
Abstract
The majority of treatments for neuropsychiatric disorders have been based on serendipitous discoveries, with little understanding of the pathogenic and pathophysiological mechanisms underlying these disorders. As many of these disorders are sensitive to stress, an understanding of the physiology of stress is important in avoiding and reversing stress-sensitive disorders. Increased understanding of the glutamatergic synapse has revealed a system that is affected by both stress and multiple neuropsychiatric treatments, suggesting a possible convergent target in these disorders. This chapter reviews how traditional neuropsychiatric treatments affect the glutamatergic synapse, and how future therapies may be developed to more directly target this system.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
17.1 Introduction
The biological and behavioral responses to stress can be beneficial or, as is the case with most neuropsychiatric illnesses, maladaptive and pathogenic. These dual effects of stress can in part be explained by the similarly dual effects of the neurotransmitter glutamate on the strength of synaptic connections between neurons. This chapter details how knowledge of stress-induced glutamatergic dysregulation can be used to develop novel therapeutics to effectively treat neuropsychiatric disorders.
The brain is both the control center for the response to stress, as well as a target for its effects. Along with refocusing energy to organs and muscles needed for escape, stress can help increase cognitive performance in the face of a challenge (Barha et al. 2007; Yuen et al. 2011, 2009) . The cognitive effects of stress can be explained by the response of the glutamatergic neurotransmitter system as glucocorticoid stress hormones are known to cause rapid increases in extracellular glutamate release (Groeneweg et al. 2011; Stein-Behrens et al. 1994; Venero and Borrell 1999) . Diverse types of behavioral stress also increase extracellular glutamate levels in the prefrontal cortex (PFC), hippocampus , and amygdala , as well as the striatum (Moghaddam 1993, 2002; Reznikov et al. 2007; Rutherford et al. 2007; Tardito et al. 2010) , and this is dependent on glucocorticoid activation (Lowy et al. 1993) .
The timing and amount of glutamate transmission is thought to influence cognitive function through the strengthening or weakening of the synapse. In certain conditions, stress-induced glutamate release is followed by increases in synaptic strength and long-term potentiation (LTP) (Luine et al. 1996) , as well as corresponding increases in glutamate receptors at the synapse in the hippocampus and PFC (Groc et al. 2008; Karst and Joëls 2005; Krugers et al. 2010; Yuen et al. 2011, 2009) . These alterations in synaptic plasticity are similarly tied to morphological changes as LTP stimulation leads to new and larger dendritic spines (Engert and Bonhoeffer 1999; Matsuzaki et al. 2004) .
While stress is an everyday part of life that can boost cognitive and physical performance, it is also a known risk factor for multiple psychiatric conditions (Anisman and Zacharko 1990; Kessler et al. 2012) . Exposure to an extreme stress can lead to symptoms of posttraumatic stress disorder (PTSD) , characterized by heightened fear memory of a stressful event and parallels increased synaptic strengthening after stress. More common though, it is exposure to chronic unpredictable stress (CUS) that is a risk factor for multiple illnesses such as depression, anxiety , bipolar, schizophrenia, addiction, among others (Caspi et al. 2003; Hammen 2005; Kendler et al. 1999a, 1999b; Lupien et al. 2009; Schneiderman et al. 2005; Sinha 2008) . While these illnesses have historically given rise to distinct treatments, their common sensitivity to chronic stress suggests underlying similarities in etiology that can be useful guides in the development of future therapies.
While acute stress increases glutamate release, the effects of chronic exposure to stress on glutamatergic transmission and synaptic strength are still poorly understood. There are complicated adaptations to additional exposures to stress that vary between, and even within, brain regions. Extracellular glutamate levels remained elevated in the hippocampus , but not PFC or striatum, after repeated tail pinch in the same day (Bagley and Moghaddam 1997; Rutherford et al. 2007) and within the PFC there are diverse responses between populations of neurons (Jackson and Moghaddam 2006) . Previous exposure to a 21-day chronic restraint stress (CRS), led to longer lasting elevations of glutamate in the face of a novel acute stress challenge. Additionally, CUS leads to reduced glutamate cycling in the PFC as measured by 13C-acetate metabolism (Banasr et al. 2010) . While acute increases in glutamatergic transmission can lead to synaptic potentiation, excessive glutamate release can lead to excitotoxicity or cell damage (Sapolsky 2000, 2003) . The potentially damaging effects of glutamate lead to a U-shaped curve of glutamate releas e on synaptic health, with acute instances of stress leading to synaptic potentiation and increased performance on some tasks, and chronic or excessive stress leads to reduced LTP, cell damage, morphological changes and behavioral deficits (Kim and Diamond 2002; Luine et al. 1996) .
Many of these changes are dependent on glutamatergic receptors, supporting the role of excessive glutamate in mediating these effects. Once released to the extracellular space, glutamate can be bound by ionotropic and metabotropic glutamate receptors. Ionotropic receptors include N-methyl-D-aspartate receptors (NMDARs), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) , and kainate receptors, while metabotropic receptors are composed of subunits mGluR1–8. Subunit composition, phosphorylation, kinetics and the location of these receptors play important roles in modulating the receptors’ effects on postsynaptic cells and synaptic plasticity.
In rodents, both CRS and CUS, as well as treatment with chronic glucocorticoids , leads to dendritic atrophy and spine loss in pyramidal cells of the CA3 region of the hippocampus (Magariños and McEwen 1995a, 1995b; Sapolsky 2000) . These functional and morphological effects of stress are blocked by drugs reducing glutamate release (Watanabe et al. 1992) and by NMDA, but not AMPAR antagonists (Kim et al. 1996; Magariños and McEwen 1995b; Martin and Wellman 2011) . Similar changes are observed in select regions of the PFC, where even relatively mild repeated stressors can lead to dendritic retraction and spine loss and this is blocked by the presence of NMDAR antagonists (Izquierdo et al. 2006; Li et al. 2010; Martin and Wellman 2011) . CUS also leads to a loss of synaptic proteins, such as the AMPAR subunit GluA1 and synaptic proteins PSD-95 and synapsin, as would be expected with a loss of spines (Li et al. 2010). These morphological changes potentially parallel the reduced neuronal size observed in patient populations (Rajkowska et al. 1999; Stockmeier et al. 2004) , but this has not been directly tested.
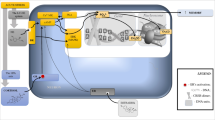
Together, this evidence suggests that dysregulation of glutamate transmission at the synapse can link chronic stress exposure to psychiatric illness and can guide future therapies. In the past, accidental discoveries with poor understanding of the true mechanisms of action have characterized the development of novel treatments for neuropsychiatric disorders. However, reexamination of traditional therapies such as monoaminergic antidepressants has revealed convergent effects on glutamatergic targets at the synapse that may reverse changes observed after stress. Similarly, drugs developed to directly target the glutamatergic system for nonpsychiatric disorders have shown off-label efficacy in many of these illnesses. This chapter summarizes how knowledge of the stressed synapse relates to established traditional neuropsychiatric therapies and what future therapies might be developed to target the glutamatergic synapse more directly (see Fig. 17.1 for overview of therapies targeting the glutamatergic synapse) .

Glutmatergic targets for antidepressant and antistress drug development. AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, EAAT excitatory amino acid transporter (1, 2, and 3), EAAC1 excitatory amino-acid carrier 1, GABA γ-aminobutyric acid, GABAR γ-aminobutyric acid receptor, GFAP glial fibrillary acidic protein, GLAST glutamate aspartate transporter, GLT1 glutamate transporter 1, mGluR2/3 metabotropic glutamate receptors 2 and 3, mGluR5 metabotropic glutamate receptor 5, MR mineralocorticoid receptor, PSD-95 postsynaptic density protein 95, THIIC N-(4-{[3-hydroxy-4-(2-methylpropanoyl)-2-(trifluoromethyl)phenoxy]methyl}benzyl)-1-methyl-1 H-imidazole-4-carboxamide, vGLUT vesicular glutamate transporters, NMDAR N-methyl-D-aspartate receptor, AMPAR amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
17.2 Therapies Regulating Presynaptic Release of Glutamate
The risk of excitotoxicity after stress suggests that therapies reducing glutamate release could ameliorate the development of stress-sensitive disorders. In fact multiple established antidepressants have now been found to reduce stimulated glutamate release, and drugs directly targeting glutamate have efficacy in neuropsychiatric disorders . However, glutamate release and stress can both play positive and negative roles in synaptic strength. Recent work on novel antidepressant therapies show the therapeutic effects of pharmacologically modulated glutamate release are complex, suggesting the timing, amplitude, and duration of glutamatergic excitation may all be critical factors in determining the relative benefits and harmful effects in relation to neuropsychiatric disorders.
17.2.1 Traditional Neuropsychiatric Therapies Modulate Presynaptic Glutamate Release
Many traditional antidepressants , such as chronic fluoxetine and desipramine, have been reassessed for effects on glutamate release and found to reduce stimulated glutamate release after chronic treatment (Bonanno et al. 2005; Musazzi et al. 2010; (for a review, see Musazzi et al. 2012) . Treatment with the atypical antidepressant tianeptine can block stress-induced glutamate release and, correspondingly, morphological changes in the hippocampus and amygdala (Czéh et al. 2001; Magariños et al. 1999; McEwen et al. 2010; Reznikov et al. 2007) , and corresponding increases in anxiety-like behavior (McEwen et al. 2010) . Similar reductions in glutamate release are seen in antidepressants of nonmonoaminergic mechanisms. The antidepressant agomelatine, which targets the melatonergic MT(1) and MT(2) receptors, as well as a 5-HT(2 C), reduces stress-induced glutamate release in the PFC (Milanese et al. 2013; Popoli 2009; Tardito et al. 2010, 2012) and can reverse the effects of prenatal stress in rats (Morley-Fletcher et al. 2011) . Treatment with chronic antidepressants also increased expression of the metabotropic glutamatergic receptor, mGluR2/3, activation of which suppresses presynaptic glutamate release (Matrisciano et al. 2002) and chronic treatment with amitriptyline, a tricyclic antidepressant, reversed decreases in mGluR2/3 observed in the hippocampus after olfactory bulbectomy (Wieroñska et al. 2001) .
Similarly, anxiolytics can reduce stress-induced increases in glutamate in the hippocampus and PFC (Bagley and Moghaddam 1997) and reduce hippocampal atrophy (Magariños et al. 1999) . Anxiolytics such as diazepam and other benzodiazepines increase GABAergic cell transmission, increasing inhibition on glutamatergic cells that effectively leads to reductions in glutamate release (see Fig. 17.1).
17.2.2 Treatments Targeting Glutamate Release Have Efficacy in Psychiatric Illnesses
Drugs originally developed to reduce stimulated glutamate release, such as anticonvulsants or treatments for amyotrophic lateral sclerosis (ALS), have demonstrated effects in preclinical rodent models and efficacy in mood disorders. In the preclinical literature, the antiepileptic drug phenytoin is known to reduce glutamate release and, when administered during chronic stress, blocks the dendritic atrophy observed in the hippocampus (Watanabe et al. 1992) , but it has not been fully investigated in clinical mood disorder trials. Other anticonvulsants, such as valproate and lamotrigine are FDA approved for use in the treatment of bipolar disorder, and are used as off-label treatments for other mood disorders (Calabrese et al. 1999; Du et al. 2007; McElroy et al. 2004; van der Loos et al. 2009) (for a review of anticonvulsants in psychiatry, see Ettinger and Argoff 2007; Mula et al. 2007) . The drug riluzole, which has anticonvulsant properties in addition to providing clinical benefit in the treatment of ALS, also appears to have clinical benefits in relation to anxiety , mood disorders , OCD in several small nonplacebo controlled clinical trials (Coric et al. 2005; Pittenger et al. 2008; Sanacora et al. 2004) . Riluzole has also been shown to have antidepressant-like properties in several rodent models (Banasr et al. 2010) , and the details of these studies are discussed below. Riluzole is known to reduce glutamatergic transmission, though it is not clear if it works on presynaptic glutamate release or through other mechanisms affecting extracellular glutamate levels.
