
Abstract
Purpose of Review
To summarize the fundamental role of transforming growth factor beta (TGFβ) signaling in osteocytes and highlight the physiological and pathophysiological conditions stemming from the deregulation of this pathway in osteocytes.
Recent Findings
Osteocytes perform a myriad of skeletal and extraskeletal functions, including mechanosensing, coordinating bone remodeling, local bone matrix turnover, and maintaining systemic mineral homeostasis and global energy balance. Transforming growth factor-beta (TGFβ) signaling, which is crucial for embryonic and postnatal bone development and maintenance, has been found to be essential for several osteocyte functions. There is some evidence that TGFβ might be accomplishing these functions through crosstalk with the Wnt, PTH, and YAP/TAZ pathways in osteocytes, and a better understanding of this complex molecular network can help identify the pivotal convergence points responsible for distinct osteocyte functions.
Summary
This review provides recent updates on the interwoven signaling cascades coordinated by TGFβ signaling within osteocytes to support their skeletal and extraskeletal functions and highlights physiological and pathophysiological conditions implicating the role of TGFβ signaling in osteocytes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The mammalian skeleton is one of the largest organs in the human body, accounting for 15% of body weight [1]. Bones serve as the building blocks of the skeleton and provide a structural framework for the body. The roles of bone are diverse; these include skeletal functions, like providing mechanical support, movement, blood cell production, and mineral storage, as well as extraskeletal functions, like endocrine regulation of mineral metabolism and whole-body energy metabolism, that are integral to a better quality of life [2,3,4]. Bone mechanical integrity is determined by the bone mass and quality. Bone mass is dynamically regulated by three primary cell types: osteoclasts, which dissolve and degrade the mineralized bone matrix; osteoblasts, which synthesize and secrete proteins forming the bone matrix; and osteocytes, which act as mechanosensory cells orchestrating activities of osteoblasts and osteoclasts [5]. Bone quality is dynamically regulated by osteocytes. The term bone quality encompasses all the parameters other than bone mass that influence bone’s resistance to fractures, including trabecular microarchitecture, bone matrix composition and material properties, collagen quality, and mineralization [6].
Osteocytes, which represent the majority of adult skeletal cells, have the longest lifespan of any bone cells and are profoundly ingrained in bone [7]. Although derived from osteoblasts, osteocytes have a distinct stellate-shaped morphology similar to the neurons in the brain [8]. The osteocyte cell body resides within tiny cavities called lacunae in the calcified bone matrix, and their emanating dendritic projections extend through narrow tunnels called canaliculi [9]. Within the confines of the lacunocanalicular network (LCN), osteocytes occupy more than 215 m2 of the total bone surface area [10]. The extensive LCN enables an osteocyte to connect with neighboring osteocytes as well as the cells lining the surfaces of bone (i.e., osteoblasts and osteoclasts), the vasculature, the adjacent marrow space, and articular cartilage. The intricate LCN also gives osteocytes the ability to sense the changes in the mechanical environment that stimulate fluid flow through the network [11]. Apart from the LCN, osteocytes are also endowed with cytoskeletal components, focal adhesions, gap junctions, the primary cilium, ion channels, and the glycocalyx [12] that collectively provide osteocytes enhanced capabilities of perceiving physical cues and initiating biochemical signals to modulate osteoblast and osteoclast behavior. Apart from mechanosensation, the extensive osteocyte LCN surface area also serves as a site of calcium release from bone [13]. Although the essential role of transforming growth factor-beta (TGFβ) in the skeleton has been studied for decades [14,15,16,17,18,19,20], this review focuses on the more recently discovered actions of TGFβ in the skeletal and extraskeletal functions of osteocytes.
Basics of the Transforming Growth Factor Beta (TGFβ) Pathway
The TGFβ superfamily consists of an evolutionarily conserved family of proteins, including TGFβs, activins, bone morphogenetic proteins (BMPs), and other related proteins [18]. The members of the TGFβ family of proteins include three highly homologous isoforms—TGFβ1, β2, and β3 that demonstrate distinct spatial and temporal expression and exert mostly non-redundant functions. Together, the TGFβ family regulates a wide variety of biological processes, such as cellular migration, proliferation, commitment, and differentiation [21]. TGFβ ligand isoforms are secreted as polypeptide dimers formed from the tethering of conserved cysteine residues of the monomers into a single inter-chain disulfide bond [22]. The dimeric TGFβ ligand then associates with the pro-region-derived latency-associated protein (LAP), which in turn binds to latent TGFβ binding protein (LTBP) or glycoprotein-A repetitions predominant (GARP) proteins to form latent complexes [23, 24]. These latent complexes shield the active TGFβ and prevent it from binding to receptors. In bone, TGFβ1 is the most abundant isoform, and, once synthesized, it is bound to latent complexes and stored in the bone extracellular matrix (ECM) [25]. TGFβ is activated mechanically upon integrin binding to LAP [24] or by exposure to an acidic microenvironment, such as that created by osteoclasts during bone resorption [26]. Several other mechanisms of TGFβ activation exist in various tissues, and these distinct mechanisms add to the complexity and precision of regulating TGFβ signaling.
Once activated, the TGFβ ligands promote the assembly and activation of the heterotetrameric TGFβ receptor complexes consisting of two subunits of type I (TβRI or ALKs) and type II transmembrane receptors (TβRII), both of which possess serine/threonine kinase activity [14,15,16,17,18,19,20]. Activated TβRII-TβRI effectively phosphorylate Smad or non-Smad effectors to modulate target gene transcription [27]. The Smad-dependent canonical pathway of TGFβ involves the activation of three subgroups of Smad proteins by the TGFβ ligand-receptor complex: the receptor-activated Smads (R-Smads, Smad2/3 for TGFβ/activin receptors, and Smad1/5/8 for BMP receptors), the common mediator Smad (Co-Smad, Smad4) that translocates R-Smads into nuclei to control gene transcription, and the inhibitory Smads (I-Smads, Smad6, and Smad7) that dampen signal transduction by rerouting the R-Smad/Co-Smad trimeric complex towards proteasomal degradation via ubiquitin ligases such as Smurf2 [28, 29]. In the non-canonical, Smad-independent TGFβ signaling pathway, TβRI or TβRII phosphorylate non-Smad proteins, such as TGFβ activation kinase 1 (TAK1) and its binding protein (TAB1), protein kinase C (PKC), protein phosphatases 2 (PP2A), and phosphoinositide 3-kinase (PI3K) complexes that can, in turn, activate various signaling pathways to regulate cellular processes like migration, proliferation, differentiation, and apoptosis [13, 30]. Overall, the TGFβ pathway is precisely regulated at multiple levels, including ligand bioavailability and activation, permutation of receptor assembly, internalization, stabilization, selection of canonical vs. non-canonical effectors, and recruitment of I-Smads. Together, these mechanisms enable the TGFβ pathway to invariantly turn on and off gene expression programs in a cell-type specific and context-dependent manner to support diverse biological processes. Abnormalities in the regulation of the TGFβ pathways disrupt normal physiology and lead to the development of pathological diseases.
