugar spots were located by heating in an electric plate after spraying a mixture of diphenylamine/aniline/acetone/phosphate (1:1:50:10, m/v/v/v).
Results and Discussion
Chromatographic Purification of PMG
The P. microspora extract was fractionated by ion exchange chromatography on DEAE-cellulose. Alpha-galactosidase activity was concentrated in fraction D4 (Fig. 1a). The active fraction was subsequently separated into four fractions Q1, Q2, Q3, and Q4 on a Q-sepharose column which was eluted with a linear gradient of 0–1 M NaCl (Fig. 1b), Alpha-galactosidase activity resided in fraction Q4. Subsequently, fraction Q4 was purified on a Mono Q 4.6/100 PE column (Fig. 1c). Fraction Mono Q2 with alpha-galactosidase activity was then resolved on a Superdex 75 HR 10/30 column into a large peak SU1 with alpha-galactosidase activity (Fig. 1d).

Elution curves of PMG from Pseudobalsamia microspora. a Ion exchange chromatography on DEAE-cellulose (2.5 cm × 20 cm). The column had been equilibrated with 10 mM phosphate buffer (pH 6.4) and was eluted with 0.05, 0.15, 0.3, and 1 M NaCl in the sample buffer to yield fractions D2, D3, D4, and D5. b Fraction D4 from DEAE-cellulose was applied on a 0.5 cm × 10 cm Q-sepharose column. Starting buffer for eluting fraction D4: 10 mM phosphate buffer (pH 6.4). Buffer for eluting fractions D4: a linear NaCl concentration (0–1 M) gradient in the same buffer. c Ion exchange chromatography on Mono Q 4.6/100 PE. Sample: proteins obtained from fraction Q4. The column was eluted with a linear NaCl concentration (0–1 M) gradient in 10 mM NaAc-HAc buffer (pH 5.0). d Gel filtration of fraction Mono Q2 on a Superdex 75 column, which was eluted with 0.1 M NaCl in 10 mM NaAc-HAc buffer (pH 5.0). Flow rate: 0.4 ml per min. Fraction SU1 was enriched in alpha-galactosidase activity
The results of the purification showed that 1224.1-fold purification of the enzyme with a specific activity of 11,275 units/mg was achieved from P. microspora (Table 1).
Determination of Molecular Mass and Amino Acid Sequence
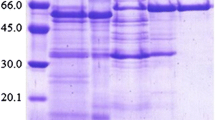
The molecular mass of PMG was deduced to be 62 kDa by SDS-PAGE (Fig. 2), which was the same as the molecular mass determined by gel filtration on Superdex 75 (Fig. 1d). It indicated that the purified alpha-galactosidase was a monomeric protein with a molecular mass of 62 kDa, which is similar to alpha-galactosidase from the culture filtrate of Mortierella vinacea, a monomeric protein with a molecular mass 51–62 kDa, as estimated by SDS-PAGE [13], but different from those of alpha-galactosidases from Coriolus versicolor (40 kDa) [14], Neosartorya fischeri P1 (49.2 kDa) [15], Aspergillus foetidus ZU-G1 (106.3, 49.7, and 109.9 kDa, respectively) [3], and Pleurotus florida (99 kDa) [16].

SDS-PAGE of purified Pseudobalsamia microspora alpha-galactosidase. Left lane: purified alpha-galactosidase (2 μg). Right lane: molecular mass standards (GE Healthcare). From top downward: phosphorylase b (94 kDa), bovine serum albumin (66 kDa), ovalbumin (43 kDa), carbonic anhydrase (30 kDa), soybean trypsin inhibitor (20 kDa), and lactalbumin (14.4 kDa)
Three inner amino acid sequences were obtained by LC–MS–MS for alpha-galactosidase: IVTAATKLNTTGLQALGYQYVNIDDCWSVK, AKSSAFPSGIKALADYVHSR, and APLLIGCDIR. Database search using BLAST indicated that these three peptide sequences showed striking similarity with alpha-galactosidases from other sources. Peptide 1 manifested 100 % identity with Talaromyces purpurogenus alpha-galactosidase (accession number BAA22992) and demonstrated 64 % identity with a probable alpha-galactosidase (accession number Q9Y865.1) from Aspergillus niger, which belongs to the glycosyl hydrolase 27 family. Peptide 2 shared 95 % homology with Ricinus communis alpha-galactosidase (accession number XP_002519009). Besides, peptide 3 also exhibited 80 % identity with Saccharomyces cerevisiae alpha-galactosidase (accession number P04824.1) belonging to GH family 27.
