
Abstract
Purpose of Review
Studies of the neurobiology and treatment of PTSD have highlighted many aspects of the pathophysiology of this disorder that might be relevant to treatment. The purpose of this review is to highlight the potential clinical importance of an often-neglected consequence of stress models in animals that may be relevant to PTSD: the stress-related loss of synaptic connectivity.
Recent Findings
Here, we will briefly review evidence that PTSD might be a “synaptic disconnection syndrome” and highlight the importance of this perspective for the emerging therapeutic application of ketamine as a potential rapid-acting treatment for this disorder that may work, in part, by restoring synaptic connectivity.
Summary
Synaptic disconnection may contribute to the profile of PTSD symptoms that may be targeted by novel pharmacotherapeutics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The field of post-traumatic stress disorder (PTSD) research has advanced significantly since the earliest attempts to embed our understanding of this disorder within a modern translational neuroscience context [1, 2]. So far, the greatest progress has been made in studies related to the regulation of fear. PTSD is associated with a bias toward viewing neutral stimuli as threat-related, increased generalization of fear, deficits in fear extinction, and altered processing of threatening contexts that are presumed to contribute to fear-related PTSD symptoms including anxious arousal (startle, hypervigilance), intrusive trauma-like symptoms (intrusive thoughts, nightmares, flashbacks, emotional/physiologic reactivity), and avoidance of thoughts and reminders of the traumas [3,4,5,6,7,8,9,10, 11•]. These advances have informed the evolution of cognitive and behavioral treatments for PTSD [12, 13], and have led to the testing of pharmacologic approaches that might enhance these processes [14, 15•, 16, 17]. However, even within this area of research, it is not entirely clear why trauma-related fear memories are often so highly resistant to extinction. For example, it is not known whether the persistence of fear-related symptoms in PTSD reflects a deficit in neuroplasticity and whether these neuroplasticity deficits, in turn, have an underlying structural component.
Toward a Synaptic Deficit Hypothesis
A model of PTSD based entirely on fear conditioning may be too narrow to explain the breadth of associated symptoms. Other PTSD symptoms listed in DSM-5 [18] may not be consequences of dysregulated fear, such as dysphoric arousal (difficulty concentrating, difficulty sleeping), anhedonia (loss of interest, detachment), and externalizing symptoms (irritability/anger, self-destructive, or reckless behavior) [3], as well as symptoms including guilt, shame, and cognitive impairments [19•]. Some of these symptoms, such as emotional/behavior disinhibition [20], apathy [21], and impaired attention [22] are also associated with neural lesions that impair the functional connectivity of the brain, as might occur in the context of traumatic brain injury or cerebrovascular disease.
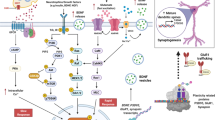
The resemblance of some PTSD symptoms to symptoms associated with impaired synaptic connectivity may be more than coincidence. It is well established that chronic stress causes neural atrophy and decreases the number of synapses within cortical and limbic circuits implicated in the regulation of mood, cognition, and behavior [23•]. Glutamate synapses are the dominant form of synaptic connectivity in these circuits. As reviewed in Fig. 1, stress compromises the integrity of signaling via glutamate synapses in several ways including by reducing signaling via brain-derived neurotrophic factor (BDNF) and impairing its downstream intracellular signaling. As a consequence, there are reductions in the number of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptors in the synapse, reductions in the size of dendritic spines, loss of dendritic spines supporting synaptic connectivity, and even loss of larger dendritic elements resulting in decreased dendritic complexity.

Stress causes neural atrophy and synapse loss. a The influence of repeated-restraint stress (7 d) on pyramidal neurons (layer V) in the medial prefrontal cortex (mPFC) of rat. The left-hand set of images shows that stress reduces the number and length of apical dendrites. The right-hand set of images shows a magnified segment of dendrite, with its spines at the point of synaptic contacts with neuronal inputs to the mPFC; repeated stress significantly decreases the number of spine synapses. b Under normal conditions, stimulation of the presynaptic neuron releases glutamate, resulting in the activation of postsynaptic glutamate AMPA receptors and depolarization; this causes activation of multiple intracellular pathways, including the BDNF-TrkB signaling pathway (and the downstream kinases Akt and ERK) and the mTORC1 pathway. These pathways are essential for regulation of synaptic plasticity, a fundamental adaptive learning mechanism that includes maturation (increased spine-head diameter) and an increase in the number of synapses. This process requires mTORC1-mediated de novo protein synthesis of synaptic proteins, including glutamate GluA1 AMPA receptors and PSD95. Repeated stress decreases BDNF and mTORC1 signaling in part via upregulation of the negative regulator REDD1 (regulated in DNA damage and repair), which decreases the synthesis of synaptic proteins and thereby contributes to a decreased number of spine synapses. Other proteins that are involved in the regulation of synaptic plasticity include GSK3 and protein phosphatase 1 (PP1) (from [23•])
Factors contributing to synaptic loss in chronic stress and PTSD may include disturbances in glucocorticoid receptor (GR) signaling, neuroinflammation, and deficits in neurotrophin signaling [24, 25, 26•, 27, 28]. Hypothalamo-pituitary adrenal axis function in PTSD is manifest at many levels including alterations in diurnal cortisol levels [29,30,31], increased GR numbers [32] or GR function [33], and alterations in GR signaling-related proteins such as the GR chaperone protein, FK506 binding protein 5 (FKBP5) [34•], and serum and glucocorticoid-regulated kinase 1 (SGK1) [35•]. With so many alterations, the impact of hypothalamo-pituitary-adrenal axis (HPA) changes may be complex, contributing to both resilience and vulnerability. This confusion has led to the testing of both GR agonists [36, 37] and antagonists [38] as treatments for PTSD. Elevations in pro-inflammatory cytokines including interleukin 1β, interleukin 6, tumor necrosis factor α, and interferon γ are associated with PTSD [39]. One consequence of the combined hypothalamo-pituitary-adrenal axis dysregulation and pro-inflammatory state may be to compromise the function of glia responsible for maintaining synaptic glutamate homeostasis, resulting in neurotoxic elevations of extrasynaptic glutamate levels [40].
To date, there are limited clinical data that directly support this hypothesis [34•]. One published pilot study of postmortem tissue evaluated 500 dendritic spines from the ventral medial frontal cortex tissue from eight PTSD cases and eight comparison subjects. They reported that the remaining dendritic spines in PTSD tended to be immature (stubby spines as opposed to mature mushroom spines). They also reported that elevated messenger RNA (mRNA) levels for FKBP5, which codes for a chaperone protein for the glucocorticoid receptor, were associated with reduced mushroom spine density.