The Biological Role of the TGFβ Pathway in Bone
A large body of work has examined the role of members of the TGFβ family in skeletal development and maintenance [14,15,16,17,18,19,20, 30,31,32,33]. Briefly, TGFβ controls bone development through endochondral and intramembranous ossification [30]. TGFβ powerfully induces chondrogenic differentiation by stimulating the recruitment, proliferation, and condensation of mesenchymal cells, as well as the formation of a cartilage template, all of which are essential for the formation of mineralized bone by endochondral ossification. Accordingly, ablation in TGFβ signaling in early development leads to severe abnormalities in the axial and appendicular skeleton. Mice deficient in TGFβ2 and TGFβ3 have short ribs and craniofacial defects [34,35,36]. Similarly, TβRII deletion inhibited the proliferation and differentiation of osteo-chondrogenic progenitors leading to defects in the development of joints and long bone [37]. TGFβ signaling is equally important for the formation and maintenance of intramembranous bones of the craniofacial skeleton and tooth eruption. Deletion of TβRII in Gli1 + osteogenic progenitors and Col1 + (3.2 kb) early osteoblasts leads to reduced alveolar bone development [38]. Inactivation of TβRII also impacts tooth development by targeting dental mesenchymal proliferation and odontoblast maturation [39].
The importance of intact TGFβ signaling for postnatal growth of the appendicular skeleton is illustrated by studies reporting low trabecular and cortical bone mass in mice exhibiting TβRII deletion in Osx + mesenchymal stem cells [38]. Despite the marked increase in CAR + osteoprogenitors, the reduced number of osteoblasts accounts for the low bone mass phenotype in these mice [38]. A drastically opposite high bone mass phenotype is observed upon expression of a dominant negative TβRII or deletion of TβRII in Ocn + mature osteoblasts [15, 40•]. Interestingly, blockade of TGFβ signaling using a pharmacologic inhibitor of TβRI-kinase or antibodies against TGFβ ligand also leads to increased bone mass [41]. Although there is a consensus regarding TGFβ’s importance in postnatal skeletal development and maintenance, the dramatic differences in bone phenotypes in different mouse models suggest that the role of TGFβ signaling in bone is highly cell type-specific and context-dependent. This has also been observed from a pathological perspective, particularly conditions of osteogenesis imperfecta and Camurati–Engelmann disease, where increased TGFβ ligand bioavailability has severe skeletal consequences, in part due to its effects on bone remodeling [42].
TGFβ signaling integrates the activity of multiple cell types involved in bone remodeling to maintain bone mass. TGFβ regulates RANKL-induced osteoclast formation and bone-resorbing activity [43]. Both TGFβ1 and TGFβ2 ligands control osteoclasts in a dose-dependent manner. Low concentrations of TGFβ ligands stimulated osteoclast development, whereas high levels of TGFβ attenuated osteoclastogenesis [44]. Time-dependent regulation of osteoclastogenesis by TGFβ signaling has been recently reported; TGFβ signaling promoted RANKL-mediated osteoclastogenesis in the early stages of differentiation and inhibited osteoclasts in the late stages [45]. Remarkably, TGFβ induces osteoclast apoptosis at later stages by upregulating Bim [46]. To maintain the close coordination between bone resorption and production, TGFβ signaling in osteoclasts couples their actions with those of osteoblasts in the bone [26].
The Function of TGFβ Signaling in Osteocytes
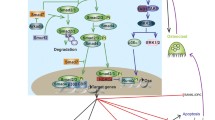
Over the last 5 years, new data has emerged that identifies the TGFβ pathway as a critical regulator of osteocyte functions [47•, 48•, 49, 50•, 51, 52•, 53]. Several breakthroughs have been made that greatly improved our knowledge of the mechanisms that upregulate or downregulate osteocytic TGFβ signaling through differential signaling pathways, as well as the pathophysiological response to multiple stimuli. These studies reinforce the notion of balanced TGFβ signaling and the context-dependent impact of TGFβ signaling in osteocytes. In this review, we summarize the several molecular mechanisms that crosstalk with TGFβ signaling in osteocytes and highlight the different skeletal and extraskeletal functions impaired in response to the ablation of TGFβ signaling within osteocytes (Table 1). In the following subsections, we review four main functions of osteocytes that TGFβ signaling contributes to, including- regulation of bone quality, mechanical loading, mineral metabolism, and energy metabolism as shown in Fig. 1.

The multifaceted role of osteocytic TGFβ signaling. Osteocytic TGFβ signaling regulates both the conventional skeletal and extraskeletal functions of bone. A In basal conditions, osteocytic TGFβ signaling supports the maintenance of bone quality; B in mechanical stress conditions, osteocytic TGFβ signaling integrates and converts mechanical cues into biological signals (such as RANKL and SOST) that modulate the number and activity of osteoclasts and osteoblasts; C in conditions of mineral metabolic stress, osteocytic TGFβ signaling controls calcium homeostasis by locally lysing and then remodeling perilacunar and canalicular bone matrix (e.g., during lactation); although untested, it is possible that osteocytic TGFβ signaling also functions in an endocrine manner by releasing factors like fibroblast growth factor 23 (FGF23), which will act on the kidney and parathyroid glands to modulate systemic calcium and phosphate homeostasis; D TGFβ signaling can also directly impact osteocyte energetics and affect the production of other cytokines like sclerostin to modulate whole-body energy metabolism through regulation of adipogenesis, muscle activity, and glucose metabolism. Figure created with BioRender.com
Regulation of Bone Quality
Bone material properties, one of several bone quality parameters, are regulated by osteocytes, at least in part through the process of perilacunar/canalicular remodeling (PLR) [47•, 48•, 54, 55]. PLR involves resorption of the mineral and proteolysis of the organic matrix lining the lacunae and canaliculi, mediated by vacuolar H + ATPases (Atp6V1G1, Atp6V0D2, and Atp6V0B), carbonic anhydrases (CA1 and CA2), tartrate-resistant acid phosphatase (TRAP), cathepsin K (CTSK), and matrix metalloproteinases (MMPs) and replenishment of the resorbed matrix by secretion of collagen I, dentin matrix protein 1 (DMP1), matrix extracellular phosphoglycoprotein (MEPE), and a phosphate-regulating gene with homologies to endopeptidases on the X chromosome essential for phosphate metabolism (PHEX) [13, 56,57,58,59,60]. Phenotypic characterization of PLR can be conducted through visualization of the enlargement of lacunar volume and canalicular diameters of osteocytes or by monitoring PLR enzyme levels, among other strategies [13, 19, 54, 61].
TGFβ is a key regulator of PLR [47•, 48•, 49]. In vitro, TGFβ promotes osteocytic expression of resorptive genes, and its blockade leads to reduced osteocyte-mediated acidification. In vivo, inhibition of TGFβ signaling using a pharmacologic inhibitor (TβRI) or in a mouse model with osteocyte-intrinsic ablation of TβRII (TβRIIocy−/−) led to suppressed PLR. Although PLR suppression did not impact bone mass, it led to a marked reduction in bone material properties and increased fragility of cortical bone of TβRIIocy−/− mice. Besides regulating PLR gene expression, TGFβ signaling also impacts the LCN. Whether through systemic pharmacologic inhibition of TGFβ signaling or in TβRIIocy−/− mice, osteocyte dendricity and canalicular length were reduced [47•, 49]. The underlying mechanism behind the disruption of osteocytic dendrites with ablation of the TGFβ pathway is unclear. Expression of at least two genes essential for dendrite formation in early osteocytes, such as podoplanin (E11/gp38) and MT1-MMP (MMP14), are controlled by TGFβ signaling and could likely be responsible for altered LCN. Whether the regulation of E11/gp38 and MMP14 by TGFβ signaling is direct requires further investigation. Several proteins that are transcriptionally regulated by TGFβ signaling, including sclerostin (SOST), parathyroid hormone receptor (PTH1R), yes-associated protein (YAP), and transcriptional coactivator with PDZ-binding motif (TAZ), have been identified as mediators of PLR and bone quality [13, 56, 62]. Additional research will be needed to determine if these factors act in an epistatic fashion to control bone quality.
