
Abstract
Purpose of Review
The sensory neuronopathies are sensory-predominant polyneuropathies that result from damage to the dorsal root and trigeminal sensory ganglia. This review explores the various causes of acquired sensory neuronopathies, the approach to diagnosis, and treatment.
Recent Findings
Diagnostic criteria have recently been published and validated to allow differentiation of sensory neuronopathies from other polyneuropathies. On the basis of serial electrodiagnostic studies, the treatment window for the acquired sensory neuronopathies has been identified as approximately 8 months. If treatment is initiated within 2 months of symptom onset, there is a better opportunity for improvement of the patient's condition.
Summary
Even though sensory neuronopathies are rare, significant progress has been made regarding characterization of their clinical, electrophysiologic, and imaging features. This does not hold true, however, for treatment. There have been no randomized controlled clinical trials to guide management of these diseases, and a standard treatment approach remains undetermined.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sensory neuronopathies (SN) are a subset of peripheral neuropathies that result from destruction of the dorsal root ganglion (DRG) and trigeminal ganglion sensory neurons. They go by many names in the literature, including dorsal root ganglionopathies and even ataxic neuropathies. These disorders can be broadly classified into acquired, or inherited and degenerative SN. An in-depth discussion of the inherited and degenerative disorders, which include Friedreich ataxia, POLG mutations, cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS), hereditary sensory and autonomic neuropathy type 1 (due to SPTLC1 mutations), and facial-onset sensory motor neuronopathy, is beyond the scope of this review [1,2,3,4,5]. The clinical presentation of these heritable and degenerative SN is distinct from that of acquired SN given their additional neurologic features. Regardless of the cause—acquired, inherited or degenerative—as a result of the rarity of these diseases, there is relatively little published information. There have been no blinded, placebo-controlled, clinical trials investigating the treatment of these conditions. Most publications are isolated case reports, retrospective case series, or reviews. We will expand on the various causes of acquired SN and their treatment strategies, with an emphasis on recent developments, followed by a recommended approach to diagnosis.
Anatomy
The DRG houses the cell bodies of the sensory pseudounipolar neurons. There are two subsets of neurons that make up the substance of the DRG. The larger cells give rise to Aβ and Aδ fibers. These carry proprioceptive and tactile sensation through the dorsal columns of the spinal cord. The smaller, unmyelinated C fibers carry thermal and nociceptive sensation through the spinothalamic tracts. The neurons of the DRG are susceptible to antibodies and toxins as a result of the loose blood–nerve barrier caused by fenestrated capillaries. The histopathologic findings in the DRG of inflammatory SN include reduced neuron density, satellite cell proliferation forming Nageotte nodules, which develop following degeneration of the ganglion cell bodies, and inflammatory infiltration (Fig. 1) [6].

Microphotography of the dorsal root ganglia. a Normal neurons. Hematoxylin and eosin (HE), ×200 magnification. b With evidence of neuronal loss and Nageotte nodules (arrows). HE, ×200 magnification. c Nageotte nodule detail (arrows). HE, ×600 magnification. d Mononuclear inflammatory infiltrate (arrows). HE, ×400 magnification. (Reprinted with permission from Colli et al. [6])
Causes
Paraneoplastic Sensory Neuronopathies
Paraneoplastic syndromes associated with anti-Hu antibodies, and less often anti-collapsin-response mediator protein 5 (anti-CRMP-5)/CV2 antibodies, are one of the commonest causes of SN [7, 8]. The typical presentation is subacute sensory ataxia; however, patients can also present with concomitant motor neuropathy, limbic encephalitis, Lambert–Eaton myasthenia gravis, and cerebellar or brainstem involvement [7, 9, 10]. Autonomic features, including bilateral tonic pupils from effects on the ciliary ganglion, gastroparesis, and pseudo-obstruction, have also been reported [11,12,13,14]. In one case series, pain was a predominant feature in 80% of the patients in whom SN had been diagnosed [15]. Anti-Yo antibodies have been reported in a monomelic SN in a patient with invasive ductal carcinoma of the breast [16]. Amphiphysin antibodies have also been described associated with SN [17]. The association between anti-Hu paraneoplastic syndromes and small-cell lung cancer is widely known [18]. There are, however, a host of other malignancies with which anti-Hu antibodies have been linked, including uterine and other gynecologic tumors, testicular tumors, bladder tumors, prostate cancer, and even a rare case of hepatocellular carcinoma [19,20,21,22,23,24].
Pathologically, anti-Hu antibodies are generated by the immune system against Hu-expressing tumor cells but also destroy Hu-expressing neurons. They can penetrate at the level of the DRG, where the blood–nerve barrier is fenestrated and permeable. However, the role of anti-Hu antibodies in direct neuronal damage has yet to be elucidated. They are not thought to be directly responsible for the immune attack; rather it appears that this is primarily a CD8 cytotoxic T cell response [25, 26]. Anti-Hu antibodies react with HuD (ELAV-like neuron-specific RNA-binding protein 4) as well as HuC and HuB, which are neuron-specific proteins that bind messenger RNA. HuD is expressed by small-cell lung cancer cells, and likely initiates the autoimmune response in these patients [27, 28]. Tumors with anti-Hu antibody production express MHC type 1 molecules, further supporting the hypothesis that the anti-Hu response is T cell mediated [29, 30].
