
Abstract
Purpose of review
More effective therapies for glioblastoma are urgently needed. Immunotherapeutic strategies appear particularly promising and are therefore intensively studied. This article reviews the current understanding of the immunosuppressive glioblastoma microenvironment, discusses the rationale behind various immunotherapies, and outlines the findings of several recently published clinical studies.
Recent findings
The results of CheckMate-143 indicated that nivolumab is not superior to bevacizumab in patients with recurrent glioblastoma. A first-in man exploratory study evaluating EGFRvIII-specific CAR T cells for patients with newly diagnosed glioblastoma demonstrated overall safety of CAR T cell therapy and effective target recognition. A pilot study evaluating treatment with adoptively transferred CMV-specific T cells combined with a CMV-specific DC vaccine was found to be safe and resulted in increased polyclonality of CMV-specific T cells in vivo.
Summary
Despite the success of immunotherapies in many cancers, clinical evidence supporting their efficacy for patients with glioblastoma is still lacking. Nevertheless, the recently published studies provide important proof-of-concept in several areas of immunotherapy research. The careful and critical interpretation of these results will enhance our understanding of the opportunities and challenges of immunotherapies for high-grade gliomas and improve the immunotherapeutic strategies investigated in future clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioblastoma is the most common malignant primary brain tumor and ~ 12,000 patients are diagnosed with this tumor in the USA each year [1]. Despite enormous research efforts to better understand the pathobiology of this incurable disease and develop more effective therapies, the established standard of care therapy has remained unchanged for more than a decade [2]. Prognosis for patients with glioblastoma remains poor with a median survival time of 14.9 months and 5-year overall survival rates of only 5.5% [1]. Fueled by the success of immunotherapies for many non-CNS cancers, the idea of harnessing the immune system to treat glioblastoma has gained great interest over the past years. Various immunotherapeutic strategies are currently evaluated including immune checkpoint blockade, anti-tumor vaccination, and cellular therapies. Here, we review the current understanding of the immunosuppressive processes in the glioblastoma tumor microenvironment and the immunotherapeutic strategies that are currently being investigated.
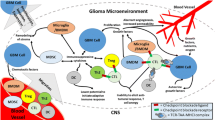
The immunosuppressive tumor microenvironment in glioblastoma
Glioblastomas create an immunosuppressive tumor microenvironment which allows tumor cells to escape from immune surveillance and effectively disrupts and modulates effector immune cell function via multiple mechanisms.
Change in the expression of surface molecules on glioma and glioma stem cells
Glioma cells downregulate the presentation of cellular antigens via the MHC class I and MHC II pathways [3, 4]. Instead of classical HLA class I molecules (HLA-A, HLA-A, and HLA-C), GBM cells express non-classical HLA class I molecules such as HLA-G and HLA-E. HLA-G affects the function of tumor-infiltrating lymphocytes [5]. The upregulation of HLA-E and lectin-like transcript-1 (LLT-1) on glioma cells impairs the function of natural killer (NK) cells [6,7,8]. In addition, glioma cells express Fas ligand (FasL, CD95) which, upon binding to the receptor Fas, induces apoptosis in tumor infiltrating lymphocytes [9].
Upregulation of STAT3 and key immunosuppressive cytokines in glioblastoma
Hypoxia-mediated upregulation of signal transducer and activator of transcription 3 (STAT3) is one of the hallmark pro-oncogenic alterations in glioblastoma. STAT3 activation in glioblastoma cells and glioma stem cells (GSCs) induces the production of key immunosuppressive cytokines including TGF-β, IL-6, IL-10, and IL-23 with important downstream effects on immune cell composition and immune cell activity in the glioma microenvironment [10,11,12]. In dendritic cells, STAT3 activation leads to reduced antigen presentation [13] and downregulation of CD40, CD80, CD86, and MHCII [14•]. Antigen presentation by these dendritic cells in the presence of IL-10 and TGF-β induces FoxP3 expression in CD4+ cells and leads to recruitment of regulatory T cells (Tregs) to the tumor [15]. STAT3 therefore represents an attractive treatment target and preclinical studies have demonstrated that STAT3 blockade results in upregulation of the co-stimulatory molecules CD80 and CD86 and increases the secretion of the pro-inflammatory cytokines [16, 17]. Unfortunately, this has not yet translated into successful clinical therapies for patients with glioblastoma although the development of small molecular inhibitors is ongoing [18,19,20].
Transforming growth factor (TGF)-β secretion is one of the key immunosuppressive cytokines secreted by glioma cells and leads to upregulation of FoxP3 in CD4+ cells thereby promoting their differentiation into Tregs [21, 22]. TGF-β-mediated activation of the SMAD/ATF1 pathways also dampens effector CD8+ T cell function by downregulating IFN-γ and granzyme B production. TGF-β has further been described to impair NK cell function via downregulation of NKG2D, a key molecule for NK cell activation. Despite encouraging preclinical work suggesting that TGF-β inhibition increases immunogenicity in murine glioma models [23, 24], the TGF-β inhibitor galunisertib was not more effective than lomustine for patients with recurrent glioblastoma in a randomized phase II study [25].
Lastly, increased interleukin-10 (IL-10) secretion in glioblastoma impairs antigen presenting cell (APC) and effector T cell function and promotes Treg activity and proliferation [26]. Other immunosuppressive cytokines include prostaglandin E2 (PGE2) which suppresses lymphocyte proliferation [27] and monocyte chemoattractant protein-1 (MCP-1, CCL2) which promotes Treg recruitment [28, 29].
Recruitment of immunosuppressive cells to the glioblastoma microenvironment
Tregs are characterized by FoxP3 and CD25 (IL-2α) expression and suppress the activity and proliferation of effector T cells via the production of the immunosuppressive cytokines TGF-β and IL-10. They comprise up to 15% of cells within the tumor [30] and the degree of tumor infiltration by Tregs has been correlated with tumor grade and prognosis [31,32,33].
Myeloid-derived suppressor cells (MDSCs) are immature myeloid cells that originate from the bone marrow and are recruited to the glioma microenvironment, mainly driven by upregulation of hypoxia-factor-1α (HIF-1α) in the tumor [34, 35]. Through increased secretion of arginase I, nitric oxide (NO), and other reactive oxygen species (ROS), MDSCs induce apoptosis of activated T cells and further stimulate the proliferation and recruitment of Tregs [36, 37].