Regulation of Osteocyte Mechanosensitivity
TGFβ signaling, which is integral for mechanical load-induced bone anabolism, can be suppressed in bone via hindlimb loading [63•]. In fact, loss of sensitivity to TGFβ signaling blunts the anabolic effects of mechanical loading on bone, as seen in mice expressing the dominant negative version of TβRII under the control of an osteocalcin promoter. Unlike loading, the role of TGFβ signaling in unloading models has been quite controversial and contradictory. Bone unloading, characterized by disuse-associated bone loss, has been studied using spinal cord injury or denervation models in rodents. In the murine spinal cord injury model, inhibition of TGFβ mitigated the disuse-associated bone loss [64]. While in the murine denervation model, exogenous administration of TGFβ partially relieved denervation-induced bone loss by supporting osteoblastic differentiation and activity and mitigating the effects of glucocorticoids on the bone [65, 66]. Which of these effects result from mechanoregulation of TGFβ, relative to innervation or other factors, remains unknown.
At the cellular level, TGFβ’s role in osteocyte mechanobiology has been attributed to several molecular cascades. Several studies show a close link between the regulation of TGFβ signaling and the expression of sclerostin in osteoblasts and osteocytes [50•, 63•, 67•]. TGFβ signaling transcriptionally regulates sclerostin expression in osteocytes and loss of sensitivity to TGFβ impairs load-induced suppression of sclerostin, thereby causing loss of bone anabolism. The deregulated sclerostin expression has also been detected in subchondral bone osteocytes of mice expressing ablated TGFβ receptor under the control of the Dmp1-Cre promoter [50•]. Apart from sclerostin, TGFβ can indirectly promote Wnt signaling in osteocytes through the suppression of microRNA-100, a negative regulator of the Wnt/βcatenin pathway [51]. Other than Wnt, TGFβ also induces expression of gap junction protein, connexin 43 (Cx43), and channel protein pannexin (Panx1) through non-canonical activation of mitogen-activated protein kinase (ERK1/2) [52•]. Both Cx43 and Panx1 are large-pore channel proteins located on the dendritic processes of osteocytes and mediate permeation of ions (i.e., Ca2 +) and key metabolites (ATP and prostaglandins), which are crucial for mechanotransduction in bone [68, 69]. Similarly, YAP and TAZ serve as key mechanosensors in various cell types. In bone, YAP/TAZ was recently found to support osteocyte dendricity; TGFβ signaling acts upstream of YAP/TAZ to modulate osteocyte behavior [62]. While TGFβ-YAP/TAZ dictates PLR, what remains unclear is how this molecular mechanism coordinates the mechanobiology of osteocytes. Studies using finite element modeling have postulated that load will induce changes in perilacunar and canalicular volume within osteocytes and that lacunar and canalicular structures will experience different strain distributions [49, 70]. Furthermore, changes in canalicular density are also predicted to alter the corresponding shear stress experienced by osteocytes [49]. Based on the predictions from the computational model, the mechanosensitivity of the osteocytes will be negatively affected as the LCN and canalicular density are known to be reduced in mice with osteocytic TGFβ-signaling ablation. Experimental studies in the TβRIIocy−/− mice will be useful in understanding the relevance of LCN in mediating cellular mechanosensitivity and load-induced bone anabolism.
Regulation of Mineral Metabolism
Osteocyte-mediated regulation of mineral metabolism is attributed to PLR [57], osteoclast governing functions of osteocytes [57, 71], and to the osteocytic secretion of FGF23, a phosphaturic hormone targeting endocrine regulation of phosphate metabolism [72]. In physiological conditions of mineral stress, such as lactation, where bouts of calcium are needed to support milk production, osteocytes rapidly resorb and release calcium and phosphate from the bone matrix lining the lacunae and canaliculi, leading to increased lacunar volume and canalicular diameter. With the cessation of mineral demand, the geometry of the osteocyte lacunar and canalicular structures fully recovers, along with an increase in bone mass [73]. While osteocytes support maternal calcium needs, deregulated PLR fails to detectably impact milk production. Maternal calcium demands are met through other compensatory mechanisms, such as increased intestinal calcium absorption [73, 74].
TGFβ signaling is intimately linked to the activation of osteocytic PLR during lactation [48•]. Under basal conditions, female TβRIIocy−/− mice do not exhibit any phenotypic differences relative to controls. However, with lactation, the typical trabecular and cortical bone loss phenotype observed in control mice is mitigated in the TβRIIocy−/−bones. The impaired lactation-induced bone loss in TβRIIocy−/− mice stems from reduced PLR and reduced type I receptor (PTH1R) expression in osteocytes. PTH1R, the common G-protein-coupled receptor for PTH and PTHrP ligand, is crucial in osteocytes for lactation-induced PLR, such that its ablation block PLR and induction of osteoclasts during lactation [13].
Although osteocyte-specific ablation of PTH1R in mice can lead to hypocalcemia [75], systemic calcium levels are maintained in mice with osteocytic TβRII deficiency [48•]. It is possible that with a deficiency in PLR, systemic mineral needs in the TβRII knockout mice are met in an endocrine manner via increased osteocytic secretion of FGF23 and vitamin D3 (1,25(OH)2D3).
Interestingly, in osteoblasts, TGFβ can stimulate FGF23 production and enhance cellular calcium levels [76]. Similarly, intense crosstalk between TGFβ and vitamin D3 signaling has been previously characterized [77], and newer studies indicate transcriptional induction of vitamin D receptors by TGFβ in non-bone cells [78, 79]. Although osteocytic TGFβ signaling is crucial for PLR-mediated regulation of systemic mineral metabolism, it is worthwhile to understand how osteocytic TGFβ signaling influences the endocrine arm of systemic mineral metabolism regulated by osteocytes when considering the crosstalk between TGFβ-FGF23-PTH-1,25(OH)2D3.
Pathological conditions associated with defective mineral metabolism, chronic kidney disease (CKD), and renal osteodystrophy have been widely studied recently. Elevated ligands, receptors, and downstream targets of TGFβ signaling have been observed in animal models of CKD and CKD patients [80]. Given the link of TGFβ with PTH and FGF23 signaling, both mechanisms could be implicated in disrupted mineral metabolism. In fact, the resistance of CKD bones to the calcemic action of PTH, reported in many studies [81, 82], could be attributed to the attenuation of PTH1R by TGFβ signaling [40•, 83]. Similarly, the increased serum FGF23 levels in CKD patients track with increased TGFβ ligands and further strengthen the notion that TGFβ signaling is upstream of FGF23 signaling and could be targeted for restoration of impaired bone turnover and mineral metabolism in CKD. Deducing the role of osteocyte-intrinsic TGFβ in the regulation of FGF23 and PTH-mediated calcium and phosphate metabolism in CKD will provide a unique opportunity of targeting a new cell type that as per new evidence, is implicated in the development of CKD and vascular calcification [84,85,86].
Regulation of Energy Metabolism
The contribution of osteocytes in the regulation of whole-body metabolism has been well-recognized. Since the function of a cell is tightly linked to its metabolism, much interest has risen in understanding the metabolic pathways and substrates in osteocytes [87].
The first evidence that TGFβ regulates glucose metabolism in the skeleton came from a study in the articular chondrocytes [88]. TGFβ stimulates glucose consumption and lactate production in human articular chondrocytes. Increased glucose consumption was attributed to increased cellular glucose transport in articular chondrocytes due to TGFβ-mediated upregulation of glucose transporter type 1 (Glut1). Apart from Glut1, TGFβ1 also induced the expression of hexokinases I and II and upregulated glycolysis-mediated lactate production in articular chondrocytes. Similarly, a recently presented abstract provided at the ASBMR 2022 annual conference also implicated the positive role of TGFβ signaling in promoting glycolysis in chondrocytes through Glut1/3 upregulation during embryonic joint development [89]. Interestingly, TGFβ mediated upregulation in glucose uptake and induction of Glut 1 and hexokinases (HKI and HKII) has also been reported in fibroblasts [90,91,92]. In murine and human lung fibroblasts, TGFβ-stimulated glycolysis is crucial for profibrotic gene expression, cell migration, colony formation, and activation of the transcription factors YAP/TAZ [91].