Effects of pH and Temperature on PMG Activity
The optimum pH curve suggests that the alpha-galactosidase had maximal activity at pH 5.0 in Mcilvaine’s buffer (Fig. 3a) and exhibited 60 and 73 % of the maximum activity at pH 4.0 and 6.0, respectively. When the pH lied between 3.0 and 7.0, there was hardly any alpha-galactosidase activity remaining. A thermostable alpha-galactosidase from Lenzites elegans had an optimal pH at 4.5 [2]. The maximal activity of purified recombinant alpha-galactosidase from acidophilic Bispora sp. MEY-1 strain exhibited at pH 3.5 [17]. Similarly, the optimal pH of P. microspora alpha-galactosidase was in accord with alpha-galactosidase cloned from Penicillium sp. F63 CGMCC [18], which was optimally active at pH 5.0, and optimum pH for alpha-galactosidase activity from Aspergillus terreus GR was also at pH 5.0 [19]. Generally speaking, alpha-galactosidases from fungi such as Penicillium purpurogenum possess a low optimal pH (pH 4.0–6.0) [20].

Effects of pH and temperature on the activity of alpha-galactosidase from Pseudobalsamia microspora. a Effect of pH on alpha-galactosidase activity. Buffer: Na2HPO4–citric acid buffers. Results represent mean ± SD (n = 3). b Effect of temperature on alpha-galactosidase activity. Results represent mean ± SD (n = 3)
The optimal temperature of the purified alpha-galactosidase was at about 55 °C (Fig. 3b), which was higher than that of Lactobacillus fermentum (45 °C) [21] and P enicillium griseoroseum (45 °C) [22] but lower than that of alpha-galactosidase from A. foetidus ZU-G1 (60 °C) [3] and A. terreus GR (65 °C). From 20 to 55 °C, the activity rose steadily from 15.6 to 100 % of the maximal activity. Beyond 60 °C, the activity underwent a decline. When the temperature was 70 °C, there was almost no activity.
Effects of Metal Ions and Chemical Modification Regents on PMG Activity
Many metallic ions were inhibitors of the alpha-galactosidase from P. microspora, among them Cd2+, Hg2+, Fe3+, and Cu2+ ions strongly inhibited the activity of PMG at 1.25 mM. Similar to our results (Table 2), a novel alpha-galactosidase purified from Grus nigricollis feces was strongly inhibited by Ag+ and Hg2+ ions [23] and moderately inhibited by other metal ions such as Zn2+, K+, Pb2+, and Al3+ ions. Moreover, the inhibitory effects of K+, Zn2+, Mn2+, Pb2+, and Al3+ ions were stronger than those of Ca2+, Fe2 +, and Mg2+ ions.
PMG was completely inactivated by the chemical modification reagent N-bromosuccinimide (NBS) at 0.1 mM concentration. This finding suggests that Trp was located at the active site and was indispensable to the activity of PMG, reminiscent of the observation that alpha-galactosidase from Bacillus stearothermophilus was also completely inhibited by NBS [24]. Besides, DL-dithiothreitol (DTT) exhibited a slight inhibitory effect. On the contrary, other chemical modification reagents (EDC, DIC, DEPC) had a slight promoting effect on PMG. Relevant data are shown in Table 3.