There are neuroimaging data that indirectly support the hypothesis of deficits in synaptic connectivity in PTSD. These findings include reductions in cortical thickness [41], decreased subcortical volumes on MRI [42,43,44,45], reduced integrity of white matter pathways on diffusion-weighted imaging (DTI) [46, 47], and reductions in cortical functional connectivity in some studies [48], but more complex patterns of functional connectivity changes in other studies [49,50,51]. In these studies, regional reductions in cortical volumes, deficits in structural connectivity, and reduced functional connectivity were associated with PTSD symptoms, cognitive impairments, and overall functional impairment, suggesting their clinical relevance.
Synaptic loss associated with PTSD may aggravate the impact of other pathophysiologic processes. PTSD is often comorbid with conditions that independently may contribute to synaptic pruning including aging [52], traumatic brain injury [53], major depression [23•], high levels of alcohol consumption [54,55,56], and other medical conditions. The connectivity deficits of PTSD may exacerbate the impact of brain injury contributing to synergy in cortical network dysregulation, cognitive dysfunction, symptoms, and functional impairment as well as increased PTSD rates the TBI population [57,58,59, 60•, 61, 62]. Similar concerns apply to comorbid alcohol use disorders [63].
In summary, there are compelling preclinical data and early clinical correlates suggesting that synaptic loss may be an important feature of the neuropathology of PTSD. The MRI findings reviewed above suggest that the structural changes occur in brain regions involved in the executive control of emotion, such as the medial prefrontal cortex; the emotional appraisal of potentially threatening contexts, such as the anterior hippocampus; the generation of emotional states, such as the amygdala and insula; and in the white matter pathways connecting these regions. In computational models of circuit function, adequate integrity of synaptic connectivity is needed to generate and maintain appropriate representations of information [64], to support adaptive neuroplasticity [23•], and to adaptively regulate circuits generating emotional states [65]. These functional roles for normally dense synaptic connectivity support the hypothesis that impairments in these functional domains might emerge from synaptic loss, contributing to PTSD symptoms.
Synaptic Deficits and Circuit Function
How might synaptic loss impair the regulation of cortical circuits? The nature of synaptic loss described in the preclinical studies is rather subtle. Generally speaking, stress reduces the richness of synaptic connections rather than obliterating the integrity of a brain region or the connections between regions of the brain, as might occur in the context of traumatic brain injury.
Computational neuroscience studies suggest that synaptic loss may impair cortical network function in subtle and, perhaps, paradoxical ways. Stress-related synaptic loss has been best studied in the hippocampus. Clinical studies suggest that anterior hippocampal structural deficits in PTSD are associated to a moderate degree with reduced functional connectivity, profiles of PTSD symptoms [44], and impaired memory [66, 67]. In animals, both chronic stress and chronic glucocorticoid administration produce apical dendrite retraction first in the CA3 region and then in other hippocampal regions [68]. Loss of dendritic connectivity in both conditions is associated with reduced neuroplasticity, as reflected by the capacity to induce long-term potentiation, and impaired memory [69,70,71]. The learning impairments associated with chronic stress are also associated with altered properties of the place cells within the hippocampus that represent spatial information. These cells become less stable and more cue-dependent in representing spatial information [71]. This reduction in the stability and integrity of the neural representation of information by the hippocampus is consistent with earlier computational models of synaptic loss [72]. Together, these studies suggest that stress-dependent synaptic loss in the hippocampus, and perhaps other regions, may compromise cortical network functions by affecting the integrity of network functions and by undermining neuroplasticity.
However, there are conflicting data about the precise nature of stress-related disturbances in hippocampal network function. While some studies do not report altered neural excitability in association hippocampal dendritic atrophy produced by chronic stress [69, 73], other studies report increases in neural excitability [74]. The latter have informed a computational hippocampal network model in which dendritic atrophy produces increased excitability that impairs neuroplasticity by saturating long-term potentiation [75]. Another study suggests that chronic stress produces a dysfunctional dyscoordination of heightened excitability and neuroplasticity in some neural compartments, and reduced neuroplasticity in others [76]. This work is at an early stage, and it is hoped that continued progress in computational approaches to network alterations in PTSD will yield deeper insights into brain-behavior relationships.
Ketamine and Synaptic Therapeutics for PTSD
One of the most important questions raised by a focus on synaptic loss in PTSD is whether the resulting perspective informs the identification of novel therapeutics for this disorder. One might first explore how environmental factors might influence synaptic connectivity. Animals raised in environments without much social, sensory, or activity-related stimulation developed reduced levels of cortical synaptic connectivity, while environmental enrichment has the opposite effect [77,78,79,80]. For example, environmental enrichment in animals protects animals against hippocampal dendritic atrophy and the associated memory impairment produced by chronic stress [81]. One form of environmental enrichment that has received particular attention is exercise. In animals, exercise has many effects that enhance neuroplasticity, including promoting neurogenesis, increasing dendritic complexity, and increasing synaptic connectivity ([82,83,84]; see Fig. 2). These effects appear to be dependent on the level of BDNF [84]. The effects of exercise are sufficient to protect against the detrimental stress-like effects of chronic corticosterone on neurogenesis and synaptic connectivity [85]. Exercise appears to have beneficial effects on symptoms severity in people with PTSD [86]. However, it is not yet clear whether these benefits are mediated by enhancements in synaptic connectivity.

Evidence that voluntary exercise increases dendritic spine density in the dentate gyrus of the hippocampus in rats. The left figure presents the number of dendritic spines per 10 μm for dentate granule cells. The asterisk symbol indicates p < .05. The right figure presents results from Golgi-impregnated dentate granule cells at ×100 magnification from control (A) and exercised (B) animals. Scale bar = 10 μM [82]
What about exercising the brain? Cognitive remediation therapies might be beneficial for PTSD. This is a relatively new area of research. Some studies suggest that psychotherapy may increase regional cortical volumes or white matter integrity (increased fractional anisotropy in diffusion weighted imaging) in PTSD, but these increases are not universally associated with clinical improvement [87•, 88, 89]. As opposed to cognitive therapies, which aim to address aberrant thought patterns, cognitive remediation therapies are a form of “brain exercise” that aims to engage experience-dependent neuroplasticity in order to restore or enhance functional connectivity [90, 91]. This approach has been studied extensively in the field of schizophrenia research [92], but it has received relatively little attention as a treatment for PTSD symptoms.