Although the mechanism underlying TGFβ-stimulated aerobic glycolysis is an active line of investigation, it will be interesting to if conservation in TGFβ’s action on cellular metabolism persists across different skeletal tissues. It is remarkable to consider that both chondrocytes and mature osteocytes sustain in a hypoxic environment [93,94,95]. Remarkably, hypoxia is a known inducer of TGFβ signaling, and this may be integral for metabolic reprogramming in hypoxic environments. Further studies need to be conducted to understand the relevance of aerobic glycolysis in osteocyte survival and function.
Whether an osteocyte’s metabolic program (normoxic vs. hypoxic environment) is affected by where they are located inside the cortical bone (closer to the bone surface vs. deeply embedded in the calcified bone matrix) needs to be investigated. Notably, elevated glucose levels stimulate TGFβ signaling in non-osteocyte cells [89]. A positive feedback loop may exist to support the upregulation of TGFβ signaling in conditions of high glucose to mediate glucose uptake and continue cell reliance on glycolysis. Whether such a feedback loop exists in osteocytes and the relevance of TGFβ mediated metabolic reprogramming for osteocyte function in physiological and pathological conditions requires further investigation. Unlike osteoblasts or osteoclasts, where the relationship of glucose utilization to cell differentiation and function is clear [96,97,98,99], the functional consequences of control of osteocytic glucose utilization remain unresolved. The sequestration of osteocytes within the mineralized bone matrix makes it challenging to characterize their metabolism in vivo. As a result, we rely primarily on ex vivo metabolic profiling techniques despite their inability to recapitulate the osteocytic environment. With these challenges, many gaps remain regarding the role of the osteocytic TGFβ pathway in regulating cellular fatty acids, glucose, and glutamine metabolism, and the impact of changes in the cellular metabolism of osteocytes on the regulation of whole-body energy metabolism.
The notion that bone is a driver of energy metabolism has been strengthened by the growing list of bone-derived factors coordinating systemic energy intake and expenditure. Recent reports in conference proceedings have opened the possibility that TGFβ signaling within bone cells could contribute to the maintenance of whole-body (organismal) energy metabolism [53, 100, 101]. Although this is still a subject of active investigation, it is not far-fetched to construe that the effects of bone intrinsic TGFβ signaling on energy metabolism could possibly be mediated through its molecular partners like sclerostin. Sclerostin, a direct target of the TGFβ pathway, has been shown to increase in the bones of mice that were fed a high-fat diet (a model of type 2 diabetes) [102]. In addition, sclerostin overexpression results in increased adiposity and impaired glucose homeostasis in mice [103, 104]. Moreover, both TGFβ and sclerostin are recognized as mediators of bone-muscle crosstalk [105,106,107]. In fact, the regulation of skeletal muscles by bone-derived TGFβ has been elegantly described in the context of cancer cachexia following tumor metastasis to bone. Cancer metastasis increases TGFβ release from the bone extracellular matrix and upregulates NADPH oxidase 4-mediated RyR1 oxidation in muscle cells. Oxidized RyR1 leads to calcium leak from the sarcoplasmic reticulum and impairs muscle contraction and muscle wasting. Inhibition of TGFβ signaling using TGFβ receptor I kinase inhibitor (SD-208) or TGFβ neutralizing antibody (1D11) improved muscle weight and function and increased body weight [105, 108]. Similarly, osteocytic Cx43, which is a target of TGFβ, has also been recognized as a regulator of bone-muscle crosstalk [109]. While these studies highlight the role of bone-derived TGFβ signaling in regulating muscle mass and function, the key point to take away is that some of the effects of TGFβ signaling on whole-body energy metabolism could be mediated directly or indirectly through the regulation of skeletal muscle and adipose tissue functions. Apart from cancer, increased TGFβ signaling has been reported in the pathophysiology of obesity and type 2 diabetes. Bridging the gap between how TGFβ signaling within bone cells drives whole-body energy metabolism could contribute to improving declining metabolic health associated with obesity and T2D.
Conclusion and Future Perspectives
All the preclinical studies highlighted in this review emphasize the multifaceted role played by osteocytic TGFβ signaling in bone, where its regulation is linked to the maintenance of bone mass and quality, bone anabolic response to loading, and facilitating systemic mineral metabolism (Fig. 1). We believe that the interactions of the TGFβ pathway with SOST, PTH1R, Cx43, and YAP/TAZ serve at the interface of many of these functions and epistatically fine-tune osteocyte responses. Our understanding regarding the choice or the sequence of the molecular partners of TGFβ signaling during a particular physiological or pathophysiological context (Table 1) is very limited.
The emerging line of research implicating TGFβ signaling in the coordination of cellular energy metabolism adds another level of complexity to the already confounding effects of TGFβ in bone. We believe that understanding how TGFβ signaling within osteocytes regulates metabolic flexibility will help us comprehend, to some extent, the context-dependent effects of the TGFβ pathway on bone during homeostasis vs. in a physically (exercise) or metabolically (lactation) challenging situation. It is worth noting that cellular energy metabolism also actively regulates TGFβ signaling. This has been evidently shown in the context of cancer, where intracellular metabolites and metabolic proteins affect the production or bioactivity of TGFβ ligands, influence the expression of TGFβ receptors, and regulate the activation and abundance of Smad proteins [110,111,112,113,114]. With the recent push towards mapping metabolomic signatures linked to poor skeletal health, delineating metabolites associated with deregulated osteocytic TGFβ signaling could offer newer options for improving skeletal health [115,116,117]. For example, in obesity where lipid metabolites are in abundance, understanding the impact of increased intracellular lipid metabolites on TGFβ signaling could help us target obesity-associated bone fragility.
Lastly, since TGFβ signaling is a growth factor with non-linear effects, a thorough mechanistic analysis of the new anticipated extraskeletal functions of osteocytic TGFβ is necessary. TGFβ ligand neutralizing antibodies and TGFβ receptor antagonists have proved to be beneficial for the restoration of skeletal health in diseases like osteogenesis imperfecta. However, in light of the new findings indicating that the effects of TGFβ can extend beyond bone, it will be crucial to carefully weigh the benefits and drawbacks of modulating this pathway on the overall physiology of the organism prior to developing novel strategies for refining TGFβ signaling.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Sommerfeldt D, Rubin C. Biology of bone and how it orchestrates the form and function of the skeleton. Eur Spine J. 2001;10:S86–95.
Su N, Yang J, Xie Y, Du X, Chen H, Hong Z, Chen L. Bone function, dysfunction and its role in diseases including critical illness. Int J Biol Sci. 2019;15:776–87.
Fulzele K, Krause DS, Panaroni C, Saini V, Barry KJ, Liu X, Lotinun S, Baron R, Bonewald L, Feng JQ, Chen M, Weinstein LS, Wu JY, Kronenberg HM, Scadden DT, Divieti PP. Myelopoiesis is regulated by osteocytes through Gsα-dependent signaling. Blood. 2013;121:930–9.
Oury F, Sumara G, Sumara O, Ferron M, Chang H, Smith CE, Hermo L, Suarez S, Roth BL, Ducy P, Karsenty G. Endocrine regulation of male fertility by the skeleton. Cell. 2011;144:796–809.
Florencio-Silva R, Sasso GRS, Sasso-Cerri E, Simões MJ, Cerri PS. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed Res Int 2015:1–17.