Substrate Specificity and Kinetics Properties of P. microspora. PMG
As shown in Table 4, PMG showed the highest specificity toward pNPGal (100 %), but it had little or no activity toward oNPGal, 4-Nitrophenyl β-d-glucuronide. Compared to synthetic substrate pNPGal, the enzyme exhibited lower activity for natural substrates such as melibiose (15.4 %), stachyose (6.4 %), and raffinose (49.9 %). Generally, alpha-galactosidase showed higher activity for composite substrates than natural substrates, consistent with the findings of Ferreira et al. [25].
Kinetic constants were calculated from the initial rate of P. microspora PMG against pNPGal. Substrate concentrations between 1 and 10 mM were employed to estimate the kinetic parameters using the Lineweaver–Burk transformation of the Michaelis–Menten equation. The Michaelis–Menten constants (K m) and maximum velocities (V max) of the alpha-galactosidase on pNPGal, as determined from the double reciprocal plot, were 0.29 mM and 0.97 μmol ml−1 min−1, respectively (Fig. 4). The calculated K m of P. microspore toward alpha-galactosidase was lower than those of alpha-galactosidases from white chickpea [26], the deep-sea bacterium Bacillus megaterium [27], and white-rot fungus P. florida [16], but was higher than its counterpart from Phaseolus coccineus seeds [28].

Determination of K m and V max by a Lineweaver–Burk plot for the Pseudobalsamia microspora alpha-galactosidase (PMG) with PNPGal as substrate
Enzymatic Hydrolysis of Oligosaccharides
A time-course of hydrolysis of raffinose and stachyose in P. microspora is shown in Fig. 5. Raffinose was almost completely hydrolyzed to galactose and sucrose in just 30 min, while only a small amount of stachyose was hydrolyzed. The ability to rapidly hydrolyze raffinose could be explained by the fact that the enzyme displayed greater substrate specificity and catalytic efficiency for this sugar as compared to stachyose, which was different from the results from Priti katrolia, showing higher binding affinity to stachyose [12]. There are some reports which showed that alpha-galactosidases from other sources degraded RFOs. Alpha-galactosidase from Aspergillus oryzae DR-5 showed complete hydrolysis of stachyose and raffinose in soymilk in 3 h at pH 5.0 and 50 °C [29]. Mulimani et al. had reported that alpha-galactosidase from Gibberella fujikuroi revealed complete hydrolysis of raffinose and stachyose in 3 h in the following composition of oligosaccharides in a local soybean variety: sucrose, 5.53 %; raffinose, 1.95 %; and stachyose, 6.1 %, by TLC [30].

Thin-layer chromatography of the hydrolysis of raffinose and stachyose by alpha-galactosidase from Pseudobalsamia microspora: a enzymatically treated stachyose; b enzymatically treated raffinose
Conclusion
In this study, a new alpha-galactosidase from P. microspora was purified and characterized. The enzyme was a monomeric protein with a molecular mass of 62 kDa and was active in an acidic pH range which broadens the application of PMG and showed the highest activity at 55 °C. Cd2+, Hg2+, Fe3+, and Cu2+ ions strongly inhibited the activity of PMG. The chemical modification reagent NBS inhibited the activity of PMG at 0.1 mM concentration suggesting that Trp was located at the active site and was indispensable to PMG. Moreover, substrate specificity and use of TLC revealed preferential degradation activity of the enzyme toward raffinose than stachyose. These results lay a foundation for development of the application of PMG in terms of degradation of non-digestible oligosaccharides from food and feed industries. The investigation will be conducted to study the effects of P. microspora as feed additive composition to improve animal digestion and increase feed efficiency to make the best use of the infectious microbe.
References
Glasscock, H. H., & Ware, W. M. (1941). Investigations on the invasion of mushroom beds by Pseudobalsamia microspora. Annals of Applied Biology, 28, 85–90.
Sampietro, D., Quiroga, E., Sgariglia, M., Soberon, J., & Vattuone, M. A. (2012). A thermostable alpha-galactosidase from Lenzites elegans (Spreng.) ex Pat. MB445947: purification and properties. Antonie Van Leeuwenhoek, 102, 257–267.