Treatment of paraneoplastic SN is challenging. The treatment strategy is based predominantly on expert opinion as there have been no randomized controlled clinical trials given the exceptionally rare nature of the disease. The mainstay of therapy lies in identifying and treating the underlying malignancy. In a retrospective study, tumor treatment was the only intervention that resulted in stabilization of the paraneoplastic syndrome in patients with anti-Hu antibodies [18]. The second principle of management is immunomodulatory treatment. We often initiate corticosteroid therapy (intravenously administered methylprednisolone) or intravenous immune globulin (IVIG) therapy while the malignancy is being evaluated. A limited window of opportunity for disease stabilization exists, and following the inflammatory reaction, there is irreversible neuronal damage, at which point therapeutic intervention is likely futile [31••, 32]. A number of immunosuppressant and immunomodulatory therapies have been used in uncontrolled trials, including corticosteroid therapy, IVIG therapy, plasma exchange, and rituximab therapy [33,34,35,36,37]. Cyclophosphamide may be beneficial in refractory disease when a tumor is not identified [32]. Most recently, de Jongste et al. [38] published a prospective, open-label study using sirolimus to treat anti-Hu paraneoplastic syndromes. Sirolimus is an immunosuppressive drug that inhibits activated T cells (cytokine-mediated T-cell proliferation). In this study of 17 patients with progressive anti-Hu antibody mediated paraneoplastic neurologic syndromes, nine had SN. Of the 17, ten had stabilization of their disease, six had progression of their disease, and one patient had improvement in function, as measured by the modified Rankin scale. Efficacy was not compared among the various neurologic syndromes [38]. These results support the use of immunomodulating therapies, but further study is needed to determine the use of one therapy versus another. The third principle of management is that of symptomatic treatment. Gabapentin, pregabalin, duloxetine, venlafaxine, and amitriptyline are all effective in treating neuropathic pain.
Autoimmune Causes
In addition to the paraneoplastic SN, the other large category of acquired SN is that of systemic autoimmune disease. The most commonly reported is Sjögren’s syndrome, and its association with SN has been identified since the 1980s [39]. Although less common than other neurologic complications of Sjögren’s syndrome, SN is more specific to Sjögren’s syndrome, and tends to be more disabling [40,41,42,43,44]. Patients usually have a sensory ataxia, areflexia, and autonomic dysfunction [45, 46]. Brainstem involvement, likely with disease affecting the ciliary ganglion, has also been reported [47]. Not much is known about the pathologic basis of SN in Sjögren’s syndrome, although there is loss of neuronal cell bodies, and T-cell-mediated infiltration in the DRG has been demonstrated [48]. In one autopsy of an 88-year-old woman with Sjögren’s syndrome associated SN, there were diminished numbers of DRG neurons in the cervical, thoracic, and lumbar segments, and diminished numbers of sympathetic ganglion cells across all segments [46]. CD8 T cell infiltrates were visualized.
Treatment strategies for Sjögren’s syndrome associated SN are based on case series and case reports, as there have been no randomized clinical trials. Successful response has been reported with the use of corticosteroids in association with mycophenolate mofetil [41•]. Other treatment strategies include plasma exchange, rituximab therapy, cyclophosphamide therapy, and azathioprine therapy [49,50,51,52]. IVIG therapy has had variable reports of success, with some studies suggesting that SN is less responsive to IVIG than are small-fiber sensory or sensorimotor neuropathies [53]. Others found that the long-term use of IVIG in ataxic SN with Sjögren’s syndrome led to abatement of neurologic symptoms [54]. One case report by Caroyer et al. [55] demonstrated clinical and electrophysiologic improvement in the condition of a patient with SN due to Sjögren’s syndrome after 12 weeks of therapy with infliximab, and the improvements remained after 48 months. A recent study by Goodman [56] suggests that autonomic symptoms related to Sjögren’s syndrome may respond to repeated, ongoing, or adjunctive immunomodulation.
SN have also been identified in celiac disease, with or even without enteropathy, although this is somewhat controversial [57]. In celiac disease, the autoimmune response to gliadin results in small bowel inflammation, which resolves with removal of gluten from the diet [58]. Autopsy and biopsy data in these patients have demonstrated humoral immune activation in the peripheral nerves and DRG as well as other portions of the nervous system [57]. The commonest neurologic complication of gluten sensitivity is cerebellar ataxia, termed gluten ataxia, but various forms of peripheral neuropathy, including cases of SN, have also been reported [57, 59, 60]. In the hallmark article from 2010 by Hadjivassiliou et al. [57], 53 of 409 patients (13%) with chronic neuropathies were found to have SN [57]. SN was defined as patchy sensory loss with or without sensory ataxia, which was supported by electrodiagnostic evidence of a patchy, non-length-dependent sensory fiber involvement. Seventeen percent of the SN patients had serologic evidence of gluten sensitivity as determined by antigliadin antibodies. Of the 17 patients with SN and gluten sensitivity, only seven had enteropathy on biopsy ,and the rate of false positive results for antibodies was high. In all patients, cerebellar ataxia (gluten ataxia) had been excluded. Four patients demonstrated length-dependent sensory fiber involvement without motor involvement. Many would classify this as a length-dependent sensory axonopathy as opposed to SN. It has also been reported that celiac patients have a small-fiber neuronopathy [61]. In 2014, McKeon et al. [62] investigated the association of gliadin antibody positivity (without celiac disease) and neurologic dysfunction (including cerebellar ataxia, neuropathy, and other presentations). They determined that in most gliadin antibody positive patients without celiac disease, alternative causes of neurologic dysfunction, including nutritional deficiency and coexisting autoimmunity, could result in the neurologic manifestations. Reports of treatment response differ, with some neurologic symptoms stabilizing [57] or abating with initiation of a gluten-free diet [57, 61]. However, there is some suggestion that initiation of a gluten-free diet long after onset of neurologic symptoms does not produce significant abatement of symptoms [63].