Compston J. Bone quality: what is it and how is it measured? Arq Bras Endocrinol Metabol. 2006;50:579–85.
Kennedy OD, Schaffler MB. The roles of osteocyte signaling in bone. J Am Acad Orthop Surg. 2012;20:670–1.
Sugawara Y, Kamioka H, Honjo T, Tezuka K, Takano-Yamamoto T. Three-dimensional reconstruction of chick calvarial osteocytes and their cell processes using confocal microscopy. Bone. 2005;36:877–83.
Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011;26:229–38.
Buenzli PR, Sims NA. Quantifying the osteocyte network in the human skeleton. Bone. 2015;75:144–50.
Weinbaum S, Cowin SC, Zeng Y. A model for the excitation of osteocytes by mechanical loading-induced bone fluid shear stresses. J Biomech. 1994;27:339–60.
Qin L, Liu W, Cao H, Xiao G. Molecular mechanosensors in osteocytes. Bone Res. 2020;8:23.
Qing H, Ardeshirpour L, Divieti Pajevic P, Dusevich V, Jähn K, Kato S, Wysolmerski J, Bonewald LF. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res. 2012;27:1018–29.
Bonewald LF, Mundy GR. Role of transforming growth factor-beta in bone remodeling. Clin Orthop Relat Res 1990:261–76.
Filvaroff E, Erlebacher A, Ye J-Q, Gitelman SE, Lotz J, Heillman M, Derynck R. Inhibition of TGF-β receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development. 1999;126:4267–79.
Janssens K, ten Dijke P, Janssens S, Van Hul W. Transforming growth factor-β1 to the bone. Endocr Rev. 2005;26:743–74.
Crane JL, Xian L, Cao X. Role of TGF-β signaling in coupling bone remodeling. In: Feng XH, Xu P, Lin X, editors. TGF-β Signaling. Methods in Molecular Biology. Humana Press 2016:1344:287–300.
MacFarlane EG, Haupt J, Dietz HC, Shore EM. TGF-β family signaling in connective tissue and skeletal diseases. Cold Spring Harb Perspect Biol. 2017;9:a022269.
Tang SY, Alliston T. Regulation of postnatal bone homeostasis by TGFβ. Bonekey Rep. 2013;2:255.
Erlebacher A, Filvaroff EH, Ye J-Q, Derynck R. Osteoblastic responses to TGF-β during bone remodeling. Mol Biol Cell. 1998;9:1903–18.
Chang H, Brown CW, Matzuk MM. Genetic analysis of the mammalian transforming growth factor-β Superfamily. Endocr Rev. 2002;23:787–823.
Hinck AP, Mueller TD, Springer TA. Structural biology and evolution of the TGF-β family. Cold Spring Harb Perspect Biol. 2016;8:a022103.
Wang R, Zhu J, Dong X, Shi M, Lu C, Springer TA. GARP regulates the bioavailability and activation of TGFβ. Mol Biol Cell. 2012;23:1129–39.
Hinz B. The extracellular matrix and transforming growth factor-β1: tale of a strained relationship. Matrix Biol. 2015;47:54–65.
Poniatowski ŁA, Wojdasiewicz P, Gasik R, Szukiewicz D. Transforming growth factor beta family: insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediators Inflamm. 2015;2015:1–17.
Weivoda MM, Ruan M, Pederson L, Hachfeld C, Davey RA, Zajac JD, Westendorf JJ, Khosla S, Oursler MJ. Osteoclast TGF-β receptor signaling induces wnt1 secretion and couples bone resorption to bone formation. J Bone Miner Res. 2016;31:76–85.
Hata A, Chen Y-G. TGF-β Signaling from receptors to smads. Cold Spring Harb Perspect Biol. 2016;8:a022061.
Miyazawa K, Miyazono K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb Perspect Biol. 2017;9:a022095.
Derynck R, Zhang YE. Smad-dependent and smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–84.
Wu M, Chen G, Li Y-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016;4:16009.
Dallas SL, Alliston T, Bonewald LF. Chapter 53: transforming growth factor-ß. In: Bilezikian J, Martin TJ, Clemens T, Rosen C, editors. Principles of Bone Biology, 4th ed. 2007:1145–1166.
Alliston T, Piek E, Derynck R. TGF-β family signaling in skeletal development, maintenance, and disease. In: Cold Spring Harbor Monograph Series. 2008:667–723.
Rys JP, Monteiro DA, Alliston T. Mechanobiology of TGFβ signaling in the skeleton. Matrix Biol. 2016;52–54:413–25.
Sanford LP, Ormsby I, Groot ACG, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development. 1997;124:2659–70.
Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MWJ, Doetschman T. Transforming growth factor–β3 is required for secondary palate fusion. Nat Genet. 1995;11:409–14.
Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF–β3 indicates defects of epithelial–mesenchymal interaction. Nat Genet. 1995;11:415–21.
Seo H-S, Serra R. Deletion of Tgfbr2 in Prx1-cre expressing mesenchyme results in defects in development of the long bones and joints. Dev Biol. 2007;310:304–16.
Abou-Ezzi G, Supakorndej T, Zhang J, Anthony B, Krambs J, Celik H, Karpova D, Craft CS, Link DC. TGF-β Signaling plays an essential role in the lineage specification of mesenchymal stem/progenitor cells in fetal bone marrow. Stem Cell Reports. 2019;13:48–60.
Oka S, Oka K, Xu X, Sasaki T, Bringas P, Chai Y. Cell autonomous requirement for TGF-β signaling during odontoblast differentiation and dentin matrix formation. Mech Dev. 2007;124:409–15.
• Qiu T, Wu X, Zhang F, Clemens TL, Wan M, Cao X. TGF-β type II receptor phosphorylates PTH receptor to integrate bone remodelling signalling. Nat Cell Biol. 2010;12:224–34. (This paper demonstrates that PTH induces an interaction between TβRII and PTH1R to regulate bone remodeling.)
Mohammad KS, Chen CG, Balooch G, Stebbins E, McKenna CR, Davis H, Niewolna M, Peng XH, Nguyen DH, Ionova-Martin SS, Bracey JW, Hogue WR, Wong DH, Ritchie RO, Suva LJ, Derynck R, Guise TA, Alliston T. Pharmacologic inhibition of the TGF-β type I receptor kinase has anabolic and anti-catabolic effects on bone. PLoS ONE. 2009;4:e5275.
Grafe I, Yang T, Alexander S, Homan EP, Lietman C, Jiang MM, Bertin T, Munivez E, Chen Y, Dawson B, Ishikawa Y, Weis MA, Sampath TK, Ambrose C, Eyre D, Bächinger HP, Lee B. Excessive transforming growth factor-β signaling is a common mechanism in osteogenesis imperfecta. Nat Med. 2014;20:670–5.
Itonaga I, Sabokbar A, Sun SG, Kudo O, Danks L, Ferguson D, Fujikawa Y, Athanasou NA. Transforming growth factor-β induces osteoclast formation in the absence of RANKL. Bone. 2004;34:57–64.
Karst M, Gorny G, Galvin RJS, Oursler MJ. Roles of stromal cell RANKL, OPG, and M-CSF expression in biphasic TGF-β regulation of osteoclast differentiation. J Cell Physiol. 2004;200:99–106.
Lee B, Oh Y, Jo S, Kim T-H, Ji JD. A dual role of TGF-β in human osteoclast differentiation mediated by Smad1 versus Smad3 signaling. Immunol Lett. 2019;206:33–40.
Houde N, Chamoux E, Bisson M, Roux S. Transforming growth factor-β1 (TGF-β1) induces human osteoclast apoptosis by up-regulating bim. J Biol Chem. 2009;284:23397–404.