Liu, C. Q., & He, G. Q. (2012). Multiple alpha-galactosidases from Aspergillus foetidus ZU-G1: purification, characterization and application in soybean milk hydrolysis. European Food Research and Technology, 234, 743–751.
Shen, W., Li, Y., Chen, H., Jin, Z., Xu, X., Zhao, J., & Xie, Z. (2009). Purification and application of alpha-galactosidase from germinating coffee beans (Coffea arabica). European Food Research and Technology, 228, 969–974.
Cox, T. M. (1996). The metabolic and molecular bases of inherited disease: Vols I, II and III (7th edn). Trends in Genetics, 12, 78–79.
Liu, Q. P., Sulzenbacher, G., Yuan, H., Bennett, E. P., Pietz, G., Saunders, K., Spence, J., Nudelman, E., Levery, S. B., White, T., Neveu, J. M., Lane, W. S., Bourne, Y., Olsson, M. L., Henrissat, B., & Clausen, H. (2007). Bacterial glycosidases for the production of universal red blood cells. Nature Biotechnology, 25, 454–464.
Singh, N., & Kayastha, A. M. (2013). A novel application of Cicer alpha-galactosidase in reduction of raffinose family oligosaccharides in soybean flour. Journal of Plant Biochemistry and Biotechnology, 22, 353–356.
Brown, R. E., Jarvis, K. L., & Hyland, K. J. (1989). Protein measurement using bicinchoninic acid—elimination of interfering substances. Analytical Biochemistry, 180, 136–139.
Laemmli, U. K., & Favre, M. (1973). Maturation of head of bacteriophage-T4. 1. DNA packaging events. Journal of Molecular Biology, 80, 575–599.
Miller, G. L. (1959). Use of dinitrosalicylic acid reagent for determination of reducing sugar. Analytical Chemistry, 31, 426–428.
Dwevedi, A., & Kayastha, A. M. (2009). Stabilization of beta-galactosidase (from peas) by immobilization onto amberlite MB-150 beads and its application in lactose hydrolysis. Journal of Agricultural and Food Chemistry, 57, 682–688.
Katrolia, P., Jia, H., Yan, Q., Song, S., Jiang, Z., & Xu, H. (2012). Characterization of a protease-resistant alpha-galactosidase from the thermophilic fungus Rhizomucor miehei and its application in removal of raffinose family oligosaccharides. Bioresource Technology, 110, 578–586.
Shibuya, H., Kobayashi, H., Sato, T., Kim, W. S., Yoshida, S., Kaneko, S., Kasamo, K., & Kusakabe, I. (1997). Purification, characterization, and cDNA cloning of a novel alpha-galactosidase from Mortierella vinacea. Bioscience Biotechnology and Biochemistry, 61, 592–598.
Du, F., Liu, Q., Wang, H., & Ng, T. (2014). Purification an alpha-galactosidase from Coriolus versicolor with acid-resistant and good degradation ability on raffinose family oligosaccharides. World Journal of Microbiology and Biotechnology, 30, 1261–1267.
Wang, H., Shi, P., Luo, H., Huang, H., Yang, P., & Yao, B. (2014). A thermophilic alpha-galactosidase from Neosartorya fischeri P1 with high specific activity, broad substrate specificity and significant hydrolysis ability of soymilk. Bioresource Technology, 153, 361–364.
Ramalingam, Saraswathy, N., Sadasivam, S., Subha, K., & Poorani, N. (2007). Purification and properties of alpha-galactosidase from white-rot fungus Pleurotus florida. Indian Journal of Biochemistry & Biophysics, 44, 76–81.
Wang, H., Luo, H., Li, J., Bai, Y., Huang, H., Shi, P., Fan, Y., & Yao, B. (2010). An alpha-galactosidase from an acidophilic Bispora sp. MEY-1 strain acts synergistically with beta-mannanase. Bioresource Technology, 101, 8376–8382.