Many other autoimmune diseases have been linked to SN. There are several case reports of SN in patients with autoimmune hepatitis [64,65,66]. Unfortunately, the patients’ neurologic symptoms did not significantly respond to immunomodulating therapies. Unlike in Sjögren’s syndrome, the association between SN and systemic lupus erythematosus is quite rare, with only a few case reports [67, 68]. A newly described association is that of livedoid vasculopathy and SN [69]. Livedoid vasculopathy is a cutaneous skin disorder that results in purpuric lesions that progress to ulcerations of the limbs. Healing of the ulcerations results in atrophie blanche (white atrophied scars). There is a report of a 32-year-old patient with livedoid vasculopathy who developed SN, initially presenting with gait ataxia and dysesthesia [69]. Alternative causes were excluded, and she was treated with mycophenolate mofetil and prednisolone. When the SN progressed, treatment with rituximab was added, which resulted in clinical and electrodiagnostic stabilization.
Anti-fibroblast growth factor receptor 3 (anti-FGFR3) antibodies have recently been associated with SN [70••]. In 2015, Antoine et al. [70••] studied 106 patients with pure sensory neuropathy, 72 of whom fulfilled the diagnostic criteria for SN. Anti-FGFR3 antibodies were identified in 17 patients (16 with a sensory neuropathy and 1 with systemic lupus erythematosus and no reported neuropathy). Antoine et al. proposed that the target of the antibodies is likely the DRG given that 87% of the antibody-positive patients had a non-length-dependent pattern of sensory disturbance and 82% fulfilled the diagnostic criteria for probable SN. The characteristic features include severe pain and trigeminal nerve involvement. One patient had severe dysautonomia. Nerve biopsy demonstrated fiber loss without regenerating clusters, supporting an SN over an axonopathy. Further, electromyography results were similar to those in SN. Antoine et al. reported that anti-FGFR3 antibodies were present in 19% of patients with idiopathic SN or SN associated with another autoimmune disorder in their series. These results suggest that identification of anti-FGFR3 antibodies may aid in diagnosis of SN.
Toxic Causes
Pyridoxine (vitamin B6)-induced SN was first described in the 1980s, and many cases have been reported since then [71, 72]. Pyridoxine exerts a dose-dependent effect on the DRG, with irreversible damage at higher doses despite cessation of intake of the vitamin [73]. In humans, SN has been reported with dosages anywhere from 200 to 6000 mg per day [72, 74]. Patients typically experience a severe sensory ataxia due to necrosis of the DRG and degeneration of their central and peripheral projections [75]. The exact mechanism of this necrosis has yet to be determined.
Another well-described toxic cause of SN is the platinum-based chemotherapeutic agents, including cisplatin, carboplatin, and oxaliplatin [76]. Cisplatin and oxaliplatin are considered more neurotoxic than carboplatin, with neurotoxicity reported in up to 90% of patients treated with oxaliplatin [77]. The effect is dose dependent, and with cisplatin, a cumulative dose higher than 300 mg/m2 has been demonstrated to result in electrophysiologic evidence of damage to large-fiber neurons, leading to SN-related symptoms [78, 79]. Platinum-containing drugs are known to result in peripheral nervous system toxicity through nuclear and mitochondrial DNA damage, inducing oxidative stress, and disturbing ion channels [80]. Cisplatin, in particular, is known to induce apoptosis in the DRG by binding to nuclear DNA and mitochondrial DNA, resulting in DNA damage and activation of p53 and Bax-mediated apoptosis [81]. Recent work by Maj et al. [82] suggests that prevention of the p53 accumulation with small-molecule pifithrin-μ may be neuroprotective. The “coasting effect” refers to the emergence of a symptomatic neuropathy up to 2 months after cessation of the chemotherapy and clearance of the drug [76]. For this reason, it is important to stop use of the offending agent at the first sign of neuronal dysfunction.
Another potential toxic cause has recently been described in the literature. Novak et al. [83] reported on the association between a non-length-dependent, subclinical, small-fiber neuropathy and statin use. In this study, the skin and sweat gland biopsy specimens of 80 statin users were compared with those of age-matched controls. Novak et al. found that compared with the biopsy specimens of non-statin users, the biopsy specimens of statin users had significantly reduced epidermal and sweat gland nerve fiber densities at the thigh but not at the calf. Of importance, there were no significant differences between the two groups with regard to autonomic testing scores, or subjective pain or numbness scale scores. They concluded that this non-length-dependent process represents a new association between sensory and autonomic ganglionopathy and statin use.
Infectious Causes
Several infectious causes have been implicated in the development of SN. The commonest is HIV/AIDS, but others include Epstein–Barr virus, varicella–zoster virus, and human T-lymphotropic virus [84,85,86,87,88,89]. Other infectious associations include leprosy, particularly in countries such as India, where the disease is commoner [90]. Recently, Chiu et al. [91] reported on a case of a 3-year-old girl in whom enterovirus infection had been diagnosed [91]. Nine days later she developed dorsal column dysfunction and absent sensory nerve action potentials (SNAP) on nerve conduction studies. This is the youngest reported case of SN, and the only case associated with enterovirus to date.