17.2.3 Novel Neuropsychiatric Treatments and Glutamate Release
In sum, the evidence suggests a number of therapies with diverse structures, but seemingly convergent effects on presynaptic glutamate release in regions implicated in neuropsychiatric disorders, posses antidepressant-like properties in rodent models and in the clinic. While this may suggest presynaptic glutamate release as an ideal target for many of these stress-sensitive disorders, a new class of effective antidepressants suggests that the story is more complicated. Efforts to create fast-acting therapies that directly target the glutamatergic system have led to the discovery of the antidepressant properties of drugs that appear to acutely increase glutamate release such as the NMDAR antagonist ketamine .
Evidence suggesting that antidepressants downregulate NMDAR expression led to the testing of NMDAR antagonists in preclinical models of depression (Trullas and Skolnick 1990) . NMDAR antagonists have been found to have fast-acting antidepressant activity in preclinical and clinical studies (Diazgranados et al. 2010; Ibrahim et al. 2011; Skolnick et al. 2001, 2009) and the NMDAR antagonist ketamine has in particular demonstrated efficacy in clinical trials (Berman et al. 2000; Zarate et al. 2006a) (for a review of ketamine in depression, see Mathews and Zarate 2013) . The subanesthetic doses at which ketamine has been shown to have antidepressant-like effects are also known to induce a sharp increase of glutamate efflux in the PFC and hippocampus as measured in microdialysis (Moghaddam et al. 1997) . More recently these same doses were found to increase glutamate cycling in the PFC (Chowdhury et al. 2012) and to stimulate a series of cellular processes that are associated with changes in synaptic plasticity (Autry et al. 2011; Li et al. 2010) . Preclinical work has suggested that these antidepressant-like effects are dependent on AMPA/kainate receptor activity, indicating a requirement for increased synaptic transmission (Autry et al. 2011; Koike et al. 2011; Maeng et al. 2008) . Interestingly, a similar mechanism involving a rapid increase in glutamate release and activation of AMPA receptors has also been shown to be related to the rapid antidepressant-like effects of scopolamine (Voleti et al. 2013) . However, it is critical to note that the increased glutamate efflux produced by these treatments appears to be of short duration, and to have completely dissipated by the time the antidepressant-like behavioral effects are observed. Recent work demonstrating that ketamine treatment reduces expression of presynaptic release machinery over a period of hours (Müller et al. 2013) , suggests the overall effect of the treatments on glutamate release is complex and may vary with time .
The complex role of glutamate release in antidepressant therapies is further demonstrated in the case of mGluR2/3-related treatments. The metabotropic mGluR2/3-containing glutamate receptor is predominantly located presynaptically (Tamaru et al. 2001) and its activation exerts negative feedback on additional glutamate release (Anwyl 1999; Cartmell and Schoepp 2000; Tamaru et al. 2001) . mGluR2/3 expression is altered in depressed patients and preclinical models of depression (Feyissa et al. 2010; Matrisciano et al. 2008; Wierońska et al. 2008) and had been proposed as a novel target for depression (Sanacora et al. 2008; Witkin et al. 2007) . As noted earlier, treatment with chronic monoaminergic-based antidepressants increases mGlur2/3 expression (Matrisciano et al. 2002). However, pharmacological strategies both increasing or decreasing mGluR2/3 activation have demonstrated preclinical efficacy as anxiolytics and antidepressants (Palucha and Pilc 2007; Pilc et al. 2008) . For a review of metabotropic receptors in psychiatry, see Chaki et al. (2013) .
Potentiating these receptors can dampen excessive glutamate release and therefore may be beneficial in mediating stress-induced pathophysiology. Administration of a low dose mGluR2/3 agonist shortens the latency to therapeutic effects of chronic antidepressant treatments in preclinical models (Matrisciano et al. 2005, 2007) and agonists of the mGluR2/3 receptor have antidepressant-like efficacy in preclinical tests (DD and Marek 2002; Swanson et al. 2005) . A new positive allosteric modulator of mGluR2/3 called N-(4-{[3-hydroxy-4-(2-methylpropanoyl)-2-(trifluoromethyl)phenoxy]methyl}benzyl)-1-methyl-1 H-imidazole-4-carboxamide (THIIC) has robust preclinical antidepressant-like effects (Fell et al. 2011; Johnson et al. 2005) . THIIC and other allosteric modulators only activate metabotropic glutamate receptors under conditions of excessive glutamate release to reduce glutamate release (Johnson et al. 2005) .
However, treatments with opposing effects on the mGluR2/3 receptor have similar antidepressant-like efficacy. Various mGluR2/3 antagonists including MGS0039, LY341495, and RO4491533 have demonstrated efficacy in the rodent forced swim test (FST) (Chaki et al. 2004; Pałucha-Poniewiera et al. 2010; Yoshimizu et al. 2006) . Similar to ketamine , these preclinical effects are dependent on AMPAR throughput suggesting that an increase in glutamatergic transmission is necessary for its effects (Dwyer et al. 2012; Koike et al. 2011) .
While the efficacy of NMDAR and mGluR2/3-based treatments supports a role of glutamatergic transmission in antidepressant therapy, it casts doubt on the hypothesis that a simple stress-induced hyperglutamatergic state is the sole contributor to the pathophysiology of mood disorders, and that simply reducing presynaptic glutamate release is necessary and sufficient to mitigate and reverse stress-sensitive disorders. Instead, it now appears that different pathophysiological processes may predominate at different stages in the evolution of the illness. It is possible that the early phases in the evolution of stress sensitive disorders are associated with excessive glutamate efflux and sustained elevation of extracellular glutamate concentrations. This is consistent with reports showing elevated glutamate to be associated with hippocampal toxicity (Sapolsky 2000) . However, once the disorder has developed, compensatory changes resulting in diminished synaptic glutamatergic neurotransmission may dominate in relation to the cognitive, behavioral, and emotional symptoms associated with the disorder. This may explain why in the case of some fast-acting antidepressants , decreased glutamate release may actually block any therapeutic effect. As the number of ketamine clinical trials grows, some data suggest that drugs reducing glutamate release, such as with anesthetics, may reduce the efficacy of ketamine (Abdallah et al. 2012) . However, this remains to be confirmed in more definite studies. As will be further discussed below, while blocking NMDARs has antidepressant action, therapies that instead boost AMPAR throughput may be associated with similar types of antidepressant-like therapeutic effects (Chappell et al. 2007; Knapp et al. 2002; Li et al. 2001; Lindholm et al. 2012; Nations et al. 2012) . Correspondingly stress-induced pyramidal cell atrophy can be reduced by blocking NMDAR activation, but not by blocking AMPA receptors (Magariños and McEwen 1995b) . Together these studies suggest that the stress-induced increase in glutamate release is not in itself harmful, but its subsequent postsynaptic effects resulting from the relative activation of the various glutamatergic receptors may be more important for determining the physiological or pathophysiological consequences of the enhanced release .
17.3 Therapies Regulating Extracellular Glutamate Uptake
The ubiquitous nature of glutamate, along with its ability to cause excitotoxicity, necessitates a tightly regulated system controlling its release and extracellular levels. Once released to the extracellular space, glutamate is not broken down but is instead taken up by neighboring glia or neurons via excitatory amino acid transporters (EAAT1–5 in humans) (O’Shea 2002) . EAAT 1 and 2 (GLAST and GLT1 in rodents) mainly transport glutamate to astrocytes where it can be converted to glutamine, while EAAT3 (EAAC1 in rodents), transports glutamate to neurons (Anderson and Swanson 2000; Arriza et al. 1994) . EAATs and astrocytes placed near the synapse play a critical role in regulating extracellular glutamate levels and risk of excitotoxicity (Arriza et al. 1994; Shigeri et al. 2004; Zarate et al. 2002; Zheng et al. 2008) .
While the role of glia cells in neuropsychiatry has been understudied in the past, there is now an increased understanding of their complexity and roles in glutamatergic transmission and dysfunction. Astrocytes are ideally placed to help restrict extracellular glutamate to the synapse and limit glutamate to spillover to peri- or extrasynaptic sites, where transmission is thought to weaken synaptic strength and damage the cell (Hardingham and Bading 2010) . While normally associated with glutamate uptake, astrocytes may also release glutamate to the extracellular space in certain conditions (Malarkey and Parpura 2008) , leading to increased glutamate at these potentially damaging extrasynaptic locations (Talantova et al. 2013) . As a single astrocyte can cover multiple synapses, the disruption of individual astrocytes can have wide-reaching effects (Bushong et al. 2002) .
Glial cells also express glutamatergic receptors though their subunit composition, expression patterns, and function are not well studied (Hansson and Rönnbäck 2004; Verkhratsky and Kirchhoff 2007) . For example, unlike neuronal NMDARS, astrocytic NMDARs are unblocked by magnesium at baseline, suggesting NMDARS containing NR3 subunits (Palygin et al. 2011) , while NR2B containing receptors may be expressed after injury or stress such as ischemia (Krebs et al. 2003) . A weak magnesium blockade at baseline suggests that these receptors are more sensitive than neurons to increases in extracellular glutamate (Lalo et al. 2006; Palygin et al. 2011). The expression of metabotropic glutamatergic receptors on glia is also debated, with recent evidence that mGluR5 receptors are only expressed in younger animals (Sun et al. 2013) . Functionally, the unique properties of glial NMDARs likely explains the differential effects of various NMDAR antagonists on glia and neurons, with the NR2B-selective antagonist ifenprodil selectively blocking an inward current and Ca2+ influx in neurons, but not astrocytes, and memantine and MK-801 blocking both cell types (Palygin et al. 2011).
Interestingly, a reduction in glial density and number is observed in the PFC of major depressive disorder (MDD) patients (Cotter 2001; Cotter et al. 2002; Ongur et al. 1998; Rajkowska and Miguel-Hidalgo 2007; Rajkowska et al. 1999; Uranova et al. 2004) (for recent reviews, see Rajkowska and Stockmeier 2013; Sanacora and Banasr 2013) . These reductions have been observed in depression , bipolar disorder, and in some cases, schizophrenia, in regions implicated in these disorders such as Brodmanns area 24, the orbitofrontal cortex, and the dorsolateral PFC (Gittins and Harrison 2011; Ongur et al. 1998; Rajkowska et al. 1999) . Glial fibrillary acidic protein (GFAP), a marker of astrocytes , reveals reductions in the hippocampus , PFC and amygdala , with the effect in the PFC is the most consistent in patient populations (Altshuler et al. 2010; Johnston-Wilson et al. 2000; Müller et al. 2013; Webster et al. 2001) . Changes in glutamate transporters such as EAAT 2 are similarly observed in depressed patients (Bernard et al. 2011; Choudary et al. 2005; McCullumsmith and Meador-Woodruff 2002; Sequeira et al. 2009) .