• Dole NS, Mazur CM, Acevedo C, Lopez JP, Monteiro DA, Fowler TW, Gludovatz B, Walsh F, Regan JN, Messina S, Evans DS, Lang TF, Zhang B, Ritchie RO, Mohammad KS, Alliston T. Osteocyte-intrinsic TGF-β signaling regulates bone quality through perilacunar/canalicular remodeling. Cell Rep. 2017;21:2585–96. (This research article showcases TGFβ as a key regulator of perilacunar/canalicular remodeling.)
• Dole NS, Yee CS, Mazur CM, Acevedo C, Alliston T. TGFβ regulation of perilacunar/canalicular remodeling is sexually dimorphic. J Bone Miner Res. 2020;35:1549–61. (This paper highlights the link between TGFβ signaling and osteocytic perilacunar/canalicular remodeling during lactation.)
Schurman CA, Verbruggen SW, Alliston T. Disrupted osteocyte connectivity and pericellular fluid flow in bone with aging and defective TGF-β signaling. Proc Natl Acad Sci USA. 2021;118:e2023999118.
• Bailey KN, Nguyen J, Yee CS, Dole NS, Dang A, Alliston T. Mechanosensitive control of articular cartilage and subchondral bone homeostasis in mice requires osteocytic transforming growth factor β signaling. Arthritis Rheumatol. 2021;73:414–25. (This research article demonstrates that osteocytic TGFβ signaling is required for subchondral bone mechanosensation and its subsequent regulation of sclerostin.)
Dole NS, Yoon J, Monteiro DA, Yang J, Mazur CM, Kaya S, Belair CD, Alliston T. Mechanosensitive miR-100 coordinates TGFβ and Wnt signaling in osteocytes during fluid shear stress. FASEB J. 2021;35:e21883.
• Liu W, Zhang D, Li X, Zheng L, Cui C, Cui Y, Sun J, Xie J, Zhou X. TGF-β1 facilitates cell–cell communication in osteocytes via connexin43- and pannexin1-dependent gap junctions. Cell Death Discov. 2019;5:141. (This research article chronicles the regulation of Cx43 and Panx1 by osteocytic TGFβ.)
Dole NS, Yee C, Alliston T. Osteocyte-specific TGFβ signaling mitigates obesity-induced deregulated energy metabolism and compromised bone quality. J Endocr Soc. 2021;5:A441–2.
Fowler TW, Acevedo C, Mazur CM, Hall-Glenn F, Fields AJ, Bale HA, Ritchie RO, Lotz JC, Vail TP, Alliston T. Glucocorticoid suppression of osteocyte perilacunar remodeling is associated with subchondral bone degeneration in osteonecrosis. Sci Rep. 2017;7:44618.
Kerschnitzki M, Kollmannsberger P, Burghammer M, Duda GN, Weinkamer R, Wagermaier W, Fratzl P. Architecture of the osteocyte network correlates with bone material quality. J Bone Miner Res. 2013;28:1837–45.
Arnett TR. Osteocytes: regulating the mineral reserves? J Bone Miner Res. 2013;28:2433–5.
Qing H, Bonewald LF. Osteocyte remodeling of the perilacunar and pericanalicular matrix. Int J Oral Sci. 2009;1:59–65.
Inoue K, Mikuni-Takagaki Y, Oikawa K, Itoh T, Inada M, Noguchi T, Park JS, Onodera T, Krane SM, Noda M, Itohara S. A crucial role for matrix metalloproteinase 2 in osteocytic canalicular formation and bone metabolism. J Biol Chem. 2006;281:33814–24.
Holmbeck K, Bianco P, Pidoux I, Inoue S, Billinghurst RC, Wu W, Chrysovergis K, Yamada S, Birkedal-Hansen H, Poole AR. The metalloproteinase MT1-MMP is required for normal development and maintenance of osteocyte processes in bone. J Cell Sci. 2005;118:147–56.
Kulkarni RN, Bakker AD, Gruber EV, Chae TD, Veldkamp JBB, Klein-Nulend J, Everts V. MT1-MMP modulates the mechanosensitivity of osteocytes. Biochem Biophys Res Commun. 2012;417:824–9.
Kaya S, Basta-Pljakic J, Seref-Ferlengez Z, Majeska RJ, Cardoso L, Bromage TG, Zhang Q, Flach CR, Mendelsohn R, Yakar S, Fritton SP, Schaffler MB. Lactation-induced changes in the volume of osteocyte lacunar-canalicular space alter mechanical properties in cortical bone tissue. J Bone Miner Res. 2017;32:688–97.
Kegelman CD, Coulombe JC, Jordan KM, Horan DJ, Qin L, Robling AG, Ferguson VL, Bellido TM, Boerckel JD. YAP and TAZ mediate osteocyte perilacunar/canalicular remodeling. J Bone Miner Res. 2020;35:196–210.
• Nguyen J, Tang SY, Nguyen D, Alliston T. Load regulates bone formation and sclerostin expression through a TGFβ-dependent mechanism. PLoS ONE. 2013;8: e53813. (This research article investigates the role of TGFβ in regulating sclerostin and load-induced bone formation.)
Sahbani K, Cardozo CP, Bauman WA, Tawfeek HA. Inhibition of TGF-β signaling attenuates disuse-induced trabecular bone loss after spinal cord injury in male mice. Endocrinology 2022:163.
Yu Z, Li Y, Wang Y, Chen Y, Wu M, Wang Z, Song M, Lu F, Lu X, Dong Z. TGF-β prevents the denervation-induced reduction of bone formation and promotes the bone regeneration through inhibiting ubiquitin-proteasome pathway. Biosci Rep 2019:39.
Li Y, Jie L, Tian AY, Zhong S, Tian MY, Zhong Y, Wang Y, Li H, Li J, Sun X, Du H. Transforming growth factor beta is regulated by a glucocorticoid-dependent mechanism in denervation mouse bone. Sci Rep. 2017;7:9925.
• Loots GG, Keller H, Leupin O, Murugesh D, Collette NM, Genetos DC. TGF-β regulates sclerostin expression via the ECR5 enhancer. Bone. 2012;50:663–9. (This paper establishes a key target of TGFβ used to enhance sclerostin.)
Aguilar-Perez A, Pacheco-Costa R, Atkinson EG, Deosthale P, Davis HM, Essex AL, Dilley JE, Gomez L, Rupert JE, Zimmers TA, Thompson RJ, Allen MR, Plotkin LI. Age- and sex-dependent role of osteocytic pannexin1 on bone and muscle mass and strength. Sci Rep. 2019;9:13903.
Plotkin LI, Speacht TL, Donahue HJ. Cx43 and mechanotransduction in bone. Curr Osteoporos Rep. 2015;13:67–72.
Rath Bonivtch A, Bonewald LF, Nicolella DP. Tissue strain amplification at the osteocyte lacuna: a microstructural finite element analysis. J Biomech. 2007;40:2199–206.
Miller SC, Bowman BM. Rapid inactivation and apoptosis of osteoclasts in the maternal skeleton during the bone remodeling reversal at the end of lactation. Anat Rec. 2007;290:65–73.
Agoro R, Ni P, Noonan ML, White KE. Osteocytic FGF23 and its kidney function. Front Endocrinol 2020:11.
Kovacs CS. Maternal mineral and bone metabolism during pregnancy, lactation, and post-weaning recovery. Physiol Rev. 2016;96:449–547.
Ardeshirpour L, Dumitru C, Dann P, Sterpka J, VanHouten J, Kim W, Kostenuik P, Wysolmerski J. OPG treatment prevents bone loss during lactation but does not affect milk production or maternal calcium metabolism. Endocrinology. 2015;156:2762–73.