Idiopathic Causes
Despite extensive workup, about half of all SN are deemed to be idiopathic [86, 92]. This is obviously a diagnosis of exclusion, although it is thought that these presentations are likely of autoimmune or possibly toxic cause [93]. Unlike most other subacute presentations described thus far, idiopathic SN are typically indolent and slowly progressive [92].
Diagnostic Evaluation
In 2009, Camdessanché et al. [94] proposed a set of diagnostic criteria for SN on the basis of a retrospective of a study population so as to help distinguish SN from other sensory neuropathies [94]. These criteria were later validated in a study of 210 patients at 15 referral centers [95•]. In this validation study, the SN criteria were 90.3% sensitive and 85.2% specific, and had a positive predictive value of 91.9% and a negative predictive value of 82.5%. The criteria were validated against expert diagnosis as opposed to biopsy of DRG. See Table 1 for the score form for the SN diagnostic criteria.
When workup is being performed on a patient with suspected SN, electrophysiologic testing can be the most helpful study to further support the diagnosis, and is strongly recommended. Non-length-dependent sensory abnormalities are the hallmark, and often the upper extremities are affected disproportionately [86]. These abnormalities are typically widespread and asymmetric [96]. Although classically motor nerve conduction study findings are normal, motor nerve conduction study abnormalities have been reported for most causes [45, 64, 87, 92, 97, 98]. Needle electromyography is remarkable for incomplete activation of muscles due to loss of sensory input and the resultant incomplete interference pattern [99]. Recently, serial nerve conduction study findings were studied in 86 inflammatory SN patients [31••]. There was a monthly reduction of SNAP amplitudes that worsened in the first 2 months, then slowed down after 7 months, and finally stabilized after 10 months. The decline in SNAP amplitudes correlated with disability progression and lymphocytic reaction in the cerebrospinal fluid (CSF). On the basis of this observation, the study authors proposed that treatment should be initiated within the first 8 months if possible. The authors recommend treatment within 2 months of symptom onset during which the inflammatory reaction and decline in sensory response amplitude are at their greatest. The radial and ulnar nerves seemed to be the most sensitive to change.
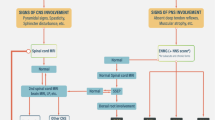
Depending on the specific clinical picture and history of exposures, serum and CSF analysis can aid in the diagnosis of the specific autoimmune or toxic causes. In general, we recommend the study of the following for initial diagnostic evaluation: anti-Hu and anti-CRMP-5/CV2 antibodies, antinuclear antibodies, anti-SSA and anti-SSB antibodies, vitamin B6 level (if there is a history of supplementation), and HIV. Should the study findings be unremarkable, then a more extensive workup is warranted, including additional laboratory and imaging studies as outlined in Fig. 2. In the case of a paraneoplastic syndrome, the sensitivity and specificity of serum anti-Hu antibodies are 82% and 99% respectively [100]. Some patients with anti-Hu antibodies also have anti-CRMP-5/CV2 antibodies, and they may have a mix of SN and superimposed demyelinating sensorimotor neuropathy. Anti-CRMP-5/CV2 antibodies in isolation are associated with a mixed axonal and demyelinating polyneuropathy [8]. Notably, nearly 20% of paraneoplastic sensory neuropathies are seronegative [100]. In this case, characteristic CSF sampling may be useful, and demonstrates pleocytosis, elevated protein levels, and oligoclonal bands [18, 94, 101]. Importantly, neurologic symptoms are often the presenting symptom of a malignancy, and precede the cancer diagnosis by 3–8 months, and care should be taken to look for evidence of malignancy if a paraneoplastic syndrome is diagnosed [10, 18, 24, 102]. A chest computed tomography scan should be performed, and even fluorodeoxyglucose positron emission tomography should be considered if the initial imaging findings are negative and clinical suspicion remains high [103, 104]. If underlying malignancy is not found and the patient harbors paraneoplastic antibodies, patients should undergo repeated screening in 3–6 months, and then every 6 months thereafter for 4 years [105].

Score form for the diagnosis of sensory neuronopathy. MRI magnetic resonance imaging. (Modified with permission from Camdessanché et al. [94])
Magnetic resonance imaging (MRI) of the spinal cord can also be helpful in the diagnosis of an SN as T2 hyperintensity in the dorsal columns may be observed [106]. Multiple-echo data image combination (MEDIC) and turbo inversion recovery magnitude (TIRM) imaging techniques have recently been described to reveal characteristic findings in SN. MEDIC imaging demonstrates higher signal intensity in the DRG and posterior columns. Conversely, TIRM imaging demonstrated a smaller spinal cord area and decreased nerve root diameter than in healthy controls [107]. Diffusion tensor imaging (DTI) has been investigated as a tool to differentiate SN patients from diabetic sensorimotor polyneuropathy patients and healthy individuals. In this study, MRI findings were compared among 28 patients with SN, 20 healthy controls, and 14 patients with diabetic distal sensorimotor polyneuropathy. It was found that DTI abnormalities in SN patients preceded the T2 hyperintensity in the dorsal columns on MRI, and the study authors proposed that it may be more sensitive. The study authors hypothesized that the DTI abnormalities are secondary to axonal loss and gliosis of the cuneate and gracile fasciculi, representing damage of the central projections in SN. They concluded that DTI imaging could be an earlier, noninvasive, in vivo test used in the diagnosis of SN and could also help differentiate between SN and distal neuropathies [108].