Mi, S., Meng, K., Wang, Y., Bai, Y., Yuan, T., Luo, H., & Yao, B. (2007). Molecular cloning and characterization of a novel alpha-galactosidase gene from Penicillium sp. F63 CGMCC 1669 and expression in Pichia pastoris. Enzyme and Microbial Technology, 40, 1373–1380.
Shankar, S. K., Dhananjay, S. K., & Mulimani, V. H. (2009). Purification and characterization of thermostable alpha-galactosidase from Aspergillus terreus GR. Applied Biochemistry and Biotechnology, 152, 275–285.
Shibuya, H., Nagasaki, H., Kaneko, S., Yoshida, S., Park, G. G., Kusakabe, I., & Kobayashi, H. (1998). Cloning and high-level expression of alpha-galactosidase cDNA from Penicillium purpurogenum. Applied and Environmental Microbiology, 64, 4489–4494.
Garro, M. S., DeValdez, G. F., Oliver, G., & DeGiori, G. S. (1996). Purification of alpha-galactosidase from Lactobacillus fermentum. Journal of Biotechnology, 45, 103–109.
Falkoski, D. L., Guimaraes, V. M., Callegari, C. M., Reis, A. P., de Barros, E. G., & de Rezende, S. T. (2006). Processing of soybean products by semipurified plant and microbial alpha-galactosidases. Journal of Agricultural and Food Chemistry, 54, 10184–10190.
Zhou, J., Pan, L., Li, J., Tang, X., & Huang, Z. (2012). A novel alpha-galactosidase from Arthrobacter sp. GN14 isolated from Grus nigricollis feces: gene cloning, heterologous expression and characterization. Wei Sheng Wu Xue Bao = Acta Microbiologica Sinica, 52, 611–619.
Gote, M. M., Khan, M. I., & Khire, J. M. (2007). Active site directed chemical modification of α-galactosidase from Bacillus stearothermophilus (NCIM 5146): involvement of lysine, tryptophan and carboxylate residues in catalytic site. Enzyme and Microbial Technology, 40, 1312–1320.
Ferreira, J. G., Reis, A. P., Guimaraes, V. M., Falkoski, D. L., Fialho, L. D. S., & de Rezende, S. T. (2011). Purification and characterization of Aspergillus terreus alpha-galactosidases and their use for hydrolysis of soymilk oligosaccharides. Applied Biochemistry and Biotechnology, 164, 1111–1125.
Singh, N., & Kayastha, A. M. (2012). Purification and characterization of alpha-galactosidase from white chickpea (Cicer arietinum). Journal of Agricultural and Food Chemistry, 60, 3253–3259.
Xu, H., Qin, Y., Huang, Z., & Liu, Z. (2014). Characterization and site-directed mutagenesis of an alpha-galactosidase from the deep-sea bacterium Bacillus megaterium. Enzyme and Microbial Technology, 56, 46–52.
Du, F., Zhu, M., Wang, H., & Ng, T. (2013). Purification and characterization of an alpha-galactosidase from Phaseolus coccineus seeds showing degrading capability on raffinose family oligosaccharides. Plant Physiology and Biochemistry, 69, 49–53.
Goundar, R., & Mulimani, V. H. (2004). Purification and characterization of guar galactomannan degrading alpha-galactosidase from Aspergillus oryzae DR-5. Journal of Microbiology and Biotechnology, 14, 863–867.
Mulimani, V. H., & Ramalingam (1995). Enzymic hydrolysis of raffinose and stachyose in soymilk by alpha-galactosidase from Gibberella fujikuroi. Biochemistry and Molecular Biology International, 36, 897–905.
Acknowledgments
This work was financially supported by National Grants of China (Biomass dissociation and low-molecular fragment green monomerization and transformation, 2010CB732202).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Yang, D., Tian, G., Du, F. et al. A Fungal Alpha-Galactosidase from Pseudobalsamia microspora Capable of Degrading Raffinose Family Oligosaccharides. Appl Biochem Biotechnol 176, 2157–2169 (2015). https://doi.org/10.1007/s12010-015-1705-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-015-1705-0