Powell WF, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst FR, Divieti PP. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol. 2011;209:21–32.
Feger M, Hase P, Zhang B, Hirche F, Glosse P, Lang F, Föller M. The production of fibroblast growth factor 23 is controlled by TGF-β2. Sci Rep. 2017;7:4982.
Subramaniam N, Leong GM, Cock T-A, Flanagan JL, Fong C, Eisman JA, Kouzmenko AP. Cross-talk between 1,25-dihydroxyvitamin D3 and transforming growth factor-β signaling requires binding of VDR and Smad3 proteins to their cognate DNA recognition elements. J Biol Chem. 2001;276:15741–6.
Fiz C, Apprato G, Ricca C, Aillon A, Bergandi L, Silvagno F. TGF beta induces vitamin D receptor and modulates mitochondrial activity of human pancreatic cancer cells. Cancers. 2021;13:2932.
Wang F, Chang H-M, Yi Y, Lin Y-M, Li H, Leung PCK. TGF-β1 promotes vitamin D-induced prostaglandin E2 synthesis by upregulating vitamin D receptor expression in human granulosa-lutein cells. Am J Physiol Endocrinol. 2020;318:E710–22.
Iwasaki Y, Yamato H, Fukagawa M. TGF-beta signaling in bone with chronic kidney disease. Int J Mol Sci. 2018;19:2352.
Iwasaki-Ishizuka Y, Yamato H, Nii-Kono T, Kurokawa K, Fukagawa M. Downregulation of parathyroid hormone receptor gene expression and osteoblastic dysfunction associated with skeletal resistance to parathyroid hormone in a rat model of renal failure with low turnover bone. Nephrol Dial Transplant. 2005;20:1904–11.
Evenepoel P, Bover J, Ureña TP. Parathyroid hormone metabolism and signaling in health and chronic kidney disease. Kidney Int. 2016;90:1184–90.
Jongen JW, Willemstein-Van Hove EC, Van der Meer JM, Bos MP, Jüppner H, Segre GV, Abou-Samra AB, Feyen JH, Herrmann-Erlee MP. Down-regulation of the receptor for parathyroid hormone (PTH) and PTH-related peptide by transforming growth factor-beta in primary fetal rat osteoblasts. Endocrinology. 1995;136:3260–6.
Wesseling-Perry K. Osteocyte dysfunction and renal osteodystrophy: not just calcium and phosphorus anymore. Kidney Int. 2017;91:1276–8.
Dussold C, Gerber C, White S, Wang X, Qi L, Francis C, Capella M, Courbon G, Wang J, Li C, Feng JQ, Isakova T, Wolf M, David V, Martin A. DMP1 prevents osteocyte alterations, FGF23 elevation and left ventricular hypertrophy in mice with chronic kidney disease. Bone Res. 2019;7:12.
Agoro R, Nookaew I, Noonan ML, Marambio YG, Liu S, Chang W, Gao H, Hibbard LM, Metzger CE, Horan D, Thompson WR, Xuei X, Liu Y, Zhang C, Robling AG, Bonewald LF, Wan J, White KE. Single cell cortical bone transcriptomics define novel osteolineage gene sets altered in chronic kidney disease. Front Endocrinol 2023:14.
Karthik V, Guntur AR. Energy metabolism of osteocytes. Curr Osteoporos Rep. 2021;19:444–51.
Wang C, Silverman RM, Shen J, O’Keefe RJ. Distinct metabolic programs induced by TGF-β1 and BMP2 in human articular chondrocytes with osteoarthritis. J Orthop Translat. 2018;12:66–73.
Song C, Long F. 2022. Glucose metabolism is essential for TGFβ mediated joint development. J Bone Miner Res 38 (Suppl 1). Available at https://www.asbmr.org/education/AbstractDetail?aid=f0c62847-9804-4337-86d2-2ddeb8a761e6. Accessed September 12, 2022.
Kitagawa T, Masumi A, Akamatsu Y. Transforming growth factor-beta 1 stimulates glucose uptake and the expression of glucose transporter mRNA in quiescent Swiss mouse 3T3 cells. J Biol Chem. 1991;266:18066–71.
Yin X, Choudhury M, Kang J-H, Schaefbauer KJ, Jung M-Y, Andrianifahanana M, Hernandez DM, Leof EB. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci Signal. 2019;12:eaax4067.
Inoki K, Haneda M, Maeda S, Koya D, Kikkawa R. TGF-β1 stimulates glucose uptake by enhancing GLUT1 expression in mesangial cells. Kidney Int. 1999;55:1704–12.
Jin J, Bakker AD, Wu G, Klein-Nulend J, Jaspers RT. Physicochemical niche conditions and mechanosensing by osteocytes and myocytes. Curr Osteoporos Rep. 2019;17:235–49.
Yellowley CE, Genetos DC. Hypoxia signaling in the skeleton: implications for bone health. Curr Osteoporos Rep. 2019;17:26–35.
Chen K, Zhao J, Qiu M, Zhang L, Yang K, Chang L, Jia P, Qi J, Deng L, Li C. Osteocytic HIF-1α pathway manipulates bone micro-structure and remodeling via regulating osteocyte terminal differentiation. Front Cell Dev Biol 2022:9.
Guntur AR, Gerencser AA, Le PT, DeMambro VE, Bornstein SA, Mookerjee SA, Maridas DE, Clemmons DE, Brand MD, Rosen CJ. Osteoblast-like MC3T3-E1 cells prefer glycolysis for ATP production but adipocyte-like 3T3-L1 cells prefer oxidative phosphorylation. J Bone Miner Res. 2018;33:1052–65.
Karner CM, Long F. Wnt signaling and cellular metabolism in osteoblasts. Cell Mol Life Sci. 2017;74:1649–57.
Karner CM, Long F. Glucose metabolism in bone. Bone. 2018;115:2–7.
Li B, Lee W, Song C, Ye L, Abel ED, Long F. Both aerobic glycolysis and mitochondrial respiration are required for osteoclast differentiation. FASEB J. 2020;34:11058–67.
Dole NS, Kaya S, Yee C, Yoon C, Salinas J, Miclau E, Alliston T. 2021. Ablating osteocytic TGFβ signaling mitigates the effects of obesity on systemic energy balance and bone quality. J Bone Miner Res 37 (Suppl 1). Available at https://www.asbmr.org/meetings/annualmeeting/AbstractDetail?aid=3b72065a-4732-4b1e-aac4-4f5217504e2f. Accessed October 4, 2021.
Trivedi T, Bahrami A, John S, Suresh S, Murthy S, Pagnotti G, She Y, Cao X, Mohammad K, Guise T. 2021.Excess TGFβ together with high fat diet exacerbates bone destruction and altered fat metabolism in mice with high bone turnover. J Bone Miner Res 37 (Suppl 1). Available at https://www.asbmr.org/meetings/annualmeeting/AbstractDetail?aid=02afb89c-1530-49fb-b186-681eb64fdacd. Accessed October 3, 2021.
Baek K, Hwang HR, Park H-J, Kwon A, Qadir AS, Ko S-H, Woo KM, Ryoo H-M, Kim G-S, Baek J-H. TNF-α upregulates sclerostin expression in obese mice fed a high-fat diet. J Cell Physiol. 2014;229:640–50.
Kim SP, Frey JL, Li Z, Kushwaha P, Zoch ML, Tomlinson RE, Da H, Aja S, Noh HL, Kim JK, Hussain MA, Thorek DLJ, Wolfgang MJ, Riddle RC. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc Natl Acad Sci USA. 2017;114:E11238–47.
Riddle RC. Endocrine functions of sclerostin. Curr Opin Endocr Metab Res. 2023;28:100433.