Depending on the likely underlying cause, tissue biopsy may also a useful diagnostic tool in SN. In patients with suspected Sjögren’s syndrome associated SN, a lip or salivary gland biopsy can be diagnostic should the test for autoantibodies be negative [109]. In paraneoplastic syndromes, tumor biopsy is required to confirm the pathologic features of the underlying malignancy. In patients with small-fiber-predominant SN (as in those with celiac disease), a skin biopsy may demonstrate a non-length-dependent pattern of nerve loss. The only definitive way to confirm DRG disease is by biopsy of the DRG, which is an invasive, traumatic procedure, which we do not recommend.
Conclusion
Acquired SN are a rare group of disorders that present with a non-length-dependent sensory neuropathy and early-onset ataxia. The prompt recognition and diagnostic evaluation of SN is important as an underlying malignancy is often discovered. Additionally, early identification of SN results in more immediate initiation of treatment, which has significant prognostic implications. In the future, further identification of associated autoantibodies may facilitate identification of this uncommon subset of neuropathies and may result in more successful, focused therapeutic interventions.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Koeppen AH, Mazurkiewicz JE. Friedreich ataxia: neuropathology revised. J Neuropathol Exp Neurol. 2013;72:78–90.
Lax NZ, Whittaker RG, Hepplewhite PD, Reeve AK, Blakely EL, Jaros E, et al. Sensory neuronopathy in patients harbouring recessive polymerase γ mutations. Brain. 2012;135:62–71.
Szmulewicz DJ, McLean CA, Rodriguez ML, Chancellor AM, Mossman S, Lamont D, et al. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology. 2014;82:1410–5.
Bejaoui K, Wu C, Scheffler MD, Haan G, Ashby P, Wu L, et al. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet. 2001;27:261–2.
Vucic S, Tian D, Chong PST, Cudkowicz ME, Hedley-Whyte ET, Cros D. Facial onset sensory and motor neuronopathy (FOSMN syndrome): a novel syndrome in neurology. Brain. 2006;129:3384–90.
Colli BO, Carlotti CG, Assirati JA. Lopes L da S, Marques W, Chimelli L, et al. Dorsal root ganglionectomy for the diagnosis of sensory neuropathies. Surgical technique and results. Surg Neurol. 2008;69:266–73. discussion 273
Oh SJ, Gürtekin Y, Dropcho EJ, King P, Claussen GC. Anti-Hu antibody neuropathy: a clinical, electrophysiological, and pathological study. Clin Neurophysiol. 2005;116:28–34.
Antoine JC, Honnorat J, Camdessanché JP, Magistris M, Absi L, Mosnier JF, et al. Paraneoplastic anti-CV2 antibodies react with peripheral nerve and are associated with a mixed axonal and demyelinating peripheral neuropathy. Ann Neurol. 2001;49:214–21.
Ogawa M, Nishie M, Kurahashi K, Kaimori M, Wakabayashi K. Anti-Hu associated paraneoplastic sensory neuronopathy with upper motor neurone involvement. J Neurol Neurosurg Psychiatry. 2004;75:1051–3.
Sillevis Smitt P, Grefkens J, de Leeuw B, van den Bent M, van Putten W, Hooijkaas H, et al. Survival and outcome in 73 anti-Hu positive patients with paraneoplastic encephalomyelitis/sensory neuronopathy. J Neurol. 2002;249:745–53.
Wabbels BK, Elflein H, Lorenz B, Kolling G. Bilateral tonic pupils with evidence of anti-Hu antibodies as a paraneoplastic manifestation of small cell lung cancer. Ophthalmologica. 218:141–3.
Wymenga ANM, Slebos DJ, van der Naalt J, van Putten JWG, Peters FTM. Buikklachten en neurologische symptomen als vroege manifestatie van longkanker: een uiting van het anti-Hu-syndroom. Ned Tijdschr Geneeskd. 2003;147:616–9.
Briellmann RS, Sturzenegger M, Gerber HA, Schaffner T, Hess CW. Autoantibody-associated sensory neuronopathy and intestinal pseudo-obstruction without detectable neoplasia. Eur Neurol. 1996;36:369–73.
Wildhaber B, Niggli F, Stallmach T, Willi U, Stauffer UG, Sacher P. Intestinal pseudoobstruction as a paraneoplastic syndrome in ganglioneuroblastoma. Eur J Pediatr Surg. 2002;12:429–31.
Camdessanché J-P, Antoine J-C, Honnorat J, Vial C, Petiot P, Convers P, et al. Paraneoplastic peripheral neuropathy associated with anti-Hu antibodies. A clinical and electrophysiological study of 20 patients. Brain. 2002;125:166–75.
Taieb G, Renard D, Deverdal M, Honnorat J, Labauge P, Castelnovo G. Pure monomelic sensory neuronopathy associated with anti-yo antibodies. Muscle Nerve. 2012;45:297–8.
Antoine JC, Absi L, Honnorat J, Boulesteix JM, de Brouker T, Vial C, et al. Antiamphiphysin antibodies are associated with various paraneoplastic neurological syndromes and tumors. Arch Neurol. 1999;56:172–7.