The capacity of the brain to protect and restore synaptic connectivity in the context of typical daily activity raises questions as to why synaptic deficits associated with stress persist within the context of PTSD. In fact, most people do recover, at least partly, from severe and repeated traumas. For example, the lifetime prevalence of PTSD among Vietnam veterans was approximately 30% [93]. However, the cross-sectional rate of PTSD declined over time to approximately 15% 10 years after the Vietnam War [93] and approximately 4.5% 40 years after the war [94•]. One possibility is that synaptic deficits persist in PTSD because the symptoms constitute a state of chronic stress. In other words, the persisting symptoms associated with PTSD such as fear, depression, insomnia, guilt, demoralization, shame, and numbing may evoke complex neurobiological responses that undermine synaptic integrity such as dysregulation of the hypothalamo-pituitary-adrenal (HPA) axis [95], induction of a chronic pro-inflammatory state [96], and reductions in neurotrophin signaling [23•]. Supporting this view, as noted earlier, HPA dysregulation [25], elevations of pro-inflammatory cytokines [27], and reductions in plasma BDNF levels [97, 98] have been reported in people diagnosed with PTSD. Further, volume loss on MRI appears to be a predictor of the persistence of PTSD symptoms with treatment [99, 100]. Thus, it is possible that persisting neuroendocrine dysregulation and neuroinflammation may contribute to the chronicity of PTSD via enhancing synaptic connectivity deficits and compromising neuroplasticity (see Fig. 3).

This figure illustrates a “vicious cycle” through which neuroinflammation, HPA activation, and stress-related alterations in circuit function interact to contribute to synaptic loss and how synaptic loss then contributes to the chronicity of PTSD by compromising the regulation and neuroplasticity of emotion-related neural networks (modified from [101])
Current pharmacotherapies for PTSD may work, in part, by restoring synaptic connectivity. The most commonly prescribed agents, antidepressant medications, appear to promote synaptic connectivity via raising BDNF levels, enhancing signaling via CREB, and promoting synaptic growth and neurogenesis [102, 103]. Long-term treatment with antidepressant medications appears to increase hippocampal volume and improves memory in individuals with PTSD [104], potentially reversing the deficits described in patients [43, 105, 106]. Interpreting the effects of long-term treatments are complex. Long-term therapeutic effects of antidepressant medications might be mediated by direct neural or anti-inflammatory effects [107, 108] of these drugs. However, they also might reflect the long-term effects of changes in mood or activity, as suggested by the studies of psychotherapy effects on brain function, reviewed earlier.
The emergence of the rapid-acting antidepressants has created the opportunity to study a treatment that might work to reduce PTSD symptoms, in part, by directly restoring synaptic connectivity [109, 110]. The possibility of rapid antidepressant effects produced by N-methyl-D-aspartate (NMDA) glutamate receptor antagonists was suggested by animal models [111]. This area of research was spurred by the observation that a single dose of the NMDA receptor antagonist, ketamine, produced pronounced antidepressant effects in the majority of patients with treatment-resistant symptoms of depression [40, 112]. More recently, there is preliminary evidence that ketamine produces rapid benefits in patients diagnosed with PTSD [113]. In this study, ketamine produced improvement in PTSD symptoms even when controlling for its antidepressant effects and in patients without comorbid symptoms of depression.
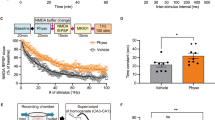
As presented in Fig. 4, ketamine may work by rapidly enhancing synaptic connectivity and by rapidly increasing dendritic spines in the apical dendrites of pyramidal neurons in superficial cortical layers, reversing the effects of stress [109, 114]. The clinical evidence supporting this hypothesis is limited currently, but it is being actively studied. In depression, reductions in cortical functional connectivity as measured by functional MRI appear to be ameliorated within 24 h by a single dose of ketamine, in conjunction with clinical improvement [115•, 116]. Similar studies are underway in PTSD patients. In animals, a single dose of ketamine causes a proliferation of functional dendritic spines. There are at least three primary hypotheses as to how ketamine might produce these effects [23•, 40]. One hypothesis suggests that exerts its effects by stimulating glutamate release. This glutamate release may trigger a chain of neural effects that begins with the stimulation of synaptic AMPA glutamate receptors and involves increased levels and release of BDNF, stimulation of tropomyosin receptor kinase B (TrkB) receptors (the receptor for BDNF), enhanced signaling via the Akt/molecular target of rapamycin (mTORC) signaling pathway, increases in the synthesis of proteins associated with dendritic spines, and the rapid emergence of new spines (see Fig. 5). Another important hypothesis is that ketamine exerts its clinical effects by blocking extrasynaptic NMDA glutamate receptors, reducing the phosphorylation of eukaryotic elongation factor-2 (eELF2), reducing the phosphorylation of the associated kinase, enhancing AMPA signaling, and increasing BDNF levels [117]. A third hypothesis, which might be viewed as a variant of the prior hypothesis, suggests that a ketamine metabolite, 2R,6R–hydroxynorketamine, enhances AMPA signaling through a mechanism that remains to be determined [118•]. Other hypotheses include the possibility that antidepressant effects of ketamine are mediated by its anti-inflammatory effects [119, 120], its effects on nitric oxide signaling [121, 122], its modulation of GABA-B receptor signaling [123], and other effects.

Confocal photomicrographs of labeled layer V pyramidal neurons in the medial prefrontal cortex. The left figure shows the low numbers of dendritic spines present in the dendrites of layer V pyramidal neurons after 21 days of chronic uncontrollable stress (CUS). The right figure illustrates the reversal by a single dose of ketamine 1 day later. Red arrows highlight dendritic spines present after ketamine

Ketamine causes a burst of glutamate that is thought to occur via disinhibition of GABA interneurons; the tonic firing of these GABA interneurons is driven by NMDA receptors, and the active, open-channel state allows ketamine to enter and block channel activity. The resulting glutamate burst stimulates AMPA receptors, which causes depolarization and activation of voltage-dependent Ca2+ channels (VDCC), leading to release of BDNF and stimulation of TrkB receptors and activation of Akt, which then increases mTORC1 signaling, leading to the increased synthesis of proteins that are required for synapse maturation and formation (i.e., GluA1 and PSD95). Under conditions in which BDNF release is blocked or neutralized, or in which mTORC1 signaling is blocked by rapamycin, the synaptic and behavioral actions of ketamine are blocked. Scopolamine also causes a glutamate burst via blockade of acetylcholine muscarinic M1 (ACh-M1) receptors on GABA interneurons. Antagonists of mGluR2/3 also produce rapid antidepressant actions via blockade of presynaptic autoreceptors that inhibit the release of glutamate. Relapse to a depressive state is associated with a decrease of synapses on mPFC neurons, which could occur via stress and imbalance of endocrine hormones (cortisol), estrogen, inflammatory cytokines, and metabolic and cardiovascular illnesses (from [23•])
If ketamine proves to produce lasting improvement in PTSD, it may serve as a prototype for other putative rapid-acting antidepressants. For example, preclinical studies suggest that muscarinic cholinergic receptor antagonists [124], metabotropic glutamate receptor-2 antagonists [125], and AMPAkines [126] might produce rapid antidepressant effects by directly or indirectly enhancing AMPA receptor signaling. Only the first of these mechanisms has been tested as an antidepressant in humans, with promising early results [127].