Glioblastomas are also heavily infiltrated by CD45high monocytes/macrophages and CD45low microglia. These cells have been reported to comprise up to 30% of the tumor mass and the extent of tumor infiltration by these cells may correlate with prognosis [38, 39]. Monocytes/macrophages and brain-resident microglia are extraordinarily difficult to distinguish phenotypically and therefore have been categorized based on their response to key cytokines: While IFN-γ and GM-CSF increase inducible nitric oxide synthase (iNOS) expression in the pro-inflammatory M1 phenotype, IL-4 and M-CSF induce secretion of arginase I, TGF-β, matrix metalloproteinase (MMP)-2, and MMP-9, IL-10, thereby characterizing the immunosuppressive M2 phenotype [40,41,42•]. In the glioblastoma environment, macrophages have been described to be predominantly shifted towards the pro-tumor M2 phenotype [43] and the binding of colony-stimulating-factor (CSF)-1 to CSF-1 receptor (CSF-1R) on macrophages appears to induce this shift from M1 to M2 [44]. It is important to note, however, that the various activity states of macrophages and microglia remain incompletely understood and in fact, the M1/M2 classification concept has recently been challenged [42•, 45]. Our understanding of the role of macrophages and microglia in glioblastoma will therefore continue to evolve.
Immune checkpoints in glioblastoma
Cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) is predominantly expressed by T cells in secondary lymphoid organs and is upregulated upon immune activation. Binding of CTLA-4 to its ligands CD 80 (B7.1) or CD86 (B7.2) leads to reduced activation and proliferation of effector T cells and increases the activation and recruitment of Tregs and MDSCs to the glioblastoma microenvironment [46].
Programmed cell death (PD)-1 is expressed and upregulated in T cells, other immune cells, and tumor cells. The PD-1 receptor has two ligands, PD-L1 (B7-homologue 1, B7-H1) and PD-L2 (B7-homologue 2, B7-H2). While PD-L1 is found on immunosuppressive Tregs, tumor-associated macrophages, and other cells within the tumor microenvironment including tumor cells [47], PD-L2 expression is restricted to reactive immune cells such as dendritic cells and macrophages [48]. Binding to of either ligand to PD-1 results in reduced function and proliferation of effector T cells [49, 50] and augments the activity, proliferation, and recruitment of Tregs to the tumor [51]. PD-L1 expression levels in glioblastoma range from 7.8 to 37.5% of tumor-resident cells and appear overall lower as compared to other cancers [52,53,54,55,56,57].
Other inhibitory checkpoint molecules in the tumor microenvironment include T cell immunoglobulin and mucin-domain containing-3 (TIM-3) and lymphocyte-activation gene 3 (LAG-3). TIM-3 is a surface marker expressed on TH1 cells and is considered a marker of T cell exhaustion which induces T cell apoptosis upon binding to its ligand galectin-9 (Gal-9) [58]. In glioblastoma, TIM-3 expression levels have been correlated with tumor grade and functional status [59, 60]. Anti-TIM-3 therapy was recently demonstrated to increase survival when combined with anti-PD-1 blockade and radiation in a murine glioma model [61]. LAG-3 is a membrane molecule upregulated on tumor infiltrating lymphocytes and LAG-3 binding to MHC class II on APCs and tumor cells results in reduced T cell and NK cell activation [62]. While LAG-3 upregulation has been reported to have important pro-oncogenic implications in other cancers including melanoma [63], there are no preclinical studies to date characterizing the role of LAG-3 in glioblastoma.
In addition to these immunosuppressive checkpoint molecules, a number of stimulatory checkpoint markers have been identified. CD137 (4-1BB) acts as a co-stimulatory protein of the T cell receptor (TCR) and promotes T cell activation and proliferation. CD137 stimulation has been demonstrated to increase survival in murine glioma models when used in combination with anti-CTLA4 antibodies and radiation [64]. Similarly, OX40L is a co-stimulatory molecule of the TCR and its expression in gliomas has been correlated with improved survival. The induction of OX40L expression improved survival in a murine glioma model [65].
IDO and tryptophan metabolism
There has been a growing interest in studying the effects of altered tumor metabolism on tumor growth and the immunosuppressive tumor environment. The indoleamine-2, 3-dioxygenase (IDO) is a key enzyme of tryptophan metabolism and metabolizes tryptophan into kynurenine. Increased kynurenine levels appear to induce T cell anergy and immune cell apoptosis [66]. IDO has been found to be upregulated in up to 90% of gliomas and is associated with higher grade, shorter survival, and increased recruitment of Tregs and MDSCs to the tumor microenvironment [67,68,69]. Simultaneous targeting of IDO, CTLA4, and PD-L1 resulted in durable treatment responses in a murine glioma model [70]. Therefore, IDO inhibitors are currently explored for patients with high-grade gliomas, either as single agent (NCT02052648) or in combination with other immune checkpoint inhibitors (NCT03192943, NCT02658890).
Immunotherapies for glioblastoma
Immune checkpoint blockade
Given the prominent expression of CTLA-4, PD-1 and their ligands in glioblastoma and fueled by the enthusiasm about the success of immune checkpoint blockade in melanoma, non-small cell lung cancer (NSCLC), and other malignancies, a multitude of clinical trials is currently evaluating the role of checkpoint inhibitors either alone or in combination for newly diagnosed and recurrent high-grade glioma. These include the anti-PD-1 antibodies nivolumab (Opdivo) and pembrolizumab (Keytruda), the anti-PD-L1 antibodies avelumab (Bavencio) and durvalumab (Imfinzi), and the anti-CTLA-4 antibodies ipilimumab (Yervoy) and tremelimumab (formerly ticilimumab). In addition, alternative checkpoint-targeting antibodies such as the anti-LAG-3 antibody urelumab and an anti-CD137 antibody are investigated, either alone or in combination with nivolumab (Table 1).
While the majority of these studies are still ongoing, the results of the first clinical study evaluating PD-1 and CTLA-4 blockade in recurrent high-grade gliomas recently became available. In the exploratory phase I part of CheckMate-143, the safety of nivolumab versus nivolumab combined with ipilimumab was investigated. Because of dose-limiting toxicities predominantly in the combination arm, the subsequent phase III part of the study investigated nivolumab alone compared to bevacizumab. Unfortunately, the primary endpoint of superior survival with nivolumab was not met in this study given that median overall survival for patients treated with nivolumab was 9.8 months compared to 10 months for those receiving bevacizumab. Response rates were 8% with nivolumab and 23% with bevacizumab [71••, 72••].
In order to understand the reasons for failure of this study, it is important to recognize some of the emerging key concepts that may influence the efficacy of checkpoint blockade in cancer. One of these is the significance of PD-1 and PD-L1 expression as a predictive biomarker of treatment response to PD-1/PD-L1 blocking therapy. In a phase I study exploring nivolumab in multiple cancer types, 36% of patients with PD-L1 positive tumors (defined as ≥ 5% of cells PD-L1+) demonstrated a treatment response whereas none of the patients with PD-L1 negative tumors had a treatment response [73]. Similarly, progression-free and overall survival were significantly prolonged in patients with NSCLC whose tumors had a proportional PD-L1 expression ≥ 50% compared to patients whose tumors had ˂ 50% proportional PD-L1 expression [74].