Changes in glutamatergic uptake and cycling are observed after stress, indicative of adaptations to increased exposure to glutamate. After CUS, rodents show decreases in glutamine cycle rate (Banasr et al. 2010), which could relate to observed CUS or corticosterone-induced loss of glia in the PFC (Alonso 2000; Banasr et al. 2010; Banasr and Duman 2007) . Some forms of stress or corticosterone exposure have been demonstrated to increase expression of GLT-1 (but not GLAST) in the PFC and hippocampus of rodents therefore increasing glutamate uptake (Autry et al. 2006; Zink et al. 2010; Zschocke et al. 2005) , possibly as a neuroprotective countermeasure to stress-induced increases in glutamate efflux. Illustrating the potential pathological behavioral effects of impaired glutamate clearance from the extracellular space, application of a glial toxin to the PFC in rodents, leads to depressive-like behaviors after one exposure to stress as opposed to weeks of chronic stress (Banasr and Duman 2008) . Providing additional support to the hypothesis that impaired glial-mediated glutamate uptake can be associated with depressive-like behaviors, rats bred for higher levels of learned helplessness showed a significantly suppressed expression of GLT1 in hippocampus and cerebral cortex compared to nonhelpless littermates (Zink et al. 2010).
17.3.1 Traditional Neuropsychiatric Therapies on Glutamate Uptake and Glia
There is evidence that traditional antidepressants have glio-protective effects and thus can influence uptake of extracellular glutamate. In preclinical studies, treatment with fluoxetine reduced the stress-induced loss of hippocampal GFAP in tree shrews, but had no effect in nonstressed animals (Czéh et al. 2006) . Similarly, chronic administration of the tricyclic antidepressant clomipramine increased GFAP expression in stressed animals (Liu et al. 2009) . Another study found that chronic fluoxetine increased GLT1 expression in the hippocampus and cortex in rats, while desipramine and an monoamine oxidase inhibitor (MAOI) showed more modest effects (Zink et al. 2011), though this study did not test these antidepressants in relation to stress. Similarly, the antidepressant paroxetine increased hippocampal GFAP expression (Sillaber et al. 2008) . In contrast, it should be noted that some studies have failed to find an ability of antidepressants to reverse stress-induced GFAP loss in the hippocampus (Araya-Callís et al. 2012) or cortex (Fatemi et al. 2008) .
17.3.2 Treatments Targeting Glial and Glutamate Uptake Have Efficacy in Psychiatric Illnesses
While traditional antidepressants have some glio-protective properties, drugs with well-established effects on glial cell function and glutamate uptake might serve as more attractive therapies in looking to reverse stress-induced glial loss. Treatment with B-lactam antibiotics such as ceftriaxone has been shown to increase GLT1 function (Rothstein et al. 2005) . Considering this property of ceftriaxone, several studies have since demonstrated its ability to modify several forms of behavior believed to be modulated by glutamatergic activation (Trantham-Davidson et al. 2012) , including reducing depressive and anxiety-like behaviors in mice (Mineur et al. 2007) . However, side effects and the difficulties related to the delivery of ceftriaxone have prohibited larger clinical trials in psychiatric patients to date.
As described earlier, the neuroprotective drug riluzole reduces glutamatergic transmission; however, the exact mechanism of action of riluzole remains unclear. More recent studies suggest much of riluzoles neuroprotective effects could be mediated through the effects on GLT1 expression (Fumagalli et al. 2008; Yoshizumi et al. 2012) . As also described earlier, multiple open label (nonplacebo controlled) clinical studies have found riluzole to be effective in the treatment of MDD, bipolar disorder (BPD), anxiety and obsessive–compulsive disorder (OCD) (Coric et al. 2005; Pittenger et al. 2008; Sanacora et al. 2004) . Additionally, riluzole was shown to alter glutamine/glutamate cycling in BPD patients (Brennan et al. 2010) . Preclinical studies show an antidepressant-like action of chronic riluzole, and an ability to reverse chronic-stress induced depression-like behaviors and loss of GLT-1 and GFAP (Banasr et al. 2010; Gourley et al. 2012) . Ongoing studies will test if GLT-1 or glial activity is necessary for the antidepressant-like activity of riluzole.
17.3.3 Novel Neuropsychiatric Treatments on Glutamate Uptake and Glia
As mentioned previously, some NMDAR antagonists such as ketamine have antidepressant efficacy, however the exact mechanism behind these treatments still remains to be elucidated. Most hypotheses have focused on a blockade of NMDARs on neuronal cells; however, Mitterauer recently suggested a key role of astrocytic NMDARs in ketamines therapeutic effect (Mitterauer 2012) . However, it should be noted that subunit-specific antagonists such as NR2B-selective antagonists, also have clinical and preclinical antidepressant-like effects (Li et al. 2010; Maeng et al. 2008; Preskorn et al. 2008) , and previous work found that the NR2B selective antagonist ifenprodil selectively affects neurons and not astrocytes (Palygin et al. 2011) . NR2B-containing receptors may only be expressed in glia after adverse events such as ischemia, possibly leading to differential activity of NMDAR antagonists in healthy and diseased states (Krebs et al. 2003) .
17.4 Therapies Regulating Postsynaptic Effects of Glutamate Release
As discussed above, effective antidepressant therapies are known to both increase, and decrease extracellular glutamate levels. In the case of NMDAR antagonists and mGluR2/3 antagonists, the antidepressant-like effect appears to require a transient increase in glutamatergic transmission through AMPARs to initiate the cascade of cellular changes that have been associated with the antidepressant-like effects (Dwyer et al. 2012; Li et al. 2010; Maeng et al. 2008) . This suggests that the type of postsynaptic glutamatergic transmission is critical in generating a rapid treatment response and possibly also in determining the responses to stress. The administration of NMDAR, but not AMPAR, antagonists blocks stress-induced morphological and plasticity-related changes in pyramidal cells in the hippocampus (Magariños and McEwen 1995b) , with similar results in the mPFC (Martin and Wellman 2011) . Administration of NMDAR-antagonists also blocks stress-induced alterations of hippocampal LTP (Kim et al. 1996) . Hippocampal CA3 and CA1 pyramidal cell atrophy caused by restraint stress was blocked by CA3 pyramidal cell-specific conditional knockout of GluN1, suggesting that these effects are mediated specifically by pyramidal cell NMDARs (Christian et al. 2011) , similarly despair behavior during chronic swim stress was reduced by a pyramidal cell knockout of NR2B in the cortex and CA1 (Kiselycznyk et al. 2011) . These findings suggest that increased postsynaptic NMDAR-mediated glutamate transmission has a critical role in mediating the effects of stress, and AMPAR throughput is necessary for an antidepressant response.
The importance of postsynaptic NMDAR throughput in mediating excitotoxic insults is consistent with the aforementioned work showing that excessive glutamate transmission through NMDARs, particularly, extrasynaptic sites, can lead to cell damage (for a review, see Hardingham and Bading 2010) . While activation of synaptic NMDARs leads to activation of CREB (cAMP response element-binding protein) and increases in synaptic strength, spillover of glutamate to extrasynaptic sites enables activation of extrasynaptic NMDARs that decreases CREB signaling and lead to long-term depression and activation of cell death pathways (Hardingham et al. 2002). In adulthood, extrasynaptic NMDARs are thought to be mainly NR2B-containing receptors, while synaptic NMDARs are mainly composed of NR2A receptors. Whether the location or subunit composition of NMDARs is important to cell growth pathways is unclear, however recent work suggests it is the C-terminal tail on the NR2B subunit that is responsible for activating downstream pathways mediating cell death pathways (Martel et al. 2012) . Interestingly, this C-terminal tail of NR2B can be cleaved in the presence of calpain (Guttmann et al. 2001, 2002) , a signaling molecule known to be increased by extrasynaptic NMDAR throughput (Xu et al. 2009) leaving a functional NR2B-containing NMDAR with possibly altered trafficking to extrasynaptic sites and altered interaction with downstream signaling pathways (Gladding and Raymond 2011) . Activation of synaptic versus extrasynaptic NMDARs is linked to nuclear CREB signaling through the messenger protein Jacob. Extrasynaptic receptors increase Jacob trafficking to the nucleus where Jacob triggers CREB shut-off pathways and cell death, while synaptic NMDARs phosphorylate Jacob to block its effects on CREB (Dieterich et al. 2008; Karpova et al. 2013) .
17.4.1 Traditional Antidepressants’ Effect on Postsynaptic Sites
Chronic, but not acute, treatment with some antidepressants reduces NMDAR transmission (Paul and Skolnick 2003; Reynolds and Miller 1988; Skolnick et al. 1996) . These same treatments appear to augment AMPAR transmission , as multiple chronic antidepressant treatments increased phosphorylation of the GluA1 subunit (McEwen et al. 2010; Svenningsson et al. 2007) , and synaptic GluA1 and GluA2 levels (Du et al. 2007) . Similarly, anticonvulsants with primarily antidepressant activity such as lamotrigine and riluzole increase GluA1 and GluA2 subunits in the hippocampus , while therapies with primarily antimanic properties, such as lithium and valproate, however, reduce GluA1 and GluA2 levels (Du et al. 2007) . However, the efficacy of acute imipramine in the rodent forced swim test is not blocked in mice lacking the phosphorylation sites on GluA1 that are increased after antidepressant treatment (Kiselycznyk et al. 2013) .
17.4.2 Therapies Targeting Postsynaptic Glutamatergic Receptors
As mentioned earlier, NMDAR antagonists such as ketamine act as fast-acting antidepressants in treatment-resistant patients (Berman et al. 2000; Diazgranados et al. 2010; Mathew et al. 2010; Murrough et al. 2013; Valentine et al. 2011; Zarate et al. 2006, 2012) and preclinical assays, such as the FST (Autry et al. 2011; Li et al. 2010; Maeng et al. 2008) . Compared with traditional antidepressants that can take weeks or months to reduce symptoms, ketamine is effective in a matter of hours and one infusion can reduce depressive symptoms for days to weeks in some patients. NMDAR antagonists also have been reported to have anxiety-reducing effects (Cryan and Dev 2007; Barkus et al. 2011) . In preclinical models, ketamine leads to long-term increases in synaptic strength in the PFC, increasing synaptic proteins like GluA1 and increasing spine density. These same NMDAR antagonists can reverse CUS-induced spine loss and behavioral changes (Li et al. 2010), and are known to increase expression of BDNF , and neurogenesis (Gould and Cameron 1997; Metsis et al. 1993) .
Acute systemic administration of pharmacological antagonists specific to the GluN2B-subunit is sufficient to produce the antidepressant-like effects seen with nonsubunit-selective NMDAR antagonists such as ketamine, both clinically (Preskorn et al. 2008) and preclinically (Li et al. 2010; Maeng et al. 2008). Administration of selective GluN2B antagonists such as Ro 25–6981 produces no effect on anxiety-like behavior in the mouse elevated plus maze (EPM) (Mathur et al. 2009) , but anxiolytic-like in the novelty-suppressed feeding (NSF) task (Li et al. 2010). Administration of another GluN2B antagonist, ifenprodil, was also anxiolytic-like effects in the rat EPM (Fraser et al. 1996) .
However, attempts to selectively delete NMDAR subunits with genetic techniques have not produced depression-related behaviors. Constitutive genetic deletion of the obligatory GluN1 subunit are lethal, however viable conditional knockouts of this subunit have been generated with postnatal deletion in specific regions and cell types. Mice with a restricted deletion of GluN1 to pyramidal cells of the CA3 region of the hippocampus displayed no differences from control mice in HPA-axis activation or anxiety-like behavior in the EPM (Christian et al. 2011; Cravens et al. 2006) . Similar deletion of the subunit GluN2B in corticohippocampal pyramidal cells displayed no alterations in the FST and anxiety (Kiselycznyk et al. 2011) . The lack of depression-related effect in these selective deletions could be due to alterations in cell-type, region, or age; however, it suggests that deletion of NMDA receptors is not enough to induce an antidepressant-like response.