However, the persisting potentiation of neuroplasticity by ketamine suggests a novel role in the treatment of PTSD: the enhancement of fear extinction among patients who have failed to respond to exposure-based treatments. Deficits in fear extinction in PTSD are a central challenge in treatment and modifications to traditional cognitive and behavioral therapies are a major focus of treatment development [128, 129]. There has long been an interest in developing medications that might enhance this process [130]. One strategy that of enhancing NMDA receptor signaling using D-cycloserine has not proven effective in PTSD [131]. Ketamine, perhaps by increasing synaptic connectivity, appears to increase neuroplasticity and to enhance fear extinction in animals [132•]. This raises the possibility that the therapeutic effects of ketamine in PTSD might be potentiated by using it to enhance the efficacy of progressive exposure, cognitive processing therapy, eye movement desensitization and reprocessing (EMDR), or other exposure-based therapies.
Summary and Implications
Synaptic deficits associated with PTSD may contribute to the complex profile of symptoms and functional impairments associated with this disorder. These deficits may arise in part from the neurobiology of chronic stress associated with persisting symptoms of PTSD. In turn, synaptic deficits may compromise neuroplasticity and impair resilience among individuals with PTSD, contributing to symptom chronicity and compromising clinical responses to current treatments. A wide range of interventions could be viewed as targeting impaired synaptic connectivity, such as stimulating life activities, including exercise. However, the ability to respond to these forms of self-healing may be compromised by PTSD-related neuroplasticity deficits. Long-term antidepressant treatment may contribute to clinical recovery by promoting synaptic plasticity. However, recent rapid-acting antidepressants may more directly target synaptic deficits associated with PTSD and there is now preliminary evidence of the rapid efficacy of ketamine. Further, ketamine may open a window of increased neuroplasticity where cognitive and behavioral therapies might be more effective in treating PTSD symptoms.
This review relies heavily on many sources of data that are quite preliminary, and so some of its key assertions may be vulnerable to being disproved by future research. For example, postmortem studies of brain tissue from individuals with PTSD are in their infancy. Results from studies employing PET radiotracers that might serve to quantify cortical synaptic density in vivo in PTSD before and after treatment have yet to be reported. Inferences about synaptic connectivity based on MRI-based imaging methods are likely to be risky. Further, our limited knowledge of the neurobiology of PTSD limits our ability to rigorously evaluate the applicability of findings from animal models of stress to the pathophysiology and treatment of this disorder. Thus, the purpose of this review is to draw attention to the importance of synaptic loss for PTSD, balancing the focus on fear acquisition in considerations of the pathophysiology, and treatment of PTSD, with the aim of stimulating more research in this area.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
van der Kolk B, Greenberg M, Boyd H, Krystal J. Inescapable shock, neurotransmitters, and addiction to trauma: toward a psychobiology of post traumatic stress. Biol Psychiatry. 1985;20(3):314–25.
Charney DS, Deutch AY, Krystal JH, Southwick SM, Davis M. Psychobiologic mechanisms of posttraumatic stress disorder. Arch Gen Psychiatry. 1993;50(4):295–305.
Armour C, Contractor A, Shea T, Elhai JD, Pietrzak RH. Factor structure of the PTSD checklist for DSM-5: relationships among symptom clusters, anger, and impulsivity. J Nerv Ment Dis. 2016;204(2):108–15.
• Desmedt A, Marighetto A, Piazza PV. Abnormal fear memory as a model for posttraumatic stress disorder. Biol Psychiatry. 2015;78(5):290–7. An important hypothesis related to PTSD.
VanElzakker MB, Dahlgren MK, Davis FC, Dubois S, Shin LM. From Pavlov to PTSD: the extinction of conditioned fear in rodents, humans, and anxiety disorders. Neurobiol Learn Mem. 2014;113:3–18.
Maren S, Phan KL, Liberzon I. The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci. 2013;14(6):417–28.
Mahan AL, Ressler KJ. Fear conditioning, synaptic plasticity and the amygdala: implications for posttraumatic stress disorder. Trends Neurosci. 2012;35(1):24–35.
Andero R, Ressler KJ. Fear extinction and BDNF: translating animal models of PTSD to the clinic. Genes Brain Behav. 2012;11(5):503–12.
Johansen JP, Cain CK, Ostroff LE, LeDoux JE. Molecular mechanisms of fear learning and memory. Cell. 2011;147(3):509–24.
Briscione MA, Jovanovic T, Norrholm SD. Conditioned fear associated phenotypes as robust, translational indices of trauma-, stressor-, and anxiety-related behaviors. Front Psychiatry. 2014;5:88.
• Liberzon I, Abelson JL. Context processing and the neurobiology of post-traumatic stress disorder. Neuron. 2016;92(1):14–30. A paper that highlights the importance of context processing for PTSD.
Rothbaum BO, Davis M. Applying learning principles to the treatment of post-trauma reactions. Ann N Y Acad Sci. 2003;1008:112–21.
Rauch SA, Eftekhari A, Ruzek JI. Review of exposure therapy: a gold standard for PTSD treatment. J Rehabil Res Dev. 2012;49(5):679–87.
Fitzgerald PJ, Seemann JR, Maren S. Can fear extinction be enhanced? A review of pharmacological and behavioral findings. Brain Res Bull. 2014;105:46–60.
• Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ. Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther. 2015;149:150–90. A paper that highlights the use of medications to enhance the effects of psychotherapy by increasing neuroplasticity.
Donovan E. Propranolol use in the prevention and treatment of posttraumatic stress disorder in military veterans: forgetting therapy revisited. Perspect Biol Med. 2010;53(1):61–74.
Brunet A, Orr SP, Tremblay J, Robertson K, Nader K, Pitman RK. Effect of post-retrieval propranolol on psychophysiologic responding during subsequent script-driven traumatic imagery in post-traumatic stress disorder. J Psychiatr Res. 2008;42(6):503–6.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, D.C.: American Psychiatric Press; 2013.
• Scott JC, Matt GE, Wrocklage KM, Crnich C, Jordan J, Southwick SM, et al. A quantitative meta-analysis of neurocognitive functioning in posttraumatic stress disorder. Psychol Bull. 2015;141(1):105–40. A systematic meta-analysis of cognitive impairments associated with PTSD.
Laskowski RA, Creed JA, Raghupathi R. Frontiers in neuroengineering pathophysiology of mild TBI: implications for altered signaling pathways. In: Kobeissy FH, editor. Brain Neurotrauma: molecular, neuropsychological, and rehabilitation aspects. Boca Raton: CRC Press/Taylor & Francis; 2015.