PD-L1 expression levels were also evaluated in CheckMate-143. Thirty-two percent of the tumors expressed PD-L1 in < 1% of tumor cells and only 27% of patients had expression levels ≥ 10% [71••]. Under the assumption that the level of PD-L1 expression in glioblastoma has similar implications as in other cancers, the PD-L1 expression found in the patient population of CheckMate-143 may therefore have been too low to result in a treatment response. However, if PD-1 and PD-L1 expression actually have a role as predictive biomarkers that can identify glioblastoma patients who are likely to respond to checkpoint blockade remains insufficiently understood at this time. This continues to be investigated in the correlative studies accompanying many of currently ongoing clinical trials.
A similarly important predictor of response to PD-1/PD-L1 targeting therapies is the mutational load of the individual tumor. The abundance of genetic alterations in a given tumor is thought to increase the amount of available tumor-specific neoantigens to which a robust anti-tumor immune response can be directed. This is supported by clinical evidence showing that patients with colorectal cancer respond better to pembrolizumab if their tumors demonstrate deficiency in DNA mismatch repair (MMR) enzymes [75]. Similarly, the mutational burden in melanoma or NSCLC appears to correlate with response to immune checkpoint blockade [76, 77]. Several case reports have demonstrated that dramatic responses can also be seen in patients with glioblastoma who have germline mutations in MMR genes [78] or POLE [79, 80]. Interestingly, treatment with temozolomide has been associated with mutational loss of MMR genes (MSH6, MLH1, and MSH2) at time of high-grade glioma recurrence [81,82,83]. Based on these results, several clinical trials are exploring the efficacy of immune checkpoint inhibition in patients with hypermutated glioblastoma (Table 1).
Other clinical studies are investigating checkpoint inhibition combined with radiation (Table 1) because radiation has been demonstrated to increase the pool of potential neoantigens and to improve MHC-I-mediated presentation of these antigens by tumor cells and APCs [84, 85].
Lastly, the average number of effector immune cells in the glioblastoma microenvironment, in particular CD4+ and CD8+ T cells, may be too low to result in an effective anti-tumor immune response even if the activity of these cells can be augmented via checkpoint inhibition. Therefore, several clinical studies investigate the combination of checkpoint inhibitors with vaccine or viral vector-based strategies with the goal to increase T cell recruitment to the tumor (Table 1). In addition, there may be a potential synergistic effect of immunotherapies combined with targeted therapies. For example, CDK4 inhibition appears to increase antigen presentation and decreases regulatory T cells in the tumor microenvironment and therefore may lead to an improved anti-tumor immune response when combined with PD1 inhibitors [86].
Vaccine strategies for glioblastoma
Anti-tumor vaccination strategies follow the idea that improved immunization with tumor antigens augments recruitment of antigen-specific effector T cells to the tumor. Vaccination strategies are inherently complex because they require the optimal interplay of multiple factors including appropriate antigen selection, efficient presentation of these antigens by APCs, efficient expansion and migration of immunized T cells to the tumor, and effective cytotoxic function of these T cells in the tumor microenvironment.
Tumor antigens used for vaccine development can be broadly categorized into tumor-specific antigens (TSA) and tumor-associated antigens (TAAs). TSAs are exclusively expressed on glioblastoma cells and typically elicit a quite robust tumor-specific immune response with a low risk of cross-reactivity. In high-grade gliomas, EGFRvIII represents the prototype of such a tumor-specific antigen and ~ 30% of glioblastomas express this mutation [87]. Based on the robust anti-tumor response seen in mice treated with anti-EGFRvIII vaccination [88], the EGFRvIII-peptide vaccine rindopepimut (Rintega) was developed and extensively studied in phase I through phase III clinical trials for patients with newly diagnosed and recurrent glioblastoma [89,90,91,92,93]. While the early-phase clinical studies demonstrated general safety of the vaccine and impressive EGFRvIII-specific humoral and cellular immune responses [91, 90, 93], an international multicenter phase III clinical trial failed to demonstrate a survival benefit when rindopepimut was added to standard radiochemotherapy [94••]. It has been speculated that the vaccine was ineffective because the majority of tumors lost EGFRvIII expression over time. Antigen loss and the intratumoral as well as interindividual heterogeneity are hallmark characteristics of glioblastomas and illustrate additional immune escape mechanisms.
Similarly, the IDH-1R132H mutation found in 70% of diffuse gliomas and ~ 5% of glioblastomas [95] harbors a tumor-specific neoantigen that was demonstrated to be effectively presented by professional APCs and to generate a robust mutation-specific CD4+ T cell response in a syngeneic mouse glioma model [96]. NOA-16, a phase I clinical study is therefore exploring an IDH-1R132H targeting vaccine for patients with recurrent IDH-1R132H mutant high-grade gliomas (Table 1).
In contrast, TAAs are native proteins that are overexpressed in glioblastoma cells but also have limited expression in other tissues, e.g., survivin [97], telomerase [98], HER2/neu [99], EphA2 [100, 101], and IL-13Rα [102]. The low-level expression in other body tissues typically causes a certain level of central immune tolerance and vaccination with these peptides therefore generates an overall weaker immune response but also lowers the risk of cross-reactivity and auto-immune complications. In order to augment the anti-tumor immune response and to reduce the chance of immune escape, various vaccines combine different TAAs.
SL-701 for example is a multi-peptide vaccine targeting IL-13Rα2, survivin, and EphA2. A multicenter phase I/II clinical study evaluating the safety and efficacy of this vaccine is currently under way (Table 1) [103]. ICT-107 represents a dendritic cell (DC) vaccine that is generated by pulsing DCs with a synthetic TAA cocktail containing the peptides AIM-2, MAGE-1, TRP-2, gp100, HER-2, and IL-13α2. In a single-arm phase 1 pilot study, a robust immune response to gp100 and HER2 was observed and median progression-free and overall survival appeared prolonged compared to historical controls [104]. This prompted further evaluation of ICT-107 in a multicenter double-blinded phase IIb study randomizing patients to receive DCs pulsed with the TAA cocktail or unpulsed DCs. While progression-free survival appeared prolonged in the treatment arm, the trend towards improved overall survival in the treatment group was not statistically significant. However, a possible survival benefit was seen in the subgroup of HLA-A2 positive individuals [105]. Therefore, a phase III clinical trial was launched in 2016 accruing HLA-A2 positive patients only. Unfortunately, further patient enrollment was halted in 2017 due to funding issues.
Other vaccines target viral antigens expressed in malignant gliomas such as the cytomegalovirus (CMV) antigens immediate early-1 (IE1), phosphoprotein 65 (pp65), and glycoprotein B (gB) [106,107,108]. In addition to CMV peptides, CMV mRNA can be used to load and activate DCs. In a randomized and blinded pilot study, DCs were pulsed with CMV pp65-lysosomal-associated membrane protein (LAMP) mRNA either with or without preconditioning of the vaccination site by tetanus toxoid (Td) injection. This study demonstrated prolonged survival in patients receiving the DCs in conjunction with the Td booster [109]. A multicenter, randomized, blinded, and placebo-controlled phase II study is currently underway to further investigate the efficacy of this vaccine (Table 1).