Metabotropic glutamate receptors containing mGluR5 are located postsynaptically and typically located near NMDARs (Brakeman et al. 1997; Lujan et al. 1996; Tu et al. 1999) . mGluR5 activity is tied to NMDARs and help regulate NMDAR throughput and activation of mGluR5 receptors increases NMDAR transmission, while blocking mGluR5 reduces NMDAR throughput (Attucci et al. 2001; Awad et al. 2000; Doherty et al. 2000; Pisani et al. 2001) . Similarly, repeated treatment with the mGluR5 antagonist 3-((2-Methyl-4-thiazolyl)ethynyl)pyridine (MTEP) also reduces NR1 expression (Cowen et al. 2005) . mGlur5 is decreased in depressed patients, as well as rats bred for depression-related phenotypes (Kovačević et al. 2012) . It is similarly decreased in the hippocampus after chronic treatment with corticosterone in rodents (Iyo et al. 2010) .
However, mGlu5 knockout mice appear to have reduced depression-related behavior in the FST (Li et al. 2006) . Additionally, mGluR5 antagonists such as 2-Methyl-6-(phenylethynyl)pyridine (MPEP) or MTEP have antidepressant- and anxiolytic-like efficacy in preclinical models (Belozertseva et al. 2007; Brodkin et al. 2002; Busse et al. 2004; Li et al. 2006; Molina-Hernández et al. 2006; Pałucha et al. 2005; Pilc et al. 2002; Tatarczyńska et al. 2001; Wieronska et al. 2002) . Similar antidepressant-like effects are observed with the negative allosteric modulator (GRN-529), in preclinical tests (Hughes et al. 2012) . While the majority of support for mGluR5-related therapies has been preclinical, some clinical trials have shown efficacy for the mGluR5 treatment Fenobam in anxiety (Pecknold et al. 1982; Porter et al. 2005) . As blockade of mGluR5 decreases NMDAR transmission, the antidepressant-like mGluR5 antagonists effects correspond to the efficacy of NMDAR antagonists.
Administration of drugs blocking non-NMDARs (i.e., AMPAR and kainate receptors), do not affect depression-related activity in the FST (Maeng et al. 2008) , unlike NMDAR antagonists. Administration of drugs selectively targeting AMPARs , such as GYKI 52466 or LY32635, have been found to cause anxiolytic-like (Alt et al. 2006; Kapus et al. 2008; Kotlinska and Liljequist 1998; Matheus and Guimarães 1997) , anxiogenic-like (Vekovischeva et al. n.d.) , or no changes (Fitzgerald et al. 2010; Kapus et al. 2008) in anxiety-related behaviors, depending on the rodent species tested or behavioral paradigm used. Mice lacking key phosphorylation sites on the AMPA subunit GluA1 also demonstrate decreases in anxiety (Kiselycznyk et al. 2013) .
However, as mentioned earlier, treatments like ketamine transiently increase extracellular glutamate release and are dependent on AMPAR transmission to generate the antidepressant-like response, suggesting that increased AMPAR throughput is necessary for the effect. Additionally, AMPAR potentiators or AMPAkines appear to have efficacy as antidepressants in a variety of rodent models (Knapp et al. 2002; Li et al. 2001; Lindholm et al. 2012) . In clinical studies, the AMPA potentiator LY451395 helped relieve depressive symptoms in Alzheimers patients (Chappell et al. 2007) . More recently, a small phase Ib study was completed with the AMPA potentiating drug Org 26576. While the study demonstrated good safety and tolerability of the drug, along with some numerical advantages in terms of depressive severity and cognitive functioning, the differences did not reach the level of statistical significance in this exploratory study with limited power (Nations et al. 2012) .
Together with the studies in ketamine , the findings suggest that increased AMPAR throughput while blocking NMDARs is necessary for an antidepressant-like response. Deletion of NMDARs without concurrent increased AMPAR throughput would therefore not be predicted to have antidepressant-like activity. Ketamine achieves this by increasing extracellular glutamate while blocking NMDARs and leaving AMPARs free. Traditional antidepressants, while they may reduce presynaptic glutamate release, also increase levels of AMPAR subunits and reduce NMDAR transmission. This proposed relationship between synaptic AMPARs and NMDARs (especially extrasynaptic NMDARs) in regulating synaptic strength suggests multiple new directions for the development of future therapeutics.
17.4.3 Future Directions Targeting Postsynaptic Sites
Increasing AMPAR throughput has antidepressant efficacy and could reverse stress-induced deficits, and AMPARs have a mainly synaptic localization in adulthood. Together with the knowledge that extrasynaptic, but not synaptic, NMDARs can mediate cell-death after excessive glutamate release, this suggests that the balance between synaptic and extrasynaptic glutamate transmission is a convergent target for stress-sensitive disorders. Ketamine has the benefit of both increasing glutamate release and boosting synaptic throughput while simultaneously blocking possibly extrasynaptic NMDARSs. However, knowledge of the effects of synaptic versus extrasynaptic throughput may enable the development of better strategies.
As extrasynaptic NMDARs are thought to be NR2B-rich, a potential strategy to target extrasynaptic receptors would be to use NR2B-selective compounds. NR2B-selective antagonists have preclinical and clinical efficacy (Li et al. 2010; Maeng et al. 2008; Preskorn et al. 2008) . However, while GluN2B-containing receptors may be preferentially expressed at extrasynaptic sites, it is not a clear division. The NMDAR memantine selectively targets extrasynaptic NMDARs at selective dose range (Xia et al. 2010) . Memantine has shown efficacy in preclinical (Moryl et al. 1993; Rogóz et al. 2002) and clinical studies (Muhonen et al. 2008) , though memantine had no effect in some clinical studies of depression (Zarate et al. 2006) . Recently a more selective version of memantine has been developed called nitromemantine that is reported to more selectively target extrasynaptic receptors (Lipton 2006).
Negative results in memantines antidepressant efficacy may be due to memantines lack of effect on extracellular glutamate release. As memantine is not known to increase glutamate release , its blockade of extrasynaptic receptors could reduce the negative effects of stress over time. However without the burst of synaptic throughput it would not be expected to have antidepressant-like effects at baseline. Instead, memantine could be combined with another source of stimulated glutamate release to generate a more rapidly acting treatment. Combining the selective blockade of extrasynaptic NMDARs by memantine with the glutamate release of acute stress exposure could lead to novel treatment strategies. This strategy would block cell death pathways in only regions and instances of stress, allowing for selectivity not available with current pharmacological treatments, also known as a pathologically activated therapeutic (PAT, Lipton 2006) . The combination of NMDAR antagonists such as memantine and stress has previously been shown to have effects on morphology not seen with either treatment alone. Administration of the NMDAR antagonists during CRS lead to a significant increase in PFC spine density not observed in NMDAR antagonist treatment alone (Martin and Wellman 2011) . Similarly administration of tianeptine during CUS caused hippocampal hypertrophy compared to nonstressed animals (Czéh et al. 2001) .
17.5 Conclusions
Converging evidence suggests that alterations in the glutamatergic neurotransmitter system play a key role in the pathogenesis and pathophysiology of stress-induced psychiatric disorders. The stress-induced release of glutamate, its uptake by glial cells, and its postsynaptic effects are all potential therapeutic targets (see Fig. 17.1 for overview of convergent targets). However, knowledge of both the positive and negative effects of stress and glutamate release may guide the design of therapies that better allow for prophylaxis and recovery from glutamate dysregulation after stress. We have seen that while multiple antidepressants help block stress-induced glutamate release, many new classes of antidepressants acutely have the exact opposite result and in fact appear to require increases in extracellular glutamate release to generate the antidepressant-like effect. Knowing that glutamate transmission focused to synaptic sites can activate cell growth versus cell death pathways, attempting to reduce stress-induced glutamate release would block these potentially beneficial effects and restrict recovery. However, regulation of the negative feedback on glutamate release through mGluR2/3 receptors may allow for a therapy selectively activated in cases of stress and extreme glutamate release and present another PAT, as with memantine.
Therapies targeting glutamate uptake may present better targets with fewer side effects. Supporting the health of glial cells to regulate extracellular glutamate levels could help reduce extrasynaptic activation while maintaining synaptic throughput. As loss of glial is one of the most consistent findings in depression, and a single astrocyte can affect many neurons, therapies targeting glial could have broad effects. However, it is unclear if increasing glutamate uptake itself will only block subsequent exposures to high glutamate levels or will be able to reverse effects of stress to have a fast-acting antidepressant effect. The ability of glia to release glutamate into the extracellular space could be used to induce glutamate release similar to ketamine; however, it would most likely cause an increase of glutamate release to extrasynaptic, versus synaptic areas, leading to activation of cell death versus cell death pathways.
Finally, therapies targeting postsynaptic sites present opportunities to activate both cell death and cell growth pathways depending on the activation of synaptic or extrasynaptic receptors. Therapies designed to target these postsynaptic glutamatergic receptors pose a substantial risk for side effects if selectivity cannot be achieved. Memantine, and now nitromemantine have been reported to selectively block extrasynaptic sites but have a narrow dose range to target extrasynaptic receptors. Future studies examining the effects of selective blockade of extrasynaptic NMDARs, if possible, would be extremely interesting. However, blocking extrasynaptic receptors would require concomitant increases in glutamate release to have a burst of synaptic throughput and a fast-acting antidepressant response. Alternatively, AMPARs could be targeted directly with AMPA potentiators to activate synaptic throughput, however this strategy would not allow for selectivity to regions activated in pathological scenarios.
The increased understanding of the relationship between the glutamatergic system and stress has illuminated potential pathways of regulating synaptic glutamate transmission to develop novel treatment strategies for stress-sensitive neuropsychiatric disorders . While current antidepressant therapies, such as monoaminergic-based treatments, have been tied to mediators of synaptic activity their exact mechanism of action remains unclear. Traditional antidepressant treatments leading to increased AMPAR levels may increase synaptic transmission , but not selectively in regions or cell-types activated in depression . Additionally, mechanisms reducing glutamate release could protect against the negative consequences of stress, but also block the increased synaptic transmission possibly needed for recovery. In total, a lack of understanding of the mechanisms behind current therapies could explain their lack of consistent effects that ultimately leads to the large gap between the number of patients prescribed antidepressants and those successfully treated. Instead, the converging evidence on novel glutamatergic and plasticity-related therapeutic targets supports a new generation of mechanistically based treatments that can more directly and consistently address the numerous challenges of treating stress-related neuropsychiatric illnesses.
References
Abdallah CG, Fasula M, Kelmendi B, Sanacora G, Ostroff R. Rapid antidepressant effect of ketamine in the electroconvulsive therapy setting. J ECT. 2012;28(3):157–61. doi:10.1097/YCT.0b013e31824f8296.
Alonso G. Prolonged corticosterone treatment of adult rats inhibits the proliferation of oligodendrocyte progenitors present throughout white and gray matter regions of the brain. Glia. 2000;31(3):219–31.
Alt A, Weiss B, Ogden AM, Li X, Gleason SD, Calligaro DO, Bleakman D, Witkin JM. In vitro and in vivo studies in rats with LY293558 suggest AMPA/kainate receptor blockade as a novel potential mechanism for the therapeutic treatment of anxiety disorders. Psychopharmacology (Berl). 2006;185(2):240–7. doi:10.1007/s00213-005-0292-0.