Starkstein SE, Pahissa J. Apathy following traumatic brain injury. Psychiatr Clin North Am. 2014;37(1):103–12.
Baldassarre A, Ramsey LE, Siegel JS, Shulman GL, Corbetta M. Brain connectivity and neurological disorders after stroke. Curr Opin Neurol. 2016;29(6):706–13.
• Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 2016;22(3):238–49. A paper that highlights the role of synaptic connectivity in stress, depression, and antidepressant therapeutic effects.
Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13(1):22–37.
Daskalakis NP, Lehrner A, Yehuda R. Endocrine aspects of post-traumatic stress disorder and implications for diagnosis and treatment. Endocrinol Metab Clin N Am. 2013;42(3):503–13.
• Daskalakis NP, Cohen H, Nievergelt CM, Baker DG, Buxbaum JD, Russo SJ, et al. New translational perspectives for blood-based biomarkers of PTSD: from glucocorticoid to immune mediators of stress susceptibility. Exp Neurol. 2016;284(Pt B):133–40. A review highlighting two important areas of biomarker development for PTSD.
Pace TW, Heim CM. A short review on the psychoneuroimmunology of posttraumatic stress disorder: from risk factors to medical comorbidities. Brain Behav Immun. 2011;25(1):6–13.
Golier J, Yehuda R. Neuroendocrine activity and memory-related impairments in posttraumatic stress disorder. Dev Psychopathol. 1998;10(4):857–69.
Mason JW, Giller EL, Kosten TR, Ostroff RB, Podd L. Urinary free-cortisol levels in posttraumatic stress disorder patients. J Nerv Ment Dis. 1986;174(3):145–9.
Mason JW, Wang S, Yehuda R, Lubin H, Johnson D, Bremner JD, et al. Marked lability in urinary cortisol levels in subgroups of combat veterans with posttraumatic stress disorder during an intensive exposure treatment program. Psychosom Med. 2002;64(2):238–46.
Inslicht SS, Marmar CR, Neylan TC, Metzler TJ, Hart SL, Otte C, et al. Increased cortisol in women with intimate partner violence-related posttraumatic stress disorder. Ann N Y Acad Sci. 2006;1071:428–9.
Yehuda R, Lowy MT, Southwick SM, Shaffer D, Giller EL Jr. Lymphocyte glucocorticoid receptor number in posttraumatic stress disorder. Am J Psychiatry. 1991;148(4):499–504.
Yehuda R, Southwick SM, Krystal JH, Bremner D, Charney DS, Mason JW. Enhanced suppression of cortisol following dexamethasone administration in posttraumatic stress disorder. Am J Psychiatr. 1993;150(1):83–6.
• Young KA, Thompson PM, Cruz DA, Williamson DE, Selemon LD. BA11 FKBP5 expression levels correlate with dendritic spine density in postmortem PTSD and controls. Neurobiol Stress. 2015;2:67–72. Perhaps the first paper to document synaptic deficits in PTSD based on the analysis of postmortem tissue.
• Licznerski P, Duric V, Banasr M, Alavian KN, Ota KT, Kang HJ, et al. Decreased SGK1 expression and function contributes to behavioral deficits induced by traumatic stress. PLoS Biol. 2015;13(10):e1002282. The first transcriptomic analysis of post-mortem cortical tissue in PTSD.
Zohar J, Yahalom H, Kozlovsky N, Cwikel-Hamzany S, Matar MA, Kaplan Z, et al. High dose hydrocortisone immediately after trauma may alter the trajectory of PTSD: interplay between clinical and animal studies. Eur Neuropsychopharmacol. 2011;21(11):796–809.
Yehuda R, Harvey PD, Buchsbaum M, Tischler L, Schmeidler J. Enhanced effects of cortisol administration on episodic and working memory in aging veterans with PTSD. Neuropsychopharmacology. 2007;32:2581–91.
Golier JA, Caramanica K, Michaelides AC, Makotkine I, Schmeidler J, Harvey PD, et al. A randomized, double-blind, placebo-controlled, crossover trial of mifepristone in Gulf War veterans with chronic multisymptom illness. Psychoneuroendocrinology. 2016;64:22–30.
Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, et al. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. 2015;2(11):1002–12.
Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73(12):1133–41.
Wrocklage KM, Averill LA, Cobb Scott J, Averill CL, Schweinsburg B, Trejo M, et al. Cortical thickness reduction in combat exposed U.S. veterans with and without PTSD. Eur Neuropsychopharmacology. 2017;27:515–25.
O'Doherty DC, Chitty KM, Saddiqui S, Bennett MR, Lagopoulos J. A systematic review and meta-analysis of magnetic resonance imaging measurement of structural volumes in posttraumatic stress disorder. Psychiatry Res. 2015;232(1):1–33.
Bremner JD, Randall P, Scott TM, Bronen RA, Seibyl JP, Southwick SM, et al. MRI-based measurement of hippocampal volume in patients with combat-related posttraumatic stress disorder. Am J Psychiatr. 1995;152(7):973–81.
Abdallah CG, Wrocklage KM, Averill CL, Akiki T, Schweinsburg B, Roy A, et al. Anterior hippocampal dysconnectivity in posttraumatic stress disorder: a dimensional and multimodal approach. Transl Psychiatry. 2017;7(2):e1045.
Pietrzak RH, Averill LA, Abdallah CG, Neumeister A, Krystal JH, Levy I, et al. Amygdala-hippocampal volume and the phenotypic heterogeneity of posttraumatic stress disorder: a cross-sectional study. JAMA Psychiatry. 2015;72(4):396–8.
Bazarian JJ, Donnelly K, Peterson DR, Warner GC, Zhu T, Zhong J. The relation between posttraumatic stress disorder and mild traumatic brain injury acquired during operations enduring freedom and Iraqi freedom: a diffusion tensor imaging study. J Head Trauma Rehabil. 2012;
Fani N, King TZ, Jovanovic T, Glover EM, Bradley B, Choi K, et al. White matter integrity in highly traumatized adults with and without post-traumatic stress disorder. Neuropsychopharmacology. 2012;37(12):2740–6.
Admon R, Leykin D, Lubin G, Engert V, Andrews J, Pruessner J, et al. Stress-induced reduction in hippocampal volume and connectivity with the ventromedial prefrontal cortex are related to maladaptive responses to stressful military service. Hum Brain Mapp. 2012;
Kennis M, van Rooij SJ, van den Heuvel MP, Kahn RS, Geuze E. Functional network topology associated with posttraumatic stress disorder in veterans. NeuroImage Clin. 2016;10:302–9.
King AP, Block SR, Sripada RK, Rauch S, Giardino N, Favorite T, et al. Altered default mode network (DMN) resting state functional connectivity following a mindfulness-based exposure therapy for posttraumatic stress disorder (PTSD) in combat veterans of Afghanistan and Iraq. Depress Anxiety. 2016;33(4):289–99.