Graus F, Keime-Guibert F, Reñe R, Benyahia B, Ribalta T, Ascaso C, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138–48.
Côté-Mantha E, Savard M. Paraneoplastic anti-HU syndrome associated with uterine tumor. Can J Neurol Sci. 2012;39:254–5.
Fournier CN, Kalra A, Lachance DH, Zarwan C, Srinivasan J. ANNA-1 (anti-Hu) associated sensory neuronopathy with malignant mixed mullerian tumor. Muscle Nerve. 2013;47:776–7.
Sano T, Tanaka K, Ito N. Anti-Hu-antibody-associated paraneoplastic neurological syndrome accompanying testicular cancer. Int J Urol. 2010;17:99.
Lukacs S, Szabo N, Woodhams S. Rare association of anti-Hu antibody positive paraneoplastic neurological syndrome and transitional cell bladder carcinoma. Case Rep Urol. 2012;2012:724940.
Cowley A, Pascoe S. Paraneoplastic subacute sensory neuronopathy in association with adenocarcinoma of the prostate. BMJ Case Rep. 2011; doi:10.1136/bcr.04.2011.4077.
Matsui T, Hori Y, Nagano H, Eguchi H, Marubashi S, Wada H, et al. Poorly differentiated hepatocellular carcinoma accompanied by anti-Hu antibody-positive paraneoplastic peripheral neuropathy. Pathol Int. 2015;65:388–92.
Voltz R, Dalmau J, Posner JB, Rosenfeld MR. T-cell receptor analysis in anti-Hu associated paraneoplastic encephalomyelitis. Neurology. 1998;51:1146–50.
Tomita M, Koike H, Kawagashira Y, Iijima M, Adachi H, Taguchi J, et al. Clinicopathological features of neuropathy associated with lymphoma. Brain. 2013;136:2563–78.
Pignolet BS, Gebauer CM, Liblau RS. Immunopathogenesis of paraneoplastic neurological syndromes associated with anti-Hu antibodies: a beneficial antitumor immune response going awry. Oncoimmunology. 2013;2:e27384.
Manley GT, Smitt PS, Dalmau J, Posner JB. Hu antigens: reactivity with Hu antibodies, tumor expression, and major immunogenic sites. Ann Neurol. 1995;38:102–10.
Dalmau J, Graus F, Cheung NK, Rosenblum MK, Ho A, Cañete A, et al. Major histocompatibility proteins, anti-Hu antibodies, and paraneoplastic encephalomyelitis in neuroblastoma and small cell lung cancer. Cancer. 1995;75:99–109.
Kuntzer T, Antoine J-C, Steck AJ. Clinical features and pathophysiological basis of sensory neuronopathies (ganglionopathies). Muscle Nerve. 2004;30:255–68.
•• Antoine J-C, Robert-Varvat F, Maisonobe T, Créange A, Franques J, Mathis S, et al. Identifying a therapeutic window in acute and subacute inflammatory sensory neuronopathies. J Neurol Sci. 2016;361:187–91. The authors retrospectively studied the electrophysiological study findings of 86 patients with inflammatory sensory neuronopathy. They used the sensory nerve action potentials as a surrogate marker of neuronal degeneration. The sensory responses declined rapidly in the first 2 months, the decline slowed down after 7 months, and stabilized after 10 months. They concluded that there is a brief therapeutic window of 8 months. Abatement of the disease is possible if treatment is initiated in the first 2 months.
Antoine J-C, Camdessanché J-P. Treatment options in paraneoplastic disorders of the peripheral nervous system. Curr Treat Options Neurol. 2013;15:210–23.
Oh SJ, Dropcho EJ, Claussen GC. Anti-Hu-associated paraneoplastic sensory neuropathy responding to early aggressive immunotherapy: report of two cases and review of literature. Muscle Nerve. 1997;20:1576–82.
Rosenfeld MR, Dalmau J. Diagnosis and management of paraneoplastic neurologic disorders. Curr Treat Options Oncol. 2013;14:528–38.
Uchuya M, Graus F, Vega F, Reñé R, Delattre JY. Intravenous immunoglobulin treatment in paraneoplastic neurological syndromes with antineuronal autoantibodies. J Neurol Neurosurg Psychiatry. 1996;60:388–92.
Graus F, Vega F, Delattre JY, Bonaventura I, Reñé R, Arbaiza D, et al. Plasmapheresis and antineoplastic treatment in CNS paraneoplastic syndromes with antineuronal autoantibodies. Neurology. 1992;42:536–40.
Shams’ili S, de Beukelaar J, Gratama JW, Hooijkaas H, van den Bent M, van’t Veer M, et al. An uncontrolled trial of rituximab for antibody associated paraneoplastic neurological syndromes. J Neurol. 2006;253:16–20.
de Jongste AH, van Gelder T, Bromberg JE, de Graaf MT, Gratama JW, Schreurs MW, et al. A prospective open-label study of sirolimus for the treatment of anti-Hu associated paraneoplastic neurological syndromes. Neuro Oncol. 2015;17:145–50.
Malinow K, Yannakakis GD, Glusman SM, Edlow DW, Griffin J, Pestronk A, et al. Subacute sensory neuronopathy secondary to dorsal root ganglionitis in primary Sjögren’s syndrome. Ann Neurol. 1986;20:535–7.