Altshuler LL, Abulseoud OA, Foland-Ross L, Bartzokis G, Chang S, Mintz J, Hellemann G, Vinters HV. Amygdala astrocyte reduction in subjects with major depressive disorder but not bipolar disorder. Bipolar Disord. 2010;12(5):541–9. doi:10.1111/j.1399-5618.2010.00838.x.
Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32(1):1–14.
Anisman H, Zacharko RM. Multiple neurochemical and behavioral consequences of stressors: implications for depression. Pharmacol Ther. 1990;46(1):119–36.
Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29(1):83–120.
Araya-Callís C, Hiemke C, Abumaria N, Flugge G. Chronic psychosocial stress and citalopram modulate the expression of the glial proteins GFAP and NDRG2 in the hippocampus. Psychopharmacology (Berl). 2012;224(1):209–22. doi:10.1007/s00213-012-2741-x.
Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14(9):5559–69.
Attucci S, Carlà V, Mannaioni G, Moroni F. Activation of type 5 metabotropic glutamate receptors enhances NMDA responses in mice cortical wedges. Br J Pharmacol. 2001;132(4):799–806. doi:10.1038/sj.bjp.0703904.
Autry AE, Grillo CA, Piroli GG, Rothstein JD, McEwen BS, Reagan LP. Glucocorticoid regulation of GLT-1 glutamate transporter isoform expression in the rat hippocampus. Neuroendocrinology. 2006;83(5-6):371–9. doi:10.1159/000096092.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng P, Kavalali ET, Monteggia LM. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475(7354):91–5. doi:10.1038/nature10130.
Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000;20(21):7871–9.
Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neuroscience. 1997;77(1):65–73.
Banasr M, Duman RS. Regulation of neurogenesis and gliogenesis by stress and antidepressant treatment. CNS Neurol Disord Drug Targets. 2007;6(5):311–20.
Banasr M, Duman RS. Glial loss in the prefrontal cortex is sufficient to induce depressive-like behaviors. Biol Psychiatry. 2008;64(10):863–70. doi:10.1016/j.biopsych.2008.06.008.
Banasr M, Chowdhury GMI, Terwilliger R, Newton SS, Duman RS, Behar KL, Sanacora G. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry. 2010a;15(5):501–11. doi:10.1038/mp.2008.106.
Barha CK, Pawluski JL, Galea LAM. Maternal care affects male and female offspring working memory and stress reactivity. Physiol Behav. 2007;92(5):939–50. doi:10.1016/j.physbeh.2007.06.022.
Barkus C, Feyder M, Graybeal C, Wright T, Wiedholz L, Izquierdo A, et al. Do GluA1 knockout mice exhibit behavioral abnormalities relevant to the negative or cognitive symptoms of schizophrenia and schizoaffective disorder? Neuropharmacology. 2011;62(3):1263–72. doi:10.1016/j.neuropharm.2011.06.005.
Belozertseva IV, Kos T, Popik P, Danysz W, Bespalov AY. Antidepressant-like effects of mGluR1 and mGluR5 antagonists in the rat forced swim and the mouse tail suspension tests. European Neuropsychopharmacol. 2007;17(3):172–9. doi:10.1016/j.euroneuro.2006.03.002.
Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351–4.
Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, et al. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol Psychiatry. 2011;16(6):634–46. doi:10.1038/mp.2010.44.
Bonanno G, Giambelli R, Raiteri L, Tiraboschi E, Zappettini S, Musazzi L, et al. Chronic antidepressants reduce depolarization-evoked glutamate release and protein interactions favoring formation of SNARE complex in hippocampus. J Neurosci. 2005;25(13):3270–9. doi:10.1523/JNEUROSCI.5033-04.2005.
Brakeman PR, Lanahan AA, O’Brien R, Roche K, Barnes CA, Huganir RL, Worley PF. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature. 1997;386(6622):284–8. doi:10.1038/386284a0.
Brennan BP, Hudson JI, Jensen JE, McCarthy J, Roberts JL, Prescot AP, et al. Rapid enhancement of glutamatergic neurotransmission in bipolar depression following treatment with riluzole. Neuropsychopharmacology. 2010;35(3):834–46. doi:10.1038/npp.2009.191.
Brodkin J, Busse C, Sukoff SJ, Varney MA. Anxiolytic-like activity of the mGluR5 antagonist MPEP: a comparison with diazepam and buspirone. Pharmacol Biochem Behav. 2002;73(2):359–66.
Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22(1):183–92.
Busse CS, Brodkin J, Tattersall D, Anderson JJ, Warren N, Tehrani L, et al. The behavioral profile of the potent and selective mGlu5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology. 2004;29(11):1971–9. doi:10.1038/sj.npp.1300540.
Calabrese JR, Bowden CL, Sachs GS, Ascher JA, Monaghan E, Rudd GD. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60(2):79–88.
Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75(3):889–907.
Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science(New York NY). 2003;301(5631):386–9. doi:10.1126/science.1083968.
Chaki S, Yoshikawa R, Hirota S, Shimazaki T, Maeda M, Kawashima N, et al. MGS0039: a potent and selective group II metabotropic glutamate receptor antagonist with antidepressant-like activity. Neuropharmacology. 2004;46(4):457–67. doi:10.1016/j.neuropharm.2003.10.009.
Chaki S, Ago Y, Palucha-Paniewiera A, Matrisciano F, Pilc A. mGlu2/3 and mGlu5 receptors: potential targets for novel antidepressants. Neuropharmacology. 2013;66:40–52. doi:10.1016/j.neuropharm.2012.05.022.
Chappell AS, Gonzales C, Williams J, Witte MM, Mohs RC, Sperling R. AMPA potentiator treatment of cognitive deficits in Alzheimer disease. Neurology. 2007;68(13):1008–12. doi:10.1212/01.wnl.0000260240.46070.7c.
Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc Natl Acad Sci U S A. 2005;102(43):15653–8. doi:10.1073/pnas.0507901102.
Chowdhury GMI, Behar KL, Cho W, Thomas Ma, Rothman DL, Sanacora G. 1H-[13C]-nuclear magnetic resonance spectroscopy measures of ketamine’s effect on amino acid neurotransmitter metabolism. Biol Psychiatry. 2012;71(11):1022–5. doi:10.1016/j.biopsych.2011.11.006.
Christian KM, Miracle AD, Wellman CL, Nakazawa K. Chronic stress-induced hippocampal dendritic retraction requires CA3 NMDA receptors. Neuroscience. 2011;174:26–36. doi:10.1016/j.neuroscience.2010.11.033.
Coric V, Taskiran S, Pittenger C, Wasylink S, Mathalon DH, Valentine G, et al. Riluzole augmentation in treatment-resistant obsessive-compulsive disorder: an open-label trial. Biol Psychiatry. 2005;58(5):424–8. doi:10.1016/j.biopsych.2005.04.043.
Cotter D. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58(6):545–53. doi:10.1001/archpsyc.58.6.545.
Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cerebral Cortex. 2002;12(4):386–94. (New York, N.Y. : 1991).
Cowen MS, Djouma E, Lawrence AJ. The metabotropic glutamate 5 receptor antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]-pyridine reduces ethanol self-administration in multiple strains of alcohol-preferring rats and regulates olfactory glutamatergic systems. J Pharmacol Exp Ther. 2005;315(2):590–600. doi:10.1124/jpet.105.090449.
Cravens CJ, Vargas-Pinto N, Christian KM, Nakazawa K. CA3 NMDA receptors are crucial for rapid and automatic representation of context memory. Eur J Neurosci. 2006;24(6):1771–80. doi:10.1111/j.1460-9568.2006.05044.x.
Cryan JF, Dev KK. Role of glutamate in anxiety In: Handbook of fear and anxiety. Blanchard DC, Blanchard RM (eds). 2007
Czéh B, Michaelis T, Watanabe T, Frahm J, de Biurrun G, van Kampen M, et al. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc Natl Acad Sci U S A. 2001;98(22):12796–801. doi:10.1073/pnas.211427898.
Czéh B, Simon M, Schmelting B, Hiemke C, Fuchs E. Astroglial plasticity in the hippocampus is affected by chronic psychosocial stress and concomitant fluoxetine treatment. Neuropsychopharmacology. 2006;31(8):1616–26. doi:10.1038/sj.npp.1300982.
DD DDS, Marek GJ. Preclinical pharmacology of mGlu2/3 receptor agonists: novel agents for schizophrenia? Curr Drug Targets CNS Neurol Disord. 2002;1(2):215–25.
Diazgranados N, Ibrahim L, Brutsche NE, Newberg A, Kronstein P, Khalife S, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67(8):793–802. doi:10.1001/archgenpsychiatry.2010.90.
Dieterich DC, Karpova A, Mikhaylova M, Zdobnova I, König I, Landwehr M, et al. Caldendrin-Jacob: a protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol. 2008;6(2):e34. doi:10.1371/journal.pbio.0060034.
Doherty AJ, Palmer MJ, Bortolotto ZA, Hargreaves A, Kingston AE, Ornstein PL, et al. A novel, competitive mGlu(5) receptor antagonist (LY344545) blocks DHPG-induced potentiation of NMDA responses but not the induction of LTP in rat hippocampal slices. Br J Pharmacol. 2000;131(2):239–44. doi:10.1038/sj.bjp.0703574.
Du J, Suzuki K, Wei Y, Wang Y, Blumenthal R, Chen Z, et al. The anticonvulsants lamotrigine, riluzole, and valproate differentially regulate AMPA receptor membrane localization: relationship to clinical effects in mood disorders. Neuropsychopharmacology. 2007;32(4):793–802. doi:10.1038/sj.npp.1301178.
Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR2/3 blockade. Int J Neuropsychopharmacol. 2012;15(4):429–34. doi:10.1017/S1461145711001702.
Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399(6731):66–70. doi:10.1038/19978.
Ettinger AB, Argoff CE. Use of antiepileptic drugs for nonepileptic conditions: psychiatric disorders and chronic pain. Neurotherapeutics. 2007;4(1):75–83. doi:10.1016/j.nurt.2006.10.003.
Fatemi SH, Folsom TD, Reutiman TJ, Pandian T, Braun NN, Haug K. Chronic psychotropic drug treatment causes differential expression of connexin 43 and GFAP in frontal cortex of rats. Schizophr Res. 2008;104(1–3):127–34. doi:10.1016/j.schres.2008.05.016.
Fell MJ, Witkin JM, Falcone JF, Katner JS, Perry KW, Hart J, et al. N-(4-((2-(trifluoromethyl)-3-hydroxy-4-(isobutyryl)phenoxy)methyl)benzyl)-1-methyl-1H-imidazole-4-carboxamide (THIIC), a novel metabotropic glutamate 2 potentiator with potential anxiolytic/antidepressant properties: in vivo profiling suggests a link between behavioral and central nervous system neurochemical changes. J Pharmacol Exp Ther. 2011;336(1):165–77. doi:10.1124/jpet.110.172957.
Feyissa AM, Woolverton WL, Miguel-Hidalgo JJ, Wang Z, Kyle PB, Hasler G, et al. Elevated level of metabotropic glutamate receptor 2/3 in the prefrontal cortex in major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(2):279–83. doi:10.1016/j.pnpbp.2009.11.018.
Fitzgerald PJ, Barkus C, Feyder M, Wiedholz LM, Chen Y-C, Karlsson R-M, … Holmes A. Does gene deletion of AMPA GluA1 phenocopy features of schizoaffective disorder? Neurobiol Dis. 2010;40(3):608–21. doi:10.1016/j.nbd.2010.08.005.