Nicholson AA, Sapru I, Densmore M, Frewen PA, Neufeld RW, Theberge J, et al. Unique insula subregion resting-state functional connectivity with amygdala complexes in posttraumatic stress disorder and its dissociative subtype. Psychiatry Res. 2016;250:61–72.
Mattson MP, Magnus T. Ageing and neuronal vulnerability. Nat Rev Neurosci. 2006;7(4):278–94.
Przekwas A, Somayaji MR, Gupta RK. Synaptic mechanisms of blast-induced brain injury. Front Neurol. 2016;7:2.
de la Monte SM, Kril JJ. Human alcohol-related neuropathology. Acta Neuropathol. 2014;127(1):71–90.
Freund G, Ballinger WE. Loss of synaptic receptors can precede morphologic changes induced by alcoholism. Alcohol Alcohol Suppl. 1991;1:385–91.
Maksimovskiy AL, McGlinchey RE, Fortier CB, Salat DH, Milberg WP, Oscar-Berman M. White matter and cognitive changes in veterans diagnosed with alcoholism and PTSD. J Alcohol Drug Depend. 2014;2(1):144.
Hoffman SW, Harrison C. The interaction between psychological health and traumatic brain injury: a neuroscience perspective. Clin Neuropsychol. 2009;23(8):1400–15.
Yurgil KA, Barkauskas DA, Vasterling JJ, Nievergelt CM, Larson GE, Schork NJ, et al. Association between traumatic brain injury and risk of posttraumatic stress disorder in active-duty Marines. JAMA Psychiatry. 2014;71(2):149–57.
Stein MB, McAllister TW. Exploring the convergence of posttraumatic stress disorder and mild traumatic brain injury. Am J Psychiatry. 2009;166(7):768–76.
• Spielberg JM, McGlinchey RE, Milberg WP, Salat DH. Brain network disturbance related to posttraumatic stress and traumatic brain injury in veterans. Biol Psychiatry. 2015;78(3):210–6. An examination of the interplay of PTSD and TBI effects on brain imaging.
Elliott TR, Hsiao YY, Kimbrel NA, Meyer EC, DeBeer BB, Gulliver SB, et al. Resilience, traumatic brain injury, depression, and posttraumatic stress among Iraq/Afghanistan war veterans. Rehabil Psychol. 2015;60(3):263–76.
Combs HL, Berry DT, Pape T, Babcock-Parziale J, Smith B, Schleenbaker R, et al. The effects of mild traumatic brain injury, post-traumatic stress disorder, and combined mild traumatic brain injury/post-traumatic stress disorder on returning veterans. J Neurotrauma. 2015;32(13):956–66.
Starcevic A, Dimitrijevic I, Aksic M, Stijak L, Radonjic V, Aleksic D, et al. Brain changes in patients with posttraumatic stress disorder and associated alcoholism: MRI based study. Psychiatr Danub. 2015;27(1):78–83.
Hoffman RE, McGlashan TH. Synaptic elimination, neurodevelopment, and the mechanism of hallucinated “voices” in schizophrenia. Am J Psychiatr. 1997;154(12):1683–9.
McEwen BS, Morrison JH. The brain on stress: vulnerability and plasticity of the prefrontal cortex over the life course. Neuron. 2013;79(1):16–29.
Vythilingam M, Luckenbaugh DA, Lam T, Morgan CA 3rd, Lipschitz D, Charney DS, et al. Smaller head of the hippocampus in Gulf War-related posttraumatic stress disorder. Psychiatry Res. 2005;139(2):89–99.
Woodward SH, Kaloupek DG, Grande LJ, Stegman WK, Kutter CJ, Leskin L, et al. Hippocampal volume and declarative memory function in combat-related PTSD. J Int Neuropsychol Soc. 2009;15(6):830–9.
Conrad CD, Ortiz JB, Judd JM. Chronic stress and hippocampal dendritic complexity: methodological and functional considerations. Physiol Behav. 2016;
Pavlides C, Nivon LG, McEwen BS. Effects of chronic stress on hippocampal long-term potentiation. Hippocampus. 2002;12(2):245–57.
Pavlides C, Watanabe Y, McEwen BS. Effects of glucocorticoids on hippocampal long-term potentiation. Hippocampus. 1993;3(2):183–92.
Park M, Kim CH, Jo S, Kim EJ, Rhim H, Lee CJ, et al. Chronic stress alters spatial representation and bursting patterns of place cells in behaving mice. Sci Rep. 2015;5:16235.
Hoffman RE. Computer simulations of neural information processing and the schizophrenia-mania dichotomy. Arch Gen Psychiatry. 1987;44(2):178–88.
Tomar A, Polygalov D, Chattarji S, McHugh TJ. The dynamic impact of repeated stress on the hippocampal spatial map. Hippocampus. 2015;25(1):38–50.
Conrad CD, Jackson JL, Wise LS. Chronic stress enhances ibotenic acid-induced damage selectively within the hippocampal CA3 region of male, but not female rats. Neuroscience. 2004;125(3):759–67.
Narayanan R, Chattarji S. Computational analysis of the impact of chronic stress on intrinsic and synaptic excitability in the hippocampus. J Neurophysiol. 2010;103(6):3070–83.
Xu A, Cui S, Wang JH. Incoordination among subcellular compartments is associated with depression-like behavior induced by chronic mild stress. Int J Neuropsychopharmacol. 2016;19(5). https://doi.org/10.1093/ijnp/pyv122.
Chaudhury S, Sharma V, Kumar V, Nag TC, Wadhwa S. Activity-dependent synaptic plasticity modulates the critical phase of brain development. Brain Dev. 2016;38(4):355–63.
Birch AM, McGarry NB, Kelly AM. Short-term environmental enrichment, in the absence of exercise, improves memory, and increases NGF concentration, early neuronal survival, and synaptogenesis in the dentate gyrus in a time-dependent manner. Hippocampus. 2013;23(6):437–50.
Jung CK, Herms J. Structural dynamics of dendritic spines are influenced by an environmental enrichment: an in vivo imaging study. Cereb Cortex. 2014;24(2):377–84.
Nichols JA, Jakkamsetti VP, Salgado H, Dinh L, Kilgard MP, Atzori M. Environmental enrichment selectively increases glutamatergic responses in layer II/III of the auditory cortex of the rat. Neuroscience. 2007;145(3):832–40.
McLaughlin KJ, Gomez JL, Baran SE, Conrad CD. The effects of chronic stress on hippocampal morphology and function: an evaluation of chronic restraint paradigms. Brain Res. 2007;1161:56–64.
Eadie BD, Redila VA, Christie BR. Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dendritic complexity, and spine density. J Comp Neurol. 2005;486(1):39–47.