Pavlakis PP, Alexopoulos H, Kosmidis ML, Mamali I, Moutsopoulos HM, Tzioufas AG, et al. Peripheral neuropathies in Sjögren’s syndrome: a critical update on clinical features and pathogenetic mechanisms. J Autoimmun. 2012;39:27–33.
• Pereira PR, Viala K, Maisonobe T, Haroche J, Mathian A, Hié M, et al. Sjögren sensory neuronopathy (Sjögren ganglionopathy): long-term outcome and treatment response in a series of 13 cases. Medicine (Baltimore). 2016;95:e3632. This is a retrospective study of 13 patients with Sjögren's syndrome-associated sensory neuronopathy. Patients presented most often with ataxia and areflexia. They were treated with a number of therapies, including corticosteroids, mycophenolate mofetil, hydroxychloroquine, and intravenous immune globulin. The authors concluded that Sjögren's syndrome-associated sensory neuronopathy is heterogeneous, insidious, chronic, and debilitating despite treatment. The patients treated with immunosuppressive drugs such as mycophenolate mofetil and corticosteroids did well compared with those treated with intravenous immune globulin.
Brito-Zerón P, Akasbi M, Bosch X, Bové A, Pérez-De-Lis M, Diaz-Lagares C, et al. Classification and characterisation of peripheral neuropathies in 102 patients with primary Sjögren’s syndrome. Clin Exp Rheumatol. 2013;31:103–10.
Carvajal Alegria G, Guellec D, Devauchelle-Pensec V, Saraux A. Is there specific neurological disorders of primary Sjögren’s syndrome? Joint Bone Spine. 2015;82:86–9.
Fauchais A-L, Magy L, Vidal E. Central and peripheral neurological complications of primary Sjögren’s syndrome. Presse Med. 2012;41:e485–93.
Griffin JW, Cornblath DR, Alexander E, Campbell J, Low PA, Bird S, et al. Ataxic sensory neuropathy and dorsal root ganglionitis associated with Sjögren’s syndrome. Ann Neurol. 1990;27:304–15.
Mori K, Iijima M, Koike H, Hattori N, Tanaka F, Watanabe H, et al. The wide spectrum of clinical manifestations in Sjögren’s syndrome-associated neuropathy. Brain. 2005;128:2518–34.
Damasceno A, França MC, Cury H, Nucci A. Autonomic dysfunction in non-paraneoplastic sensory neuronopathy: beyond sensory abnormalities. J Neurol. 2011;258:231–7.
Mellgren SI, Göransson LG, Omdal R. Primary Sjögren’s syndrome associated neuropathy. Can J Neurol Sci. 2007;34:280–7.
Chen WH, Yeh JH, Chiu HC. Plasmapheresis in the treatment of ataxic sensory neuropathy associated with Sjögren’s syndrome. Eur Neurol. 2001;45:270–4.
Gorson KC, Natarajan N, Ropper AH, Weinstein R. Rituximab treatment in patients with IVIg-dependent immune polyneuropathy: a prospective pilot trial. Muscle Nerve. 2007;35:66–9.
Santosa A, Lim AYN, Vasoo S, Lau TC, Teng GG. Neurosjögren: early therapy is associated with successful outcomes. J Clin Rheumatol. 2012;18:389–92.
Martinez ARM, Nunes MB, Nucci A, França MC. Sensory neuronopathy and autoimmune diseases. Autoimmune Dis. 2012;2012:873587.
Rist S, Sellam J, Hachulla E, Sordet C, Puéchal X, Hatron P, et al. Experience of intravenous immunoglobulin therapy in neuropathy associated with primary Sjögren’s syndrome: a national multicentric retrospective study. Arthritis Care Res (Hoboken). 2011;63:1339–44.
Takahashi Y, Takata T, Hoshino M, Sakurai M, Kanazawa I. Benefit of IVIG for long-standing ataxic sensory neuronopathy with Sjögren’s syndrome. IV immunoglobulin. Neurology. 2003;60:503–5.
Caroyer J-M, Manto MU, Steinfeld SD. Severe sensory neuronopathy responsive to infliximab in primary Sjögren’s syndrome. Neurology. 2002;59:1113–4.
Goodman BP. Immunoresponsive autonomic neuropathy in Sjögren syndrome—case series and literature review. Am J Ther. 2017; doi:10.1097/MJT.0000000000000583.
Hadjivassiliou M, Rao DG, Wharton SB, Sanders DS, Grünewald RA, Davies-Jones AGB. Sensory ganglionopathy due to gluten sensitivity. Neurology. 2010;75:1003–8.
Marsh MN. The natural history of gluten sensitivity: defining, refining and re-defining. QJM. 1995;88:9–13.
Hadjivassiliou M, Williamson CA, Woodroofe N. The immunology of gluten sensitivity: beyond the gut. Trends Immunol. 2004;25:578–82.
Reda H, Chin RL. Peripheral neuropathies of rheumatologic disease and gluten-related disorders. Semin Neurol. 2014;34:413–24.
Brannagan TH, Hays AP, Chin SS, Sander HW, Chin RL, Magda P, et al. Small-fiber neuropathy/neuronopathy associated with celiac disease: skin biopsy findings. Arch Neurol. 2005;62:1574–8.
McKeon A, Lennon VA, Pittock SJ, Kryzer TJ, Murray J. The neurologic significance of celiac disease biomarkers. Neurology. 2014;83:1789–96.