Regan JN, Trivedi T, Guise TA, Waning DL. The role of TGFβ in bone-muscle crosstalk. Curr Osteoporos Rep. 2017;15:18–23.
Zhou B, Zhang Q, Lin X, Hu J, Zhao D, Jiang Y, Xing X, Li M. The roles of sclerostin and irisin on bone and muscle of orchiectomized rats. BMC Musculoskelet Disord. 2022;23:1049.
Magarò MS, Bertacchini J, Florio F, Zavatti M, Potì F, Cavani F, Amore E, De Santis I, Bevilacqua A, Reggiani Bonetti L, Torricelli P, Maurel DB, Biressi S, Palumbo C. Identification of sclerostin as a putative new myokine involved in the muscle-to-bone crosstalk. Biomedicines. 2021;9:71.
Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, Chiechi A, Wright LE, Umanskaya A, Niewolna M, Trivedi T, Charkhzarrin S, Khatiwada P, Wronska A, Haynes A, Benassi MS, Witzmann FA, Zhen G, Wang X, Cao X, Roodman GD, Marks AR, Guise TA. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat Med. 2015;21:1262–71.
Li G, Zhang L, Lu Z, Yang B, Yang H, Shang P, Jiang JX, Wang D, Xu H. Connexin 43 channels in osteocytes are necessary for bone mass and skeletal muscle function in aged male mice. Int J Mol Sci. 2022;23:13506.
Gencer S, Oleinik N, Kim J, Panneer Selvam S, De Palma R, Dany M, Nganga R, Thomas RJ, Senkal CE, Howe PH, Ogretmen B. TGF-β receptor I/II trafficking and signaling at primary cilia are inhibited by ceramide to attenuate cell migration and tumor metastasis. Sci Signal. 2017;24:10.
Liu S, Hou H, Zhang P, Wu Y, He X, Li H, Yan N. Sphingomyelin synthase 1 regulates the epithelial-to-mesenchymal transition mediated by the TGF-β/Smad pathway in MDA-MB-231 cells. Mol Med Rep. 2019;19:1159–67.
Liu H, Chen YG. The interplay between TGF-β signaling and cell metabolism. Front Cell Dev Biol. 2022;9(10):846723.
Chen M, Zhao Y, Yang X, Zhao Y, Liu Q, Liu Y, Hou Y, Sun H, Jin W. NSDHL promotes triple-negative breast cancer metastasis through the TGFβ signaling pathway and cholesterol biosynthesis. Breast Cancer Res Treat. 2021;187:349–62.
Vasiukov G, Novitskaya T, Zijlstra A, Owens P, Ye F, Zhao Z, Moses HL, Blackwell T, Feoktistov I, Novitskiy SV. Myeloid cell-derived TGFβ signaling regulates ECM deposition in mammary carcinoma via adenosine-dependent mechanisms. Cancer Res. 2020;80:2628–38.
Fan J, Jahed V, Klavins K. Metabolomics in Bone Research. Metabolites. 2021;11:434.
Miyamoto T, Hirayama A, Sato Y, Koboyashi T, Katsuyama E, Kanagawa H, Fujie A, Morita M, Watanabe R, Tando T, Miyamoto K, Tsuji T, Funayama A, Soga T, Tomita M, Nakamura M, Matsumoto M. Metabolomics-based profiles predictive of low bone mass in menopausal women. Bone Rep. 2018;9:11–8.
Galvez-Fernandez M, Rodriguez-Hernandez Z, Grau-Perez M, Chaves FJ, Garcia-Garcia AB, Amigo N, Monleon D, Garcia-Barrera T, Gomez-Ariza JL, Briongos-Figuero LS, Perez-Castrillon JL, Redon J, Tellez-Plaza M, Martin-Escudero JC. Metabolomic patterns, redox-related genes and metals, and bone fragility endpoints in the Hortega Study. Free Radic Biol Med. 2023;194:52–61.
Collette NM, Genetos DC, Economides AN, Xie L, Shahnazari M, Yao W, Lane NE, Harland RM, Loots GG. Targeted deletion of Sost distal enhancer increases bone formation and bone mass. Proc Natl Acad Sci USA. 2012;109:14092–7.
Holguin N, Brodt MD, Silva MJ. Activation of Wnt signaling by mechanical loading is impaired in the bone of old mice. J Bone Miner Res. 2016;31:2215–26.
Aryana IGPS, Rini SS, Soejono CH. Importance of sclerostin as bone-muscle mediator crosstalk. Ann Geriatr Med Res. 2022;26:72–82.
Huang J, Romero-Suarez S, Lara N, Mo C, Kaja S, Brotto L, Dallas SL, Johnson ML, Jähn K, Bonewald LF, Brotto M. Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/β-catenin pathway. JBMR Plus. 2017;1:86–100.
Kim JA, Roh E, Hong SH, Lee YB, Kim NH, Yoo HJ, Seo JA, Kim NH, Kim SG, Baik S-H, Choi KM. Association of serum sclerostin levels with low skeletal muscle mass: the Korean sarcopenic obesity study (KSOS). Bone. 2019;128:115053.
O’Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, Robling AG, Bouxsein M, Schipani E, Turner CH, Jilka RL, Weinstein RS, Manolagas SC, Bellido T. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE. 2008;3:e2942.
Qiu T, Crane JL, Xie L, Xian L, Xie H, Cao X. IGF-I induced phosphorylation of PTH receptor enhances osteoblast to osteocyte transition. Bone Res. 2018;6:5.
Delgado-Calle J, Tu X, Pacheco-Costa R, McAndrews K, Edwards R, Pellegrini GG, Kuhlenschmidt K, Olivos N, Robling A, Peacock M, Plotkin LI, Bellido T. Control of bone anabolism in response to mechanical loading and pth by distinct mechanisms downstream of the PTH receptor. J Bone Miner Res. 2017;32:522–35.
Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, Olivos N, Passeri G, O’Brien CA, Bivi N, Plotkin LI, Bellido T. PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J Bone Miner Res. 2011;26:1035–46.
Uda Y, Saini V, Petty CA, Alshehri M, Shi C, Spatz JM, Santos R, Newell CM, Huang TY, Kochen A, Kim JW, Constantinou CK, Saito H, Held KD, Hesse E, Pajevic PD. Parathyroid hormone signaling in mature osteoblasts/osteocytes protects mice from age-related bone loss. Aging. 2021;13:25607–42.
Fan Y, Bi R, Densmore MJ, Sato T, Kobayashi T, Yuan Q, Zhou X, Erben RG, Lanske B. Parathyroid hormone 1 receptor is essential to induce FGF23 production and maintain systemic mineral ion homeostasis. FASEB J. 2016;30:428–40.
Zarka M, Etienne F, Bourmaud M, Szondi D, Schwartz J-M, Kampmann K, Helary C, Rannou F, Haÿ E, Cohen-Solal M. Mechanical loading activates the YAP/TAZ pathway and chemokine expression in the MLO-Y4 osteocyte-like cell line. Lab Invest. 2021;101:1597–604.
Funding
This work was supported by the Arkansas Biosciences Institute (AWD55316, NSD), NIH-NIDDK K01 (DK129404, NSD), NIH/NIGMS (1P20GM125503, NSD), the UAMS Musculoskeletal Creative Hub Pilot Award (NSD), NIH-NIDCR R01 (DE019284, TA), and NIH-NIAMS P30 (AR075055, TA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no conflicts of interest to disclose.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki Declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Carroll, M., Alliston, T. & Dole, N. The Multifaceted Effects of Osteocytic TGFβ Signaling on the Skeletal and Extraskeletal Functions of Bone. Curr Osteoporos Rep 21, 414–425 (2023). https://doi.org/10.1007/s11914-023-00802-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-023-00802-w