In addition, heat-shock protein (HSP)-96 has been demonstrated to bind TAAs in the tumor microenvironment. A vaccine targeting the HSP-96 peptide complex (HSPPC-9) has therefore been studied in early-phase clinical trials and was demonstrated to generate robust humoral and cellular antitumor responses [110, 111]. A randomized phase II clinical trial investigating this approach was recently reported as being negative (Table 1).
Lastly, personalized vaccination strategies are currently explored. The general safety and feasibility of this approach has been demonstrated in several pilot clinical studies using DC vaccines generated by DC exposure to antigens isolated from autologous tumor lysates [112, 113]. In a phase I clinical study for newly diagnosed glioblastoma patient, a personalized neoantigen cancer vaccine is manufactured by combining autologous tumor-specific peptides with the immunoadjuvant agent poly-ICLC (Table 1). In melanoma patients, this approach was recently demonstrated to result in an effective anti-tumor T cell response [114••]. While the advantages of individualized, tumor-specific anti-tumor vaccination appear obvious, the production of these vaccines is labor-, time-, and cost-intense. In addition, there is at least a theoretical risk of autoimmunity given that the tumor-lysate may contain self-antigens expressed in other body tissues.
Cellular therapies
Adoptive T cell transfer involve the isolation of tumor-specific T cells from tumor or tumor-draining lymph nodes, the expansion of these cells ex vivo followed by their intravenous infusion back to the patient [115, 116]. This concept has recently gained new momentum when it was demonstrated that therapy with ex vivo expanded CMV-specific T cells resulted in encouraging progression-free and overall survival in patients with recurrent glioblastoma [117]. Most recently, a pilot study evaluated the combination of CMV pp65-specific T cells with a CMV pp65 RNA-loaded DC vaccine compared to treatment with CMV-specific T cells alone and demonstrated encouraging survival times in the treated individuals [118••]. Further studies evaluating CMV-specific T cell therapies are under way (Table 2).
An alternative strategy to utilize tumor-specific T cells is the generation of chimeric antigen receptor (CAR) T cells. CAR T cells are genetically modified T cells that express a CAR construct consisting of an extracellular ligand recognition domain often derived from a monoclonal antibody which is linked to an intracellular signaling domain typically derived from a signaling subunit of the T cell receptor (CD3ζ,). Additional co-stimulatory endodomains enhance T cell cytokine production, expansion, and in vivo persistence. Upon recognition of the CAR-specific target antigen, CAR T cells are activated and proliferate and recruit other immune cells via cytokine and chemokine secretion. Importantly, the target recognition is independent of MHC presentation and the same CAR construct therefore can be used as an “off the shelf” product [119]. CAR T cells targeting CD19 were demonstrated to be extraordinarily successful in the treatment of B cell acute lymphoblastic leukemia (B-ALL) and chronic lymphoblastic leukemia (CLL) [120, 121].
CARs targeting glioblastoma-relevant antigens have been developed, including IL13Rα2 [122], Her2 [123], EGFRvIII [124, 125], and EphA2 [126]. In an exploratory cohort of a first-in human clinical study investigating an anti-EGFRvIII CAR, nine of ten patients had progressed at 1-year of follow-up. Evaluation of tumor tissue resected at time of recurrence demonstrated that EGFRvIII-specific CAR T cells could be detected in small amounts in the tumor environment and EGFRvIII expression was lost over time. While this provided indirect evidence for successful CAR T cell trafficking to the tumor and effective antigen targeting, it again demonstrated antigen loss to be an important mechanism of immune escape in glioblastoma. Moreover, these tumors demonstrated increased PD-L1 and IDO expression and increased tumor infiltration with Tregs [127••]. The overall safety and potential efficacy of CAR T cells targeting IL13Rα2 was demonstrated in a small case series and a single case report [128, 129]. More recently, a phase I clinical study demonstrated the overall feasibility and safety of a HER2-CAR in patients with recurrent glioblastoma [130••]. Based on these encouraging results, several ongoing clinical trials continue to investigate the safety and efficacy of CAR T cell therapy for patients with glioblastoma (Table 2).
Oncolytic viruses
This strategy exploits the ability to modify certain viral vectors such that they selectively infect tumor cells, intracellularly replicate and induce tumor cell lysis, thereby allowing the release new infectious particles to the tumor microenvironment. In addition, they frequently induce an inflammatory response and upregulate PDL1 expression [131]. Commonly used viral vectors include oncolytic herpes simplex virus (oHSV), oncolytic measles virus (oMV), reovirus, polio virus, and conditionally replicating adenoviruses (CRAdVs) [132,133,134,135,136,137]. Several ongoing clinical trials investigate the value of immunotherapy utilizing viral vectors alone or in combination with checkpoint inhibitors for patients with recurrent and newly diagnosed glioblastoma (Table 2). In addition, viral vectors can be used to introduce therapeutic genes into the tumor cells. These genes most often encode for metabolic enzymes such as the thymidine kinase (TK) which then serves to transform a subsequently administered prodrug such as ganciclovir or flucytosine into cytotoxic agents within the tumor microenvironment [138, 131]. In addition, genes encoding for pro-inflammatory cytokines such as IL-2, IL-4, IL-12, IFN-γ, and IFN-β can be transferred to the tumor using various viral vectors [139,140,141,142,143]. Viral vector therapies are explored in several ongoing clinical trials either alone or in combination with checkpoint inhibitors (Table 2).
Summary and future directions
Glioblastoma represents the most aggressive primary brain tumor and prognosis remains dismal despite the best current standard of care treatment. High-grade gliomas are well known to create an immunosuppressive tumor microenvironment through the alteration of cellular surface markers, the secretion of immunosuppressive cytokines, the activation of immune checkpoint inhibitory pathways, and the recruitment of immunosuppressive Tregs and MDSCs to the tumor. The recently published results of early-phase clinical studies exploring various immunotherapeutic strategies provide important proof-of-concept and support the continued investment in immunotherapies for high-grade gliomas. Nevertheless, the critical evaluation of these study results will be necessary to uncover conceptual and methodological difficulties and will be prerequisite for the improvement of the immunotherapeutic strategies investigated in future clinical studies.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, et al. CBTRUS Statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010–2014. Neuro-Oncology. 2017;19(suppl_5):v1–v88. https://doi.org/10.1093/neuonc/nox158.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. https://doi.org/10.1056/NEJMoa043330.
Facoetti A, Nano R, Zelini P, Morbini P, Benericetti E, Ceroni M, et al. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin Cancer Res. 2005;11(23):8304–11. https://doi.org/10.1158/1078-0432.CCR-04-2588.