Fraser CM, Cooke MJ, Fisher A, Thompson ID, Stone TW. Interactions between ifenprodil and dizocilpine on mouse behaviour in models of anxiety and working memory. Eur Neuropsychopharmacol. 1996;6(4):311–6.
Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578(2–3):171–6. doi:10.1016/j.ejphar.2007.10.023.
Gittins RA, Harrison PJ. A morphometric study of glia and neurons in the anterior cingulate cortex in mood disorder. J Affect Disord. 2011;133(1–2):328–32. doi:10.1016/j.jad.2011.03.042.
Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48(4):308–20. doi:10.1016/j.mcn.2011.05.001.
Gould E, Cameron HA. Early NMDA receptor blockade impairs defensive behavior and increases cell proliferation in the dentate gyrus of developing rats. Behav Neurosci. 1997;111(1):49–56.
Gourley SL, Espitia JW, Sanacora G, Taylor JR. Antidepressant-like properties of oral riluzole and utility of incentive disengagement models of depression in mice. Psychopharmacology (Berl). 2012;219(3):805–14. doi:10.1007/s00213-011-2403-4.
Groc L, Choquet D, Chaouloff F. The stress hormone corticosterone conditions AMPAR surface trafficking and synaptic potentiation. Nat Neurosci. 2008;11(8):868–70. doi:10.1038/nn.2150.
Groeneweg FL, Karst H, de Kloet ER, Joëls M. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J Endocrinol. 2011;209(2):153–67. doi:10.1530/JOE-10-0472.
Guttmann RP, Baker D, Seifert KM, Cohen AS, Coulter DA, Lynch DR. Specific proteolysis of the NR2 subunit—at multiple sites by calpain. Journal of Neurochemistry. 2001; 78(5)1083–93.
Guttmann RP, Sokol S, Baker D, Simpkins KL, Dong Y, Lynch DR. Proteolysis of the N-methyl-d-aspartate—receptor by calpain in situ. The Journal of pharmacology and experimental therapeutics 2002;302(3)1023–30.
Hammen C. Stress and depression. Annu Rev Clin Psychol. 2005;1:293–319. doi:10.1146/annurev.clinpsy.1.102803.143938.
Hansson E, Rönnbäck L. Altered neuronal-glial signaling in glutamatergic transmission as a unifying mechanism in chronic pain and mental fatigue. Neurochem Res. 2004;29(5):989–96.
Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11(10):682–96. doi:10.1038/nrn2911.
Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5(5):405–14. doi:10.1038/nn835.
Hughes ZA, Neal SJ, Smith DL, Sukoff Rizzo SJ, Pulicicchio CM, Lotarski S, et al. Negative allosteric modulation of metabolic glutamate receptor 5 results in broad spectrum activity relevant to treatment resistant depression. Neuropharmacology. 2012;66:202–14. doi:10.1016/j.neuropharm.2012.04.007.
Ibrahim L, Diazgranados N, Luckenbaugh DA, Machado-Vieira R, Baumann J, Mallinger AG, Zarate CA. Rapid decrease in depressive symptoms with an N-methyl-d-aspartate antagonist in ECT-resistant major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(4):1155–9. doi:10.1016/j.pnpbp.2011.03.019.
Iyo AH, Feyissa AM, Chandran A, Austin MC, Regunathan S, Karolewicz B. Chronic corticosterone administration down-regulates metabotropic glutamate receptor 5 protein expression in the rat hippocampus. Neuroscience. 2010;169(4):1567–74. doi:10.1016/j.neuroscience.2010.06.023
Izquierdo A, Wellman CL, Holmes A. Brief uncontrollable stress causes dendritic retraction in infralimbic cortex and resistance to fear extinction in mice. J Neurosci. 2006;26(21):5733–8. doi:10.1523/JNEUROSCI.0474-06.2006
Jackson ME, Moghaddam B. Distinct patterns of plasticity in prefrontal cortex neurons that encode slow and fast responses to stress. Eur J Neurosci. 2006;24(6):1702–10. doi:10.1111/j.1460-9568.2006.05054.x.
Johnson MP, Barda D, Britton TC, Emkey R, Hornback WJ, Jagdmann GE, et al. Metabotropic glutamate 2 receptor potentiators: receptor modulation, frequency-dependent synaptic activity, and efficacy in preclinical anxiety and psychosis model(s). Psychopharmacology (Berl). 2005;179(1):271–83. doi:10.1007/s00213-004-2099-9.
Johnston-Wilson NL, Sims CD, Hofmann J-P, Anderson L, Shore AD, Torrey EF, Yolken RH. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. Mol Psychiatry. 2000;5(2):142–9. doi:10.1038/sj.mp.4000696.
Kapus GL, Gacsályi I, Vegh M, Kompagne H, Hegedus E, Leveleki C, et al. Antagonism of AMPA receptors produces anxiolytic-like behavior in rodents: effects of GYKI 52466 and its novel analogues. Psychopharmacology (Berl). 2008;198(2):231–41. doi:10.1007/s00213-008-1121-z.
Karpova A, Mikhaylova M, Bera S, Bär J, Reddy PP, Behnisch T, et al. Encoding and transducing the synaptic or extrasynaptic origin of NMDA receptor signals to the nucleus. Cell. 2013;152(5):1119–33. doi:10.1016/j.cell.2013.02.002.
Karst H, Joëls M. Corticosterone slowly enhances miniature excitatory postsynaptic current amplitude in mice CA1 hippocampal cells. J Neurophysiol. 2005;94(5):3479–86. doi:10.1152/jn.00143.2005.
Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am J Psychiatry. 1999a;156(6):837–41.
Kendler KS, Karkowski LM, Prescott CA. The assessment of dependence in the study of stressful life events: validation using a twin design. Psychol Med. 1999b;29(6):1455–60.
Kessler RC, Avenevoli S, McLaughlin KA, Green JG, Lakoma MD, Petukhova M, et al. Lifetime co-morbidity of DSM-IV disorders in the US national comorbidity survey replication adolescent supplement (NCS-A). Psychol Med. 2012;42(9):1997–2010. doi:10.1017/S0033291712000025.
Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3(6):453–62. doi:10.1038/nrn849.
Kim JJ, Foy MR, Thompson RF. Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. Proc Natl Acad Sci U S A. 1996;93(10):4750–3.
Kiselycznyk C, Svenningsson P, Delpire E, Holmes A. Genetic, pharmacological and lesion analyses reveal a selective role for corticohippocampal GLUN2B in a novel repeated swim stress paradigm. Neuroscience. 2011;193:259–268. doi:10.1016/j.neuroscience.2011.06. 015.
Kiselycznyk C, Zhang X, Huganir RL, Holmes A, Svenningsson P. Reduced phosphorylation of GluA1 subunits relates to anxiety-like behaviours in mice. Int J Neuropsychopharmacol. 2013;16(4):919–24. doi:10.1017/S1461145712001174.
Knapp RJ, Goldenberg R, Shuck C, Cecil A, Watkins J, Miller C, et al. Antidepressant activity of memory-enhancing drugs in the reduction of submissive behavior model. Eur J Pharmacol. 2002;440(1):27–35.
Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224(1):107–11. doi:10.1016/j.bbr.2011.05.035.
Kotlinska J, Liljequist S. The putative AMPA receptor antagonist, LY326325, produces anxiolytic-like effects without altering locomotor activity in rats. Pharmacol Biochem Behav. 1998;60(1):119–24.
Kovačević T, Skelin I, Minuzzi L, Rosa-Neto P, Diksic M. Reduced metabotropic glutamate receptor 5 in the Flinders sensitive line of rats, an animal model of depression: an autoradiographic study. Brain Res Bull. 2012;87(4–5):406–12. doi:10.1016/j.brainresbull.2012.01.010.
Krebs C, Fernandes HB, Sheldon C, Raymond LA, Baimbridge KG. Functional NMDA receptor subtype 2B is expressed in astrocytes after ischemia in vivo and anoxia in vitro. J Neurosci. 2003;23(8):3364–72.
Krugers HJ, Hoogenraad CC, Groc L. Stress hormones and AMPA plasticity and memory. Nat Rev Neurosci. 2010;10:675–81.
Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci. 2006;26(10):2673–83. doi:10.1523/JNEUROSCI.4689-05.2006.
Li X, Tizzano JP, Griffey K, Clay M, Lindstrom T, Skolnick P. Antidepressant-like actions of an AMPA receptor potentiator (LY392098). Neuropharmacology. 2001;40(8):1028–33.
Li X, Need AB, Baez M, Witkin JM. Metabotropic glutamate 5 receptor antagonism is associated with antidepressant-like effects in mice. J Pharmacol Exp Ther. 2006;319(1):254–9. doi:10.1124/jpet.106.103143.
Li N, Lee B, Liu R-J, Banasr M, Dwyer JM, Iwata M, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329(5994):959–64. doi:10.1126/science.1190287. (New York NY).
Lindholm JSO, Autio H, Vesa L, Antila H, Lindemann L, Hoener MC, et al. The antidepressant-like effects of glutamatergic drugs ketamine and AMPA receptor potentiator LY 451646 are preserved in bdnf + /− heterozygous null mice. Neuropharmacology. 2012;62(1):391–7. doi:10.1016/j.neuropharm.2011.08.015.
Lipton SA. NMDA receptors, glial cells, and clinical medicine. Neuron. 2006;50(1):9–11.
Liu Q, Li B, Zhu H-Y, Wang Y-Q, Yu J, Wu G-C. Clomipramine treatment reversed the glial pathology in a chronic unpredictable stress-induced rat model of depression. Eur Neuropsychopharmacol. 2009;19(11):796–805. doi:10.1016/j.euroneuro.2009.06.010.
Lowy MT, Gault L, Yamamoto BK. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61(5):1957–60.
Luine V, Martinez C, Villegas M, Magariños AM, McEwen BS. Restraint stress reversibly enhances spatial memory performance. Physiol Behav. 1996;59(1):27–32.
Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8(7):1488–500.
Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10(6):434–45. doi:10.1038/nrn2639.
Maeng S, Zarate CA, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63(4):349–52. doi:10.1016/j.biopsych.2007.05.028.
Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: comparison of stressors. Neuroscience. 1995a;69(1):83–8.
Magariños AM, McEwen BS. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience. 1995b;69(1):89–98.
Magariños AM, Deslandes A, McEwen BS. Effects of antidepressants and benzodiazepine treatments on the dendritic structure of CA3 pyramidal neurons after chronic stress. Eur J Pharmacol. 1999;371(2-3):113–22.
Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52(1–2):142–54. doi:10.1016/j.neuint.2007.06.005.
Martel M-A, Ryan TJ, Bell KFS, Fowler JH, McMahon A, Al-Mubarak B, et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron. 2012;74(3):543–56. doi:10.1016/j.neuron.2012.03.021.
Martin KP, Wellman CL. NMDA receptor blockade alters stress-induced dendritic remodeling in medial prefrontal cortex. Cerebral Cortex. 2011;21(10):2366–73. doi:10.1093/cercor/bhr021. (New York, N.Y. : 1991)
Matheus MG, Guimarães FS. Antagonism of non-NMDA receptors in the dorsal periaqueductal grey induces anxiolytic effect in the elevated plus maze. Psychopharmacology (Berl). 1997;132(1):14–8.