Kempermann G, Fabel K, Ehninger D, Babu H, Leal-Galicia P, Garthe A, et al. Why and how physical activity promotes experience-induced brain plasticity. Front Neurosci. 2010;4:189.
Vaynman SS, Ying Z, Yin D, Gomez-Pinilla F. Exercise differentially regulates synaptic proteins associated to the function of BDNF. Brain Res. 2006;1070(1):124–30.
Yau SY, Li A, Zhang ED, Christie BR, Xu A, Lee TM, et al. Sustained running in rats administered corticosterone prevents the development of depressive behaviors and enhances hippocampal neurogenesis and synaptic plasticity without increasing neurotrophic factor levels. Cell Transplant. 2014;23(4–5):481–92.
Whitworth JW, Ciccolo JT. Exercise and post-traumatic stress disorder in military veterans: a systematic review. Mil Med. 2016;181(9):953–60.
• Kennis M, van Rooij SJ, do Tromp PM, Fox AS, Rademaker AR, Kahn RS, et al. Treatment outcome-related white matter differences in veterans with posttraumatic stress disorder. Neuropsychopharmacology. 2015;40(10):2434–42. A paper highlighting the clinical impact of white matter connectivity disturbances.
Laugharne J, Kullack C, Lee CW, McGuire T, Brockman S, Drummond PD, et al. Amygdala volumetric change following psychotherapy for posttraumatic stress disorder. J Neuropsychiatry Clin Neurosci. 2016; doi:https://doi.org/10.1176/appi.neuropsych.16010006.
Bossini L, Santarnecchi E, Casolaro I, Koukouna D, Caterini C, Cecchini F, et al. Morphovolumetric changes after EMDR treatment in drug-naive PTSD patients. Riv Psichiatr. 2017;52(1):24–31.
Buonomano DV, Merzenich MM. Cortical plasticity: from synapses to maps. Annu Rev Neurosci. 1998;21:149–86.
Merzenich MM, Van Vleet TM, Nahum M. Brain plasticity-based therapeutics. Front Hum Neurosci. 2014;8:385.
Radhakrishnan R, Kiluk BD, Tsai J. A meta-analytic review of non-specific effects in randomized controlled trials of cognitive remediation for schizophrenia. Psychiatry Q. 2016;87(1):57–62.
Kulka RA, Schlenger WE, Fairbank J. Trauma and the Vietnam war generation. New York: Bruner-Mazel; 1990.
• Marmar CR, Schlenger W, Henn-Haase C, Qian M, Purchia E, Li M, et al. Course of posttraumatic stress disorder 40 years after the Vietnam War: findings from the National Vietnam Veterans Longitudinal Study. JAMA Psychiatry. 2015;72(9):875–81. A classic long-term follow-up of PTSD in Vietnam Era veterans.
Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13(1):22–37.
Haroon E, Miller AH, Sanacora G. Inflammation, glutamate, and glia: a trio of trouble in mood disorders. Neuropsychopharmacology. 2017;42(1):193–215.
Dell'Osso L, Carmassi C, Del Debbio A, Catena Dell'Osso M, Bianchi C, da Pozzo E, et al. Brain-derived neurotrophic factor plasma levels in patients suffering from post-traumatic stress disorder. Prog Neuro-Psychopharmacol Biol Psychiatry. 2009;33(5):899–902.
Angelucci F, Ricci V, Gelfo F, Martinotti G, Brunetti M, Sepede G, et al. BDNF serum levels in subjects developing or not post-traumatic stress disorder after trauma exposure. Brain Cogn. 2014;84(1):118–22.
van Rooij SJ, Kennis M, Sjouwerman R, van den Heuvel MP, Kahn RS, Geuze E. Smaller hippocampal volume as a vulnerability factor for the persistence of post-traumatic stress disorder. Psychol Med. 2015;45(13):2737–46.
Colvonen PJ, Glassman LH, Crocker LD, Buttner MM, Orff H, Schiehser DM, et al. Pretreatment biomarkers predicting PTSD psychotherapy outcomes: a systematic review. Neurosci Biobehav Rev. 2017;75:140–56.
Averill LA, Purohit P, Averill CL, Boesl MA, Krystal JH, Abdallah CG. Glutamate dysregulation and glutamatergic therapeutics for PTSD: evidence from human studies. Neurosci Lett. 2017;649:147–55.
Carlezon WA Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28(8):436–45.
Duman RS. Role of neurotrophic factors in the etiology and treatment of mood disorders. NeuroMolecular Med. 2004;5(1):11–25.
Vermetten E, Vythilingam M, Southwick SM, Charney DS, Bremner JD. Long-term treatment with paroxetine increases verbal declarative memory and hippocampal volume in posttraumatic stress disorder. Biol Psychiatry. 2003;54(7):693–702.
Bremner JD, Vythilingam M, Vermetten E, Southwick SM, McGlashan T, Nazeer A, et al. MRI and PET study of deficits in hippocampal structure and function in women with childhood sexual abuse and posttraumatic stress disorder. Am J Psychiatry. 2003;160(5):924–32.
Bremner JD, Scott TM, Delaney RC, Southwick SM, et al. Deficits in short-term memory in posttraumatic stress disorder. Am J Psychiatr. 1993;150(7):1015–9.
Norden DM, Devine R, Bicer S, Jing R, Reiser PJ, Wold LE, et al. Fluoxetine prevents the development of depressive-like behavior in a mouse model of cancer related fatigue. Physiol Behav. 2015;140:230–5.
Valera E, Ubhi K, Mante M, Rockenstein E, Masliah E. Antidepressants reduce neuroinflammatory responses and astroglial alpha-synuclein accumulation in a transgenic mouse model of multiple system atrophy. Glia. 2014;62(2):317–37.
Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–64.
Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7(5):426–37.
Skolnick P, Popik P, Trullas R. Glutamate-based antidepressants: 20 years on. Trends Pharmacol Sci. 2009;30(11):563–9.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–4.
Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry. 2014;71(6):681–8.
Li N, Liu RJ, Dwyer JM, Banasr M, Lee B, Son H, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69(8):754–61.
• Abdallah CG, Averill LA, Collins KA, Geha P, Schwartz J, Averill C, et al. Ketamine treatment and global brain connectivity in major depression. Neuropsychopharmacology. 2017:42(6):1210-19. https://doi.org/10.1038/npp.2016.186. The first paper to show that the rapid antidepressant effects of ketamine are related to the rapid restoration of normal patterns of cortical functional connectivity in patients.
Murrough JW, Abdallah CG, Anticevic A, Collins KA, Geha P, Averill LA, et al. Reduced global functional connectivity of the medial prefrontal cortex in major depressive disorder. Hum Brain Mapp. 2016;37(9):3214–23.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475(7354):91–5.
• Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533(7604):481–6. A paper that suggests that the rapid antidepressant effects of ketamine are attributable to its metabolite, 2R,6R-hydroxynorketamine.
Loix S, De Kock M, Henin P. The anti-inflammatory effects of ketamine: state of the art. Acta Anaesthesiol Belg. 2011;62(1):47–58.
Machado-Vieira R, Gold PW, Luckenbaugh DA, Ballard ED, Richards EM, Henter ID, et al. The role of adipokines in the rapid antidepressant effects of ketamine. Mol Psychiatry. 2017;22(1):127–33.
Liebenberg N, Joca S, Wegener G. Nitric oxide involvement in the antidepressant-like effect of ketamine in the flinders sensitive line rat model of depression. Acta Neuropsychiatr. 2015;27(2):90–6.
Harraz MM, Tyagi R, Cortes P, Snyder SH. Antidepressant action of ketamine via mTOR is mediated by inhibition of nitrergic Rheb degradation. Mol Psychiatry. 2016;21(3):313–9.
Rosa PB, Neis VB, Ribeiro CM, Moretti M, Rodrigues AL. Antidepressant-like effects of ascorbic acid and ketamine involve modulation of GABAA and GABAB receptors. Pharmacol Rep. 2016;68(5):996–1001.
Voleti B, Navarria A, Liu RJ, Banasr M, Li N, Terwilliger R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742–9.
Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR(2)/(3) blockade. Int J Neuropsychopharmacol. 2012;15(4):429–34.
Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P. Antidepressant-like actions of an AMPA receptor potentiator (LY392098). Neuropharmacology. 2001;40(8):1028–33.
Drevets WC, Zarate CA Jr, Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156–63.
Zuj DV, Palmer MA, Hsu CM, Nicholson EL, Cushing PJ, Gray KE, et al. Impaired fear extinction associated with ptsd increases with hours-since-waking. Depress Anxiety. 2016;33(3):203–10.
Garfinkel SN, Abelson JL, King AP, Sripada RK, Wang X, Gaines LM, et al. Impaired contextual modulation of memories in PTSD: an fMRI and psychophysiological study of extinction retention and fear renewal. J Neurosci. 2014;34(40):13435–43.
Krystal JH, Tolin DF, Sanacora G, Castner SA, Williams GV, Aikins DE, et al. Neuroplasticity as a target for the pharmacotherapy of anxiety disorders, mood disorders, and schizophrenia. Drug Discov Today. 2009;14(13–14):690–7.
Rothbaum BO, Price M, Jovanovic T, Norrholm SD, Gerardi M, Dunlop B, et al. A randomized, double-blind evaluation of D-cycloserine or alprazolam combined with virtual reality exposure therapy for posttraumatic stress disorder in Iraq and Afghanistan War veterans. Am J Psychiatry. 2014;171(6):640–8.
• Girgenti MJ, Ghosal S, LoPresto D, Taylor JR, Duman RS. Ketamine accelerates fear extinction via mTORC1 signaling. Neurobiol Dis. 100:1–8. A paper that shows that ketamine might promote fear extinction.
Acknowledgements
The authors acknowledge support from the US Department of Veterans Affairs through its support for the National Center for PTSD and it support, with the US Department of Defense, of the Consortium to Alleviate PTSD. We also recognize the National Center for Advancing Translational Science for its support of the Yale Center for Clinical Investigation (UL1RR024139). In addition, the authors acknowledge the support of the US National Institute on Alcohol Abuse and Alcoholism (P50AA12870, JHK), the State of Connecticut for the Abraham Ribicoff Research Facilities of the Connecticut Mental Health Center (GS, RSD). Dr. Krystal acknowledges the following relevant financial interests. He is a co-sponsor of a patent for the intranasal administration of ketamine for the treatment of depression that was licensed by Janssen Pharmaceuticals, the maker of S-ketamine. He has a patent related to the use of riluzole to treat anxiety disorders that was licensed by Biohaven Medical Sciences. He has stock or stock options in Biohaven Medical Sciences, ARett Pharmaceuticals, Blackthorn Therapeutics, and Luc Therapeutics. He consults broadly to the pharmaceutical industry, but his annual income over the past year did not exceed $5000 for any organization. He receives over $5000 in income from the Society of Biological Psychiatry for editing the journal Biological Psychiatry. He has fiduciary responsibility for the International College of Neuropsychopharmacology as president of this organization.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Benjamin Kelmendi declares no conflict of interest.
John H. Krystal has received grants from the Department of Veterans Affairs and has received consultancy fees from Biogen, Idec, MA, Biomedisyn Corporation, Forum Pharmaceuticals, Janssen Research and Development, L.E.K. Consulting, Otsuka America Pharmaceuticals, Inc., S K Life Science, Spring Care, Inc., Sunovion Pharmaceuticals, Inc., Takeda Industries, Taisho Pharmaceutical Co., Ltd., Naurex Pharmaceuticals, and Pfizer Pharmaceuticals. Dr. Krystal owns stock in ArRETT Neuroscience, Inc., Biohaven Pharmaceuticals Medical Sciences, Blackthorn Therapeutics, Inc., Luc Therapeutics, Inc., and Spring Care, Inc.
Chadi G. Abdallah has received consultancy fees from Genentech and Janssen.
Lynette A. Averill has received grants from the Department of Veterans Affairs and the Brain and Behavior Foundation.
Ilan Harpaz-Rotem has received a grant from the Brain and Behavior Foundation.
Gerard Sanacora has received consultancy fees from Allergan, Alkermes, BioHaven Pharmaceuticals Holding Company, Janssen, Merck, Sage, Taisho Pharmaceuticals, Takeda, and Vistagen Therapeutics. Dr. Sanacora has also received grants from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Johnson and Johnson, Hoffman La-Roche, Merck, Naurex, and Servier and has received payment from Alkermes for developing educational Disease Education non-promotional material. Dr. Sanacora owns stock in BioHaven Holding Company.
Steven M. Southwick has received grants from the National Center for PSTD.
Ronald S. Duman has received consultancy fees from Johnson and Johnson and Taisho and grants from Taisho, Navitor, Relmada, Allergan, Johnson and Johnson. Dr. Duman has also received honoraria payments from Johnson and Johnson and Navitor.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
This article is part of the Topical Collection on Disaster Psychiatry: Trauma, PTSD, and Related Disorders
Rights and permissions
About this article

Cite this article
Krystal, J.H., Abdallah, C.G., Averill, L.A. et al. Synaptic Loss and the Pathophysiology of PTSD: Implications for Ketamine as a Prototype Novel Therapeutic. Curr Psychiatry Rep 19, 74 (2017). https://doi.org/10.1007/s11920-017-0829-z
Published:
DOI: https://doi.org/10.1007/s11920-017-0829-z