Mehling M, Simon P, Mittelbronn M, Meyermann R, Ferrone S, Weller M, et al. WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: does it reflect a potential immune escape mechanism? Acta Neuropathol. 2007;114(2):111–9. https://doi.org/10.1007/s00401-007-0231-8.
Wastowski IJ, Simoes RT, Yaghi L, Donadi EA, Pancoto JT, Poras I, et al. Human leukocyte antigen-G is frequently expressed in glioblastoma and may be induced in vitro by combined 5-aza-2′-deoxycytidine and interferon-gamma treatments: results from a multicentric study. Am J Pathol. 2013;182(2):540–52. https://doi.org/10.1016/j.ajpath.2012.10.021.
Wolpert F, Roth P, Lamszus K, Tabatabai G, Weller M, Eisele G. HLA-E contributes to an immune-inhibitory phenotype of glioblastoma stem-like cells. J Neuroimmunol. 2012;250(1–2):27–34. https://doi.org/10.1016/j.jneuroim.2012.05.010.
Wischhusen J, Friese MA, Mittelbronn M, Meyermann R, Weller M. HLA-E protects glioma cells from NKG2D-mediated immune responses in vitro: implications for immune escape in vivo. J Neuropathol Exp Neurol. 2005;64(6):523–8.
Roth P, Mittelbronn M, Wick W, Meyermann R, Tatagiba M, Weller M. Malignant glioma cells counteract antitumor immune responses through expression of lectin-like transcript-1. Cancer Res. 2007;67(8):3540–4. https://doi.org/10.1158/0008-5472.CAN-06-4783.
Weller M, Weinstock C, Will C, Wagenknecht B, Dichgans J, Lang F, et al. CD95-dependent T-cell killing by glioma cells expressing CD95 ligand: more on tumor immune escape, the CD95 counterattack, and the immune privilege of the brain. Cell Physiol Biochem. 1997;7(5):282–8.
Kinjyo I, Inoue H, Hamano S, Fukuyama S, Yoshimura T, Koga K, et al. Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med. 2006;203(4):1021–31. https://doi.org/10.1084/jem.20052333.
Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9(1):67–78. https://doi.org/10.1158/1535-7163.MCT-09-0734.
Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206(7):1457–64. https://doi.org/10.1084/jem.20090207.
Torres-Aguilar H, Aguilar-Ruiz SR, Gonzalez-Perez G, Munguia R, Bajana S, Meraz-Rios MA, et al. Tolerogenic dendritic cells generated with different immunosuppressive cytokines induce antigen-specific anergy and regulatory properties in memory CD4+ T cells. J Immunol. 2010;184(4):1765–75. https://doi.org/10.4049/jimmunol.0902133.
• Ferguson SD, Srinivasan VM, Heimberger AB. The role of STAT3 in tumor-mediated immune suppression. J Neuro-Oncology. 2015;123(3):385–94. https://doi.org/10.1007/s11060-015-1731-3. Excellent review article that reviews the role of STAT3 and the mannifold implications in the glioblastoma microenvironment.
Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108(5):1571–9. https://doi.org/10.1182/blood-2006-02-004747.
Zhang L, Alizadeh D, Van Handel M, Kortylewski M, Yu H, Badie B. Stat3 inhibition activates tumor macrophages and abrogates glioma growth in mice. Glia. 2009;57(13):1458–67. https://doi.org/10.1002/glia.20863.
Fujiwara Y, Komohara Y, Kudo R, Tsurushima K, Ohnishi K, Ikeda T, et al. Oleanolic acid inhibits macrophage differentiation into the M2 phenotype and glioblastoma cell proliferation by suppressing the activation of STAT3. Oncol Rep. 2011;26(6):1533–7. https://doi.org/10.3892/or.2011.1454.
Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, et al. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67(20):9630–6. https://doi.org/10.1158/0008-5472.CAN-07-1243.
Kohsaka S, Wang L, Yachi K, Mahabir R, Narita T, Itoh T, et al. STAT3 inhibition overcomes temozolomide resistance in glioblastoma by downregulating MGMT expression. Mol Cancer Ther. 2012;11(6):1289–99. https://doi.org/10.1158/1535-7163.MCT-11-0801.
de Groot J, Liang J, Kong LY, Wei J, Piao Y, Fuller G, et al. Modulating antiangiogenic resistance by inhibiting the signal transducer and activator of transcription 3 pathway in glioblastoma. Oncotarget. 2012;3(9):1036–48. https://doi.org/10.18632/oncotarget.663.
Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumor microenvironment. Nat Rev Cancer. 2013;13(11):788–99. https://doi.org/10.1038/nrc3603.
Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-beta 1, -beta 2, and -beta 3 by glioblastoma cells, astrocytes, and microglia. J Immunol. 1992;148(5):1404–10.
Ueda R, Fujita M, Zhu X, Sasaki K, Kastenhuber ER, Kohanbash G, et al. Systemic inhibition of transforming growth factor-beta in glioma-bearing mice improves the therapeutic efficacy of glioma-associated antigen peptide vaccines. Clin Cancer Res. 2009;15(21):6551–9. https://doi.org/10.1158/1078-0432.CCR-09-1067.
Uhl M, Aulwurm S, Wischhusen J, Weiler M, Ma JY, Almirez R, et al. SD-208, a novel transforming growth factor beta receptor I kinase inhibitor, inhibits growth and invasiveness and enhances immunogenicity of murine and human glioma cells in vitro and in vivo. Cancer Res. 2004;64(21):7954–61. https://doi.org/10.1158/0008-5472.CAN-04-1013.
Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O, et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro-Oncol. 2016;18(8):1146–56. https://doi.org/10.1093/neuonc/now009.
Huettner C, Paulus W, Roggendorf W. Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am J Pathol. 1995;146(2):317–22.
Wang L, Shi J, van Ginkel FW, Lan L, Niemeyer G, Martin DR, et al. Neural stem/progenitor cells modulate immune responses by suppressing T lymphocytes with nitric oxide and prostaglandin E2. Exp Neurol. 2009;216(1):177–83. https://doi.org/10.1016/j.expneurol.2008.11.017.
Desbaillets I, Tada M, de Tribolet N, Diserens AC, Hamou MF, Van Meir EG. Human astrocytomas and glioblastomas express monocyte chemoattractant protein-1 (MCP-1) in vivo and in vitro. Int J Cancer J Int du Cancer. 1994;58(2):240–7.
Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunotherapy: CII. 2008;57(1):123–31. https://doi.org/10.1007/s00262-007-0336-x.
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res. 2008;14(16):5166–72. https://doi.org/10.1158/1078-0432.CCR-08-0320.
El Andaloussi A, Lesniak MS. CD4+ CD25+ FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J Neuro-Oncol. 2007;83(2):145–52. https://doi.org/10.1007/s11060-006-9314-y.
Yue Q, Zhang X, Ye HX, Wang Y, Du ZG, Yao Y, et al. The prognostic value of Foxp3+ tumor-infiltrating lymphocytes in patients with glioblastoma. J Neuro-Oncol. 2014;116(2):251–9. https://doi.org/10.1007/s11060-013-1314-0.
Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJ, Wesseling P, et al. Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol. 2010;225(1–2):195–9. https://doi.org/10.1016/j.jneuroim.2010.05.020.
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13(3):206–20. https://doi.org/10.1016/j.ccr.2008.01.034.
Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, et al. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-Oncology. 2011;13(6):591–9. https://doi.org/10.1093/neuonc/nor042.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. https://doi.org/10.1038/nri2506.
Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989–99.
Morantz RA, Wood GW, Foster M, Clark M, Gollahon K. Macrophages in experimental and human brain tumors. Part 2: studies of the macrophage content of human brain tumors. J Neurosurg. 1979;50(3):305–11. https://doi.org/10.3171/jns.1979.50.3.0305.
Morimura T, Neuchrist C, Kitz K, Budka H, Scheiner O, Kraft D, et al. Monocyte subpopulations in human gliomas: expression of Fc and complement receptors and correlation with tumor proliferation. Acta Neuropathol. 1990;80(3):287–94.
Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008;216(1):15–24. https://doi.org/10.1002/path.2370.
Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, et al. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion—an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene. 2008;27(7):918–30. https://doi.org/10.1038/sj.onc.1210683.
• Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. https://doi.org/10.12703/P6-13. Opinion statement outlining and challenging the current concept of the bipoloar M1/M2 activation states.
Wu A, Wei J, Kong LY, Wang Y, Priebe W, Qiao W, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology. 2010;12(11):1113–25. https://doi.org/10.1093/neuonc/noq082.
Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18:519–27. https://doi.org/10.2119/molmed.2011.00217.
Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19(8):987–91. https://doi.org/10.1038/nn.4338.
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–5. https://doi.org/10.1126/science.1160062.
Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res. 2013;19(12):3165–75. https://doi.org/10.1158/1078-0432.CCR-12-3314.
Xiao Y, Yu S, Zhu B, Bedoret D, Bu X, Francisco LM, et al. RGMb is a novel binding partner for PD-L2 and its engagement with PD-L2 promotes respiratory tolerance. J Exp Med. 2014;211(5):943–59. https://doi.org/10.1084/jem.20130790.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9.
Hofmeyer KA, Jeon H, Zang X. The PD-1/PD-L1 (B7-H1) pathway in chronic infection-induced cytotoxic T lymphocyte exhaustion. J Biomed Biotechnol. 2011;2011:451694. https://doi.org/10.1155/2011/451694.
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–29. https://doi.org/10.1084/jem.20090847.
Wintterle S, Schreiner B, Mitsdoerffer M, Schneider D, Chen L, Meyermann R, et al. Expression of the B7-related molecule B7-H1 by glioma cells: a potential mechanism of immune paralysis. Cancer Res. 2003;63(21):7462–7.
Wilmotte R, Burkhardt K, Kindler V, Belkouch MC, Dussex G, Tribolet N, et al. B7-homolog 1 expression by human glioma: a new mechanism of immune evasion. Neuroreport. 2005;16(10):1081–5.
Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro-Oncology. 2009;11(4):394–402. https://doi.org/10.1215/15228517-2008-104.
Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology. 2015;17(8):1064–75. https://doi.org/10.1093/neuonc/nou307.
Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro-Oncology. 2016;18(2):195–205. https://doi.org/10.1093/neuonc/nov172.
Garber ST, Hashimoto Y, Weathers SP, Xiu J, Gatalica Z, Verhaak RG, et al. Immune checkpoint blockade as a potential therapeutic target: surveying CNS malignancies. Neuro-Oncology. 2016;18(10):1357–66. https://doi.org/10.1093/neuonc/now132.
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94. https://doi.org/10.1084/jem.20100643.
Han S, Feng S, Xu L, Shi W, Wang X, Wang H, et al. Tim-3 on peripheral CD4(+) and CD8(+) T cells is involved in the development of glioma. DNA Cell Biol. 2014;33(4):245–50. https://doi.org/10.1089/dna.2013.2306.
Liu Z, Han H, He X, Li S, Wu C, Yu C, et al. Expression of the galectin-9-Tim-3 pathway in glioma tissues is associated with the clinical manifestations of glioma. Oncol Lett. 2016;11(3):1829–34. https://doi.org/10.3892/ol.2016.4142.
Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017;23(1):124–36. https://doi.org/10.1158/1078-0432.CCR-15-1535.
Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21(4):503–13. https://doi.org/10.1016/j.immuni.2004.08.010.
Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011;186(9):5173–83. https://doi.org/10.4049/jimmunol.1002050.
Belcaid Z, Phallen JA, Zeng J, See AP, Mathios D, Gottschalk C, et al. Focal radiation therapy combined with 4-1BB activation and CTLA-4 blockade yields long-term survival and a protective antigen-specific memory response in a murine glioma model. PLoS One. 2014;9(7):e101764. https://doi.org/10.1371/journal.pone.0101764.
Shibahara I, Saito R, Zhang R, Chonan M, Shoji T, Kanamori M, et al. OX40 ligand expressed in glioblastoma modulates adaptive immunity depending on the microenvironment: a clue for successful immunotherapy. Mol Cancer. 2015;14:41. https://doi.org/10.1186/s12943-015-0307-3.
Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002;9(10):1069–77. https://doi.org/10.1038/sj.cdd.4401073.
Mitsuka K, Kawataki T, Satoh E, Asahara T, Horikoshi T, Kinouchi H. Expression of indoleamine 2,3-dioxygenase and correlation with pathological malignancy in gliomas. Neurosurgery. 2013;72(6):1031–8; discussion 8–9. https://doi.org/10.1227/NEU.0b013e31828cf945.
Platten M, Weller M, Wick W. Shaping the glioma immune microenvironment through tryptophan metabolism. CNS Oncol. 2012;1(1):99–106. https://doi.org/10.2217/cns.12.6.
Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–21. https://doi.org/10.1158/1078-0432.CCR-12-2130.
Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014;20(20):5290–301. https://doi.org/10.1158/1078-0432.CCR-14-0514.
•• Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase 1 cohorts of CheckMate 143. Neuro-Oncology. 2017; https://doi.org/10.1093/neuonc/nox208. Phase I-part results of the first clinical study of checkpoint inhibition with nivolumab and/or ipilimumab for patients with recurrent high-grade glioma.
•• Reardon DA, Omuro A, Brandes AA, Rieger J, Wick A, Sepulveda J, et al. OS10.3 Randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: CheckMate 143. Neuro-oncology. 2017;19(suppl_3):iii21-iii. https://doi.org/10.1093/neuonc/nox036.071. Phase III-part results of the first clinical study comparing nivolumab versus bevacizumab for patients with recurrent high-grade glioma.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. https://doi.org/10.1056/NEJMoa1200690.
Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–28. https://doi.org/10.1056/NEJMoa1501824.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20. https://doi.org/10.1056/NEJMoa1500596.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8. https://doi.org/10.1126/science.aaa1348.
Johnson DB, Lovly CM, Flavin M, Panageas KS, Ayers GD, Zhao Z, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res. 2015;3(3):288–95. https://doi.org/10.1158/2326-6066.CIR-14-0207.
Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016;34(19):2206–11. https://doi.org/10.1200/JCO.2016.66.6552.
Erson-Omay EZ, Caglayan AO, Schultz N, Weinhold N, Omay SB, Ozduman K, et al. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro-Oncology. 2015;17(10):1356–64. https://doi.org/10.1093/neuonc/nov027.
Ahn SM, Ansari AA, Kim J, Kim D, Chun SM, Kim J, et al. The somatic POLE P286R mutation defines a unique subclass of colorectal cancer featuring hypermutation, representing a potential genomic biomarker for immunotherapy. Oncotarget. 2016;7(42):68638–49. https://doi.org/10.18632/oncotarget.11862.
Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13(7):2038–45. https://doi.org/10.1158/1078-0432.CCR-06-2149.
Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, Nutt CL, et al. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res. 2009;15(14):4622–9. https://doi.org/10.1158/1078-0432.CCR-08-3012.
van Thuijl HF, Mazor T, Johnson BE, Fouse SD, Aihara K, Hong C, et al. Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol. 2015;129(4):597–607. https://doi.org/10.1007/s00401-015-1403-6.
Garnett CT, Palena C, Chakraborty M, Tsang KY, Schlom J, Hodge JW. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004;64(21):7985–94. https://doi.org/10.1158/0008-5472.CAN-04-1525.
Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203(5):1259–71. https://doi.org/10.1084/jem.20052494.
Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumor immunity. Nature. 2017;548(7668):471–5. https://doi.org/10.1038/nature23465.
Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89(7):2965–9.
Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. 2003;9(11):4247–54.
Heimberger AB, Sun W, Hussain SF, Dey M, Crutcher L, Aldape K, et al. Immunological responses in a patient with glioblastoma multiforme treated with sequential courses of temozolomide and immunotherapy: case study. Neuro-Oncology. 2008;10(1):98–103. https://doi.org/10.1215/15228517-2007-046.
Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology. 2011;13(3):324–33. https://doi.org/10.1093/neuonc/noq157.
Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE 2nd, Lally-Goss D, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8(10):2773–9. https://doi.org/10.1158/1535-7163.MCT-09-0124.
Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–9. https://doi.org/10.1200/JCO.2010.28.6963.
Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-Oncology. 2015;17(6):854–61. https://doi.org/10.1093/neuonc/nou348.
•• Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomized, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–85. https://doi.org/10.1016/S1470-2045(17)30517-X. The anti-EGFRvIII vaccine rindopepimut added to radiation and chemotherapy was not superior to standard of care treatment in patients with newly diagnosed glioblastoma.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73. https://doi.org/10.1056/NEJMoa0808710.
Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512(7514):324–7. https://doi.org/10.1038/nature13387.
Liu R, Mitchell DA. Survivin as an immunotherapeutic target for adult and pediatric malignant brain tumors. Cancer Immunol Immunother: CII. 2010;59(2):183–93. https://doi.org/10.1007/s00262-009-0757-9.
Komata T, Kanzawa T, Kondo Y, Kondo S. Telomerase as a therapeutic target for malignant gliomas. Oncogene. 2002;21(4):656–63. https://doi.org/10.1038/sj.onc.1205072.
Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–85. https://doi.org/10.1158/1078-0432.CCR-09-1322.
Li X, Wang Y, Wang Y, Zhen H, Yang H, Fei Z, et al. Expression of EphA2 in human astrocytic tumors: correlation with pathologic grade, proliferation and apoptosis. Tumour Biol. 2007;28(3):165–72. https://doi.org/10.1159/000103010.
Wang LF, Fokas E, Bieker M, Rose F, Rexin P, Zhu Y, et al. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol Rep. 2008;19(1):151–6.
Akiyama Y, Komiyama M, Miyata H, Yagoto M, Ashizawa T, Iizuka A, et al. Novel cancer-testis antigen expression on glioma cell lines derived from high-grade glioma patients. Oncol Rep. 2014;31(4):1683–90. https://doi.org/10.3892/or.2014.3049.
Reardon D, Peereboom D, Nabors B, Fink K, Phuphanich S, Mikkelsen T, et al. ATIM-11. Phase 2 trial of SL-701, a novel immunotherapy comprised of synthetic short peptides against GBM targets IL-13Rα2, EphA2, and survivin, in adults with second-line recurrent GBM: interim results. Neuro-Oncology. 2016;18(suppl_6):vi20-vi. https://doi.org/10.1093/neuonc/now212.076.
Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother: CII. 2013;62(1):125–35. https://doi.org/10.1007/s00262-012-1319-0.
Wen P, Reardon D, Phuphanich S, Aiken R, Landolfi J, Curry W, et al. AT-60 A randomized double blind placebo-controlled phase 2 trial of dendritic cell (DC) vaccine ICT-107 following standard treatment in newly diagnosed patients with GBM. Neuro-Oncology. 2014;16(Suppl 5):v22-v. https://doi.org/10.1093/neuonc/nou237.59.
Lucas KG, Bao L, Bruggeman R, Dunham K, Specht C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J Neuro-Oncol. 2011;103(2):231–8. https://doi.org/10.1007/s11060-010-0383-6.
Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, et al. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro-Oncology. 2012;14(3):246–55. https://doi.org/10.1093/neuonc/nor227.
Soderberg-Naucler C, Johnsen JI. Cytomegalovirus in human brain tumors: role in pathogenesis and potential treatment options. World J Exp Med. 2015;5(1):1–10. https://doi.org/10.5493/wjem.v5.i1.1.
Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–9. https://doi.org/10.1038/nature14320.
Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res. 2013;19(1):205–14. https://doi.org/10.1158/1078-0432.CCR-11-3358.
Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro-Oncology. 2014;16(2):274–9. https://doi.org/10.1093/neuonc/not203.
Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–25. https://doi.org/10.1158/1078-0432.CCR-05-0464.
Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64(14):4973–9. https://doi.org/10.1158/0008-5472.CAN-03-3505.
• Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547(7662):217–21. https://doi.org/10.1038/nature22991. This study provides proof-of concept that an effective antitumor T cell response can be generated using an individualized tumor-specific neoantigen vaccine.
Plautz GE, Miller DW, Barnett GH, Stevens GH, Maffett S, Kim J, et al. T cell adoptive immunotherapy of newly diagnosed gliomas. Clin Cancer Res. 2000;6(6):2209–18.
Tsuboi K, Saijo K, Ishikawa E, Tsurushima H, Takano S, Morishita Y, et al. Effects of local injection of ex vivo expanded autologous tumor-specific T lymphocytes in cases with recurrent malignant gliomas. Clin Cancer Res. 2003;9(9):3294–302.
Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014;74(13):3466–76. https://doi.org/10.1158/0008-5472.CAN-14-0296.
•• Reap EA, Suryadevara CM, Batich KA, Sanchez-Perez L, Archer GE, Schmittling RJ, et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res. 2018;78(1):256–64. https://doi.org/10.1158/0008-5472.CAN-17-0469. This study demonstrates proof-of concept of an improved polyclonal CMV-specific T cell response against recurrent glioblastoma by vaccination with a CMV-specific DC vaccine.
Krebs S, Rodriguez-Cruz TG, Derenzo C, Gottschalk S. Genetically modified T cells to target glioblastoma. Frontiers Oncol. 2013;3:322. https://doi.org/10.3389/fonc.2013.00322.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5(177):177ra38. https://doi.org/10.1126/scitranslmed.3005930.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33. https://doi.org/10.1056/NEJMoa1103849.
Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, et al. Suppression of human glioma xenografts with second-generation IL13R-specific chimeric antigen receptor-modified T cells. Clin Cancer Res. 2012;18(21):5949–60. https://doi.org/10.1158/1078-0432.CCR-12-0319.
Jin J, Joo KM, Lee SJ, Jo MY, Kim Y, Jin Y, et al. Synergistic therapeutic effects of cytokine-induced killer cells and temozolomide against glioblastoma. Oncol Rep. 2011;25(1):33–9.
Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23(10):1043–53. https://doi.org/10.1089/hum.2012.041.
Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7(275):275ra22. https://doi.org/10.1126/scitranslmed.aaa4963.
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21(3):629–37. https://doi.org/10.1038/mt.2012.210.
•• O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399) https://doi.org/10.1126/scitranslmed.aaa0984. First results from an exploratory cohort of patients treated with EGFRvIII-CAR T cells for recurrent glioblastoma.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–72. https://doi.org/10.1158/1078-0432.CCR-15-0428.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–9. https://doi.org/10.1056/NEJMoa1610497.
•• Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–101. https://doi.org/10.1001/jamaoncol.2017.0184. First results from a phase I study investigating treatment with autologous CMV-specific T cells genetically modified to express a HER2-CAR for recurrent glioblastoma.
Speranza MC, Passaro C, Ricklefs F, Kasai K, Klein SR, Nakashima H, et al. Preclinical investigation of combined gene-mediated cytotoxic immunotherapy and immune checkpoint blockade in glioblastoma. Neuro-Oncology. 2018;20(2):225–35. https://doi.org/10.1093/neuonc/nox139.
Kim JW, Kane JR, Young JS, Chang AL, Kanojia D, Morshed RA, et al. A Genetically Modified Adenoviral Vector with a Phage Display-Derived Peptide Incorporated into Fiber Fibritin Chimera Prolongs Survival in Experimental Glioma. Hum Gene Ther. 2015;26(9):635–46. https://doi.org/10.1089/hum.2015.008.
Forsyth P, Roldan G, George D, Wallace C, Palmer CA, Morris D, et al. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16(3):627–32. https://doi.org/10.1038/sj.mt.6300403.
Nandi S, Ulasov IV, Tyler MA, Sugihara AQ, Molinero L, Han Y, et al. Low-dose radiation enhances survivin-mediated virotherapy against malignant glioma stem cells. Cancer Res. 2008;68(14):5778–84. https://doi.org/10.1158/0008-5472.CAN-07-6441.
Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10(5):958–66. https://doi.org/10.1016/j.ymthe.2004.07.021.
Lang FF, Conrad C, Gomez-Manzano C, Tufaro F, Yung WKA, Sawaya R, et al. First-in-human phase I clinical trial of oncolytic delta-24-RGD (DNX-2401) with biological endpoints: implications for viro-immunotherapy. Neuro-oncology. 2014;16(Suppl 3):iii39-iii. https://doi.org/10.1093/neuonc/nou208.61.
Fueyo J, Gomez-Manzano C, Villalobos P, Rodriguez-Canales J, Wistuba I, Hess KR, et al. ATIM-08. immunomarkers in the DNX-2401 (DELTA-24-RGD) oncolytic virus phase I clinical trial. Neuro-oncology. 2017;19(suppl_6):vi27-vi. https://doi.org/10.1093/neuonc/nox168.104.
Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11(17):2389–401. https://doi.org/10.1089/104303400750038499.
Colombo F, Barzon L, Franchin E, Pacenti M, Pinna V, Danieli D, et al. Combined HSV-TK/IL-2 gene therapy in patients with recurrent glioblastoma multiforme: biological and clinical results. Cancer Gene Ther. 2005;12(10):835–48. https://doi.org/10.1038/sj.cgt.7700851.
Okada H, Pollack IF, Lotze MT, Lunsford LD, Kondziolka D, Lieberman F, et al. Gene therapy of malignant gliomas: a phase I study of IL-4-HSV-TK gene-modified autologous tumor to elicit an immune response. Hum Gene Ther. 2000;11(4):637–53. https://doi.org/10.1089/10430340050015824.
Wakabayashi T, Natsume A, Hashizume Y, Fujii M, Mizuno M, Yoshida J. A phase I clinical trial of interferon-beta gene therapy for high-grade glioma: novel findings from gene expression profiling and autopsy. J Gene Med. 2008;10(4):329–39. https://doi.org/10.1002/jgm.1160.
Chiocca EA, Smith KM, McKinney B, Palmer CA, Rosenfeld S, Lillehei K, et al. A phase I trial of Ad.hIFN-beta gene therapy for glioma. Mol Ther. 2008;16(3):618–26. https://doi.org/10.1038/sj.mt.6300396.
Saha D, Martuza RL, Rabkin SD. Macrophage polarization contributes to glioblastoma eradication by combination immunovirotherapy and immune checkpoint blockade. Cancer Cell. 2017;32(2):253–67 e5. https://doi.org/10.1016/j.ccell.2017.07.006.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Sylvia C. Kurz declares that she has no conflict of interest. Patrick Y. Wen receives research support from Agios, Angiochem, Astra Zeneca, Exelixis, Genentech/Roche, GlaxoSmith Kline, Karyopharm, Novartis, Sanofi-Aventis, Regeneron Pharmaceutical Inc., and Vascular Biogenics; he is on the advisory board for Abbvie, Cavion, Celldex, Genentech/Roche, Midatech, Momenta, Novartis, Novocure, SigmaTau, and Vascular Biogenics and is part of the Speaker’s Bureau for Merck.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Neuro-oncology
Rights and permissions
About this article

Cite this article
Kurz, S.C., Wen, P.Y. Quo Vadis—Do Immunotherapies Have a Role in Glioblastoma?. Curr Treat Options Neurol 20, 14 (2018). https://doi.org/10.1007/s11940-018-0499-0
Published:
DOI: https://doi.org/10.1007/s11940-018-0499-0