Mathew SJ, Murrough JW, aan het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment-resistant depression: a pilot randomized, placebo-controlled continuation trial. Int J Neuropsychopharmacol. 2010;13(1):71–82. doi:10.1017/S1461145709000169.
Mathews DC, Zarate J. Current status of ketamine and related compounds for depression. J Clin Psychiatry. 2013;74(05):516–7. doi:10.4088/JCP.13ac08382.
Mathur P, Graybeal C, Feyder M, Davis MI, Holmes A. Fear memory impairing effects of systemic treatment with the NMDA NR2B subunit antagonist, Ro 25-6981, in mice: attenuation with ageing. Pharmacol Biochem Behav. 2009;91(3):453–60. doi:10.1016/j.pbb.2008.08.028.
Matrisciano F, Storto M, Ngomba RT, Cappuccio I, Caricasole A, Scaccianoce S, et al. Imipramine treatment up-regulates the expression and function of mGlu2/3 metabotropic glutamate receptors in the rat hippocampus. Neuropharmacology. 2002;42(8):1008–15.
Matrisciano F, Scaccianoce S, Del Bianco P, Panaccione I, Canudas AM, Battaglia G, et al. Metabotropic glutamate receptors and neuroadaptation to antidepressants: imipramine-induced down-regulation of beta-adrenergic receptors in mice treated with metabotropic glutamate 2/3 receptor ligands. J Neurochem. 2005;93(5):1345–52. doi:10.1111/j.1471-4159.2005.03141.x.
Matrisciano F, Panaccione I, Zusso M, Giusti P, Tatarelli R, Iacovelli L, et al. Group-II metabotropic glutamate receptor ligands as adjunctive drugs in the treatment of depression: a new strategy to shorten the latency of antidepressant medication? Mol Psychiatry. 2007;12(8):704–6. doi:10.1038/sj.mp.4002005.
Matrisciano F, Caruso A, Orlando R, Marchiafava M, Bruno V, Battaglia G, et al. Defective group-II metaboropic glutamate receptors in the hippocampus of spontaneously depressed rats. Neuropharmacology. 2008;55(4):525–31. doi:10.1016/j.neuropharm.2008.05.014.
Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429(6993):761–6. doi:10.1038/nature02617.
McCullumsmith RE, Meador-Woodruff JH. Striatal excitatory amino acid transporter transcript expression in schizophrenia, bipolar disorder, and major depressive disorder. Neuropsychopharmacology. 2002;26(3):368–75. doi:10.1016/S0893-133X(01)00370-0.
McElroy SL, Zarate CA, Cookson J, Suppes T, Huffman RF, Greene P, Ascher J. A 52-week, open-label continuation study of lamotrigine in the treatment of bipolar depression. J Clin Psychiatry. 2004;65(2):204–10.
McEwen BS, Chattarji S, Diamond DM, Jay TM, Reagan LP, Svenningsson P, Fuchs E. The neurobiological properties of tianeptine (Stablon): from monoamine hypothesis to glutamatergic modulation. Mol Psychiatry. 2010;15(3):237–49. doi:10.1038/mp.2009.80.
Metsis M, Timmusk T, Arenas E, Persson H. Differential usage of multiple brain-derived neurotrophic factor promoters in the rat brain following neuronal activation. Proc Natl Acad Sci U S A. 1993;90(19):8802–6.
Milanese M, Tardito D, Musazzi L, Treccani G, Mallei A, Bonifacino T, et al. Chronic treatment with agomelatine or venlafaxine reduces depolarization-evoked glutamate release from hippocampal synaptosomes. BMC Neurosci. 2013;14:75. doi:10.1186/1471-2202-14-75.
Mineur YS, Picciotto MR, Sanacora G. Antidepressant-like effects of ceftriaxone in male C57BL/6J mice. Biol Psychiatry. 2007;61(2):250–2. doi:10.1016/j.biopsych.2006.04.037.
Mitterauer BJ. Ketamine may block NMDA receptors in astrocytes causing a rapid antidepressant effect. Front Synaptic Neurosci. 2012;4:8. doi:10.3389/fnsyn.2012.00008.
Moghaddam B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem. 1993;60(5):1650–7.
Moghaddam B. Stress activation of glutamate neurotransmission in the prefrontal cortex: implications for dopamine-associated psychiatric disorders. Biol Psychiatry. 2002;51(10):775–87.
Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17(8):2921–7.
Molina-Hernández M, Tellez-Alcántara NP, Pérez-García J, Olivera-Lopez JI, Jaramillo MT. Antidepressant-like and anxiolytic-like actions of the mGlu5 receptor antagonist MTEP, microinjected into lateral septal nuclei of male Wistar rats. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):1129–35. doi:10.1016/j.pnpbp.2006.04.022.
Morley-Fletcher S, Mairesse J, Soumier A, Banasr M, Fagioli F, Gabriel C,et al. Chronic agomelatine treatment corrects behavioral, cellular, and biochemical abnormalities induced by prenatal stress in rats. Psychopharmacology (Berl). 2011;217(3):301–13. doi:10.1007/s00213-011-2280-x.
Moryl E, Danysz W, Quack G. Potential antidepressive properties of amantadine, memantine and bifemelane. Pharmacol Toxicol. 1993;72(4–5):394–7. doi:10.1111/j.1600-0773.1993.tb01351.x.
Muhonen LH, Lönnqvist J, Juva K, Alho H. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. J Clin Psychiatry. 2008;69(3):392–9.
Mula M, Pini S, Cassano GB. The role of anticonvulsant drugs in anxiety disorders: a critical review of the evidence. J Clin Psychopharmacol. 2007;27(3):263–72. doi:10.1097/jcp.0b013e318059361a.
Müller HK, Wegener G, Liebenberg N, Zarate CA, Popoli M, Elfving B. Ketamine regulates the presynaptic release machinery in the hippocampus. J Psychiatr Res. 2013;47(7):892–9. doi:10.1016/j.jpsychires.2013.03.008.
Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CM, Perez AM, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170(10):1134–42. doi:10.1176/appi.ajp.2013.13030392.
Musazzi L, Milanese M, Farisello P, Zappettini S, Tardito D, Barbiero VS, et al. Acute stress increases depolarization-evoked glutamate release in the rat prefrontal/frontal cortex: the dampening action of antidepressants. PloS ONE. 2010;5(1):e8566. doi:10.1371/journal.pone.0008566.
Musazzi L, Treccani G, Mallei A, Popoli M. The action of antidepressants on the glutamate system: regulation of glutamate release and glutamate receptors. Biol Psychiatry. 2012;73(12):1180–8. doi:10.1016/j.biopsych.2012.11.009.
Nations KR, Dogterom P, Bursi R, Schipper J, Greenwald S, Zraket D, et al. Examination of Org 26576, an AMPA receptor positive allosteric modulator, in patients diagnosed with major depressive disorder: an exploratory, randomized, double-blind, placebo-controlled trial. J Psychopharmacol. 2012;26(12):1525–39. doi:10.1177/0269881112458728.
O’Shea RD. Roles and regulation of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol. 2002;29(11):1018–23.
Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95(22):13290–5. doi:10.1073/pnas.95.22.13290.
Palucha A, Pilc A. Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol Ther. 2007;115(1):116–47. doi:10.1016/j.pharmthera.2007.04.007.
Pałucha A, Brański P, Szewczyk B, Wierońska JM, Kłak K, Pilc A. Potential antidepressant-like effect of MTEP, a potent and highly selective mGluR5 antagonist. Pharmacol Biochem Behav. 2005;81(4):901–6. doi:10.1016/j.pbb.2005.06.015.
Pałucha-Poniewiera A, Wierońska JM, Brański P, Stachowicz K, Chaki S, Pilc A. On the mechanism of the antidepressant-like action of group II mGlu receptor antagonist, MGS0039. Psychopharmacology (Berl). 2010;212(4):523–35. doi:10.1007/s00213-010-1978-5.
Palygin O, Lalo U, Pankratov Y. Distinct pharmacological and functional properties of NMDA receptors in mouse cortical astrocytes. Br J Pharmacol. 2011;163(8):1755–66. doi:10.1111/j.1476-5381.2011.01374.x.
Paul IA, Skolnick P. Glutamate and depression: clinical and preclinical studies. Ann N Y Acad Sci. 2003;1003:250–72.
Pecknold JC, McClure DJ, Appeltauer L, Wrzesinski L, Allan T. Treatment of anxiety using fenobam (a nonbenzodiazepine) in a double-blind standard (diazepam) placebo-controlled study. J Clin Psychopharmacol. 1982;2(2):129–33.
Pilc A, Kłodzińska A, Brański P, Nowak G, Pałucha A, Szewczyk B, et al. Multiple MPEP administrations evoke anxiolytic- and antidepressant-like effects in rats. Neuropharmacology. 2002;43(2):181–7.
Pilc A, Chaki S, Nowak G, Witkin JM. Mood disorders: regulation by metabotropic glutamate receptors. Biochem Pharmacol. 2008;75(5):997–1006. doi:10.1016/j.bcp.2007.09.021.
Pisani A, Gubellini P, Bonsi P, Conquet F, Picconi B, Centonze D, et al. Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-D-aspartate responses in medium spiny striatal neurons. Neuroscience. 2001;106(3):579–87.
Pittenger C, Coric V, Banasr M, Bloch M, Krystal JH, Sanacora G. Riluzole in the treatment of mood and anxiety disorders. CNS Drugs. 2008;22(9):761–86.
Popoli M. Agomelatine: innovative pharmacological approach in depression. CNS Drugs. 2009;23(Suppl 2):27–34. doi:10.2165/11318640-000000000-00000.
Porter RHP, Jaeschke G, Spooren W, Ballard TM, Büttelmann B, Kolczewski S, et al. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharmacol Exp Ther. 2005;315(2):711–21. doi:10.1124/jpet.105.089839.
Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-methyl-D-aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. J Clin Psychopharmacol. 2008;28(6):631–7. doi:10.1097/JCP.0b013e31818a6cea.
Rajkowska G, Miguel-Hidalgo JJ. Gliogenesis and glial pathology in depression. CNS Neurol Disord Drug Targets. 2007;6(3):219–33.
Rajkowska G, Stockmeier CA. Astrocyte pathology in major depressive disorder: insights from human postmortem brain tissue. Curr Drug Targets. 2013;14(11):1225–36.
Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, … Stockmeier CA (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 45(9):1085–98. doi:10.1016/S0006-3223(99)00041-4.
Reynolds IJ, Miller RJ. Tricyclic antidepressants block N-methyl-D-aspartate receptors: similarities to the action of zinc. Br J Pharmacol. 1988;95(1):95–102.
Reznikov LR, Grillo CA, Piroli GG, Pasumarthi RK, Reagan LP, Fadel J. Acute stress-mediated increases in extracellular glutamate levels in the rat amygdala: differential effects of antidepressant treatment. Eur J Neurosci. 2007;25(10):3109–14. doi:10.1111/j.1460-9568.2007.05560.x.
Rogóz Z, Skuza G, Maj J, Danysz W. Synergistic effect of uncompetitive NMDA receptor antagonists and antidepressant drugs in the forced swimming test in rats. Neuropharmacology. 2002;42(8):1024–30.
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73–7. doi:10.1038/nature03180.
Rutherford EC, Pomerleau F, Huettl P, Strömberg I, Gerhardt GA. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J Neurochem. 2007;102(3):712–22. doi:10.1111/j.1471-4159.2007.04596.x.
Sanacora G, Banasr M. From pathophysiology to novel antidepressant drugs: glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry. 2013;73(12):1172–9. doi:10.1016/j.biopsych.2013.03.032.
Sanacora G, Kendell SF, Fenton L, Coric V, Krystal JH. Riluzole augmentation for treatment-resistant depression. Am J Psychiatry. 2004;161(11):2132. doi:10.1176/appi.ajp.161.11.2132.
Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7(5):426–37. doi:10.1038/nrd2462.
Sapolsky RM. The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol Psychiatry. 2000;48(8):755–65.
Sapolsky RM. Neuroprotective gene therapy against acute neurological insults. Nat Rev Neurosci. 2003;4(1):61–9. doi:10.1038/nrn1006.
Schneiderman N, Ironson G, Siegel SD. Stress and health: psychological, behavioral, and biological determinants. Annu Rev Clin Psychol. 2005;1:607–28. doi:10.1146/annurev.clinpsy.1.102803.144141.
Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PloS ONE. 2009;4(8):e6585. doi:10.1371/journal.pone.0006585.
Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev. 2004;45(3):250–65. doi:10.1016/j.brainresrev.2004.04.004.
Sillaber I, Panhuysen M, Henniger MSH, Ohl F, Kühne C, Pütz B, et al. Profiling of behavioral changes and hippocampal gene expression in mice chronically treated with the SSRI paroxetine. Psychopharmacology (Berl). 2008;200(4):557–72. doi:10.1007/s00213-008-1232-6.
Sinha R. Chronic stress, drug use, and vulnerability to addiction. Ann N Y Acad Sci. 2008;1141:105–30. doi:10.1196/annals.1441.030.
Skolnick P, Layer RT, Popik P, Nowak G, Paul IA, Trullas R. Adaptation of N-methyl-D-aspartate (NMDA) receptors following antidepressant treatment: implications for the pharmacotherapy of depression. Pharmacopsychiatry. 1996;29(1):23–6. doi:10.1055/s-2007-979537.
Skolnick P, Legutko B, Li X, Bymaster FP. Current perspectives on the development of non-biogenic amine-based antidepressants. Pharmacol Res. 2001;43(5):411–23. doi:10.1006/phrs.2000.0806.
Skolnick P, Popik P, Trullas R. Glutamate-based antidepressants: 20 years on. Trends Pharmacol Sci. 2009;30(11):563–9. doi:10.1016/j.tips.2009.09.002.
Stein-Behrens B, Mattson MP, Chang I, Yeh M, Sapolsky R. Stress exacerbates neuron loss and cytoskeletal pathology in the hippocampus. J Neurosci. 1994;14(9):5373–80.
Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, et al. Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry. 2004;56(9):640–50. doi:10.1016/j.biopsych.2004.08.022.
Sun W, McConnell E, Pare J-F, Xu Q, Chen M, Peng W, et al. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science. 2013;339(6116):197–200. doi:10.1126/science.1226740. (New York NY).
Svenningsson P, Bateup H, Qi H, Takamiya K, Huganir RL, Spedding M, et al. Involvement of AMPA receptor phosphorylation in antidepressant actions with special reference to tianeptine. Eur J Neurosci. 2007;26(12):3509–17. doi:10.1111/j.1460-9568.2007.05952.x.
Swanson CJ, Bures M, Johnson MP, Linden A-M, Monn JA, Schoepp DD. Metabotropic glutamate receptors as novel targets for anxiety and stress disorders. Nat Rev Drug Discov. 2005;4(2):131–44. doi:10.1038/nrd1630.
Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, et al. A induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110(27):E2518–27. doi:10.1073/pnas.1306832110.
Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106(3):481–503.
Tardito D, Milanese M, Bonifacino T, Musazzi L, Grilli M, Mallei A, et al. Blockade of stress-induced increase of glutamate release in the rat prefrontal/frontal cortex by agomelatine involves synergy between melatonergic and 5-HT2C receptor-dependent pathways. BMC Neurosci. 2010;11:68. doi:10.1186/1471-2202-11-68.
Tardito D, Molteni R, Popoli M, Racagni G. Synergistic mechanisms involved in the antidepressant effects of agomelatine. Eur Neuropsychopharmacol. 2012;22(Suppl 3):S482–6. doi:10.1016/j.euroneuro.2012.06.016.
Tatarczyńska E, Klodzińska A, Chojnacka-Wójcik E, Palucha A, Gasparini F, Kuhn R, Pilc A. Potential anxiolytic- and antidepressant-like effects of MPEP, a potent, selective and systemically active mGlu5 receptor antagonist. Br J Pharmacol. 2001;132(7):1423–30. doi:10.1038/sj.bjp.0703923.
Trantham-Davidson H, LaLumiere RT, Reissner KJ, Kalivas PW, Knackstedt LA. Ceftriaxone normalizes nucleus accumbens synaptic transmission, glutamate transport, and export following cocaine self-administration and extinction training. J Neurosci. 2012;32(36):12406–10. doi:10.1523/JNEUROSCI.1976-12.2012.
Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol. 1990;185(1):1–10.
Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23(3):583–92.
Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley neuropathology consortium. Schizophr Res. 2004;67(2–3), 269–75. doi:10.1016/S0920-9964(03)00181-6.
Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, et al. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [(1)H]-MRS. Psychiatry Res. 2011;191(2):122–7. doi:10.1016/j.pscychresns.2010.10.009.
Van der Loos MLM, Mulder PGH, Hartong EGTM, Blom MBJ, Vergouwen AC, de Keyzer HJUEM, et al. Efficacy and safety of lamotrigine as add-on treatment to lithium in bipolar depression: a multicenter, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(2):223–31.
Vekovischeva OY, Aitta-aho T, Verbitskaya E, Sandnabba K, Korpi ER. Acute effects of AMPA-type glutamate receptor antagonists on intermale social behavior in two mouse lines bidirectionally selected for offensive aggression. Pharmacol Biochem Behav. n.d.;87(2):241–9. doi:10.1016/j.pbb.2007.04.020.
Venero C, Borrell J. Rapid glucocorticoid effects on excitatory amino acid levels in the hippocampus: a microdialysis study in freely moving rats. Eur J Neurosci. 1999;11(7):2465–73.
Verkhratsky A, Kirchhoff F. NMDA receptors in glia. Neuroscientist. 2007;13(1):28–37. doi:10.1177/1073858406294270.
Voleti B, Navarria A, Liu R-J, Banasr M, Li N, Terwilliger R, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742–9. doi:10.1016/j.biopsych.2013.04.025.
Watanabe Y, Gould E, Cameron HA, Daniels DC, McEwen BS. Phenytoin prevents stress- and corticosterone-induced atrophy of CA3 pyramidal neurons. Hippocampus. 1992;2(4):431–5. doi:10.1002/hipo.450020410.
Webster MJ, Knable MB, Johnston-Wilson N, Nagata K, Inagaki M, Yolken RH. Immunohistochemical localization of phosphorylated glial fibrillary acidic protein in the prefrontal cortex and hippocampus from patients with schizophrenia, bipolar disorder, and depression. Brain Behav Immun. 2001;15(4):388–400. doi:10.1006/brbi.2001.0646.
Wieroñska JM, Brañski P, Szewczyk B, Papp M, Gruca P, Moryl E, Pilc A. Preliminary communication changes in the expression of metabotropic glutamate receptor 5 (mglur5) in the rat hippocampus in an animal model of depression. Polish Journal of Pharmacology 2001;5:5–8.
Wieronska JM, Szewczyk B, Branski P, Palucha A, Pilc A. Antidepressant-like effect of MPEP, a potent, selective and systemically active mGlu5 receptor antagonist in the olfactory bulbectomized rats. Amino Acids. 2002;23(1–3):213–6. doi:10.1007/s00726-001-0131-5.
Wierońska JM, Legutko B, Dudys D, Pilc A. Olfactory bulbectomy and amitriptyline treatment influences mGlu receptors expression in the mouse brain hippocampus. Pharmacol Rep. 2008;60(6):844–55.
Witkin JM, Marek GJ, Johnson BG, Schoepp DD. Metabotropic glutamate receptors in the control of mood disorders. CNS Neurol Disord Drug Targets. 2007;6(2):87–100.
Xia P, Chen HV, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30(33):11246–50. doi:10.1523/JNEUROSCI.2488-10.2010.
Xu J, Kurup P, Zhang Y, Goebel-Goody SM, Wu PH, Hawasli AH, et al. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci. 2009;29(29):9330–43. doi:10.1523/JNEUROSCI.2212-09.2009.
Yoshimizu T, Shimazaki T, Ito A, Chaki S. An mGluR2/3 antagonist, MGS0039, exerts antidepressant and anxiolytic effects in behavioral models in rats. Psychopharmacology (Berl). 2006;186(4):587–93. doi:10.1007/s00213-006-0390-7.
Yoshizumi M, Eisenach JC, Hayashida K. Riluzole and gabapentinoids activate glutamate transporters to facilitate glutamate-induced glutamate release from cultured astrocytes. Eur J Pharmacol. 2012;677(1–3):87–92. doi:10.1016/j.ejphar.2011.12.015.
Yuen EY, Liu W, Karatsoreos IN, Feng J, McEwen BS, Yan Z. Acute stress enhances glutamatergic transmission in prefrontal cortex and facilitates working memory. Proc Natl Acad Sci U S A. 2009;106(33):14075–9. doi:10.1073/pnas.0906791106.
Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, Yan Z. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry. 2011;16(2):156–70. doi:10.1038/mp.2010.50.
Zarate CA, Quiroz J, Payne J, Manji HK. Modulators of the glutamatergic system: implications for the development of improved therapeutics in mood disorders. Psychopharmacol Bull. 2002;36(4):35–83.
Zarate CA, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006a;63(8):856–64. doi:10.1001/archpsyc.63.8.856.
Zarate CA, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry. 2006b;163(1):153–5. doi:10.1176/appi.ajp.163.1.153.
Zarate CA, Brutsche NE, Ibrahim L, Franco-Chaves J, Diazgranados N, Cravchik A, et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. 2012;71(11):939–46. doi:10.1016/j.biopsych.2011.12.010.
Zheng K, Scimemi A, Rusakov DA. Receptor actions of synaptically released glutamate: the role of transporters on the scale from nanometers to microns. Biophys J. 2008;95(10):4584–96. doi:10.1529/biophysj.108.129874.
Zink M, Vollmayr B, Gebicke-Haerter PJ, Henn FA. Reduced expression of glutamate transporters vGluT1, EAAT2 and EAAT4 in learned helpless rats, an animal model of depression. Neuropharmacology. 2010;58(2):465–73. doi:10.1016/j.neuropharm.2009.09.005.
Zink M, Rapp S, Donev R, Gebicke-Haerter PJ, Thome J. Fluoxetine treatment induces EAAT2 expression in rat brain. J Neural Trans. 2011;118(6):849–55. doi:10.1007/s00702-010-0536-y. (Vienna, Austria: 1996).
Zschocke J, Bayatti N, Clement AM, Witan H, Figiel M, Engele J, Behl C. Differential promotion of glutamate transporter expression and function by glucocorticoids in astrocytes from various brain regions. J Biol Chem. 2005;280(41):34924–32. doi:10.1074/jbc.M502581200.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Kiselycznyk, C., Sanacora, G. (2014). Using Our Understanding of Stress-Related Effects on Glutamate Neurotransmission to Guide the Development of Novel Treatment Strategies. In: Popoli, M., Diamond, D., Sanacora, G. (eds) Synaptic Stress and Pathogenesis of Neuropsychiatric Disorders. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1056-4_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1056-4_17
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1055-7
Online ISBN: 978-1-4939-1056-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)