Volta U, De Giorgio R. Gluten sensitivity: an emerging issue behind neurological impairment? Lancet Neurol. 2010;9:233–5.
Merchut MP, Adams EM, Morrissey M. Sensory neuronopathy in autoimmune chronic active hepatitis. Neurology. 1993;43:2410–1.
Magy L, Bassez G, Chassande B, Poynard T, Léger JM. Neuronopathie sensitive associee a une hepatite chronique auto-immune. Rev Neurol (Paris). 1997;153:70–2.
Liedholm LJ, Månsson A, Holmgren H. Subakuta sensoriska neuropatier. Nord Med. 1994;109:296–7. 309
Navinan MR, Piranavan P, Akram AUA, Yudhishdran J, Kandeepan T, Kulatunga A. Sensory neuronopathy complicating systemic lupus erythematosus: a case report. J Med Case Rep. 2014;8:141.
Wang J-C, Lin Y-C, Yang T-F, Lin H-Y. Ataxic sensory neuronopathy in a patient with systemic lupus erythematosus. Lupus. 2012;21:905–9.
Alix JJP, Hadjivassiliou M, Ali R, Slater D, Messenger AG, Rao DG. Sensory ganglionopathy with livedoid vasculopathy controlled by immunotherapy. Muscle Nerve. 2015;51:296–301.
•• Antoine J-C, Boutahar N, Lassablière F, Reynaud E, Ferraud K, Rogemond V, et al. Antifibroblast growth factor receptor 3 antibodies identify a subgroup of patients with sensory neuropathy. J Neurol Neurosurg Psychiatry. 2015;86:1347–55. The authors identified anti-fibroblast growth factor receptor 3 antibodies in 17 patients, 16 of whom had a sensory neuropathy. The authors concluded that these antibodies may be seen in patients with an underlying autoimmune disorder affecting the dorsal root and trigeminal nerve ganglia as 87% of patients had a non-length-dependent neuropathy consistent with sensory neuronopathy.
Kulkantrakorn K. Pyridoxine-induced sensory ataxic neuronopathy and neuropathy: revisited. Neurol Sci. 2014;35:1827–30.
Schaumburg H, Kaplan J, Windebank A, Vick N, Rasmus S, Pleasure D, et al. Sensory neuropathy from pyridoxine abuse. A new megavitamin syndrome. N Engl J Med. 1983;309:445–8.
Xu Y, Sladky JT, Brown MJ. Dose-dependent expression of neuronopathy after experimental pyridoxine intoxication. Neurology. 1989;39:1077–83.
Parry GJ, Bredesen DE. Sensory neuropathy with low-dose pyridoxine. Neurology. 1985;35:1466–8.
Perry TA, Weerasuriya A, Mouton PR, Holloway HW, Greig NH. Pyridoxine-induced toxicity in rats: a stereological quantification of the sensory neuropathy. Exp Neurol. 2004;190:133–44.
Windebank AJ, Grisold W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst. 2008;13:27–46.
Balayssac D, Ferrier J, Descoeur J, Ling B, Pezet D, Eschalier A, et al. Chemotherapy-induced peripheral neuropathies: from clinical relevance to preclinical evidence. Expert Opin Drug Saf. 2011;10:407–17.
Glendenning JL, Barbachano Y, Norman AR, Dearnaley DP, Horwich A, Huddart RA. Long-term neurologic and peripheral vascular toxicity after chemotherapy treatment of testicular cancer. Cancer. 2010;116:2322–31.
Krarup-Hansen A, Helweg-Larsen S, Schmalbruch H, Rørth M, Krarup C. Neuronal involvement in cisplatin neuropathy: prospective clinical and neurophysiological studies. Brain. 2007;130:1076–88.
Kerckhove N, Collin A, Condé S, Chaleteix C, Pezet D, Balayssac D. Long-term effects, pathophysiological mechanisms, and risk factors of chemotherapy-induced peripheral neuropathies: a comprehensive literature review. Front Pharmacol. 2017;8:86.
Podratz JL, Knight AM, Ta LE, Staff NP, Gass JM, Genelin K, et al. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011;41:661–8.
Maj MA, Ma J, Krukowski KN, Kavelaars A, Heijnen CJ. Inhibition of mitochondrial p53 accumulation by PFT-μ prevents cisplatin-induced peripheral neuropathy. Front Mol Neurosci. 2017;10:108.
Novak P, Pimentel DA, Sundar B, Moonis M, Qin L, Novak V. Association of statins with sensory and autonomic ganglionopathy. Front Aging Neurosci. 2015;7:191.
Rance NE, McArthur JC, Cornblath DR, Landstrom DL, Griffin JW, Price DL. Gracile tract degeneration in patients with sensory neuropathy and AIDS. Neurology. 1988;38:265–71.
Scaravilli F, Sinclair E, Arango JC, Manji H, Lucas S, Harrison MJ. The pathology of the posterior root ganglia in AIDS and its relationship to the pallor of the gracile tract. Acta Neuropathol. 1992;84:163–70.
Sghirlanzoni A, Pareyson D, Lauria G. Sensory neuron diseases. Lancet Neurol. 2005;4:349–61.
Rubin DI, Daube JR. Subacute sensory neuropathy associated with Epstein-Barr virus. Muscle Nerve. 1999;22:1607–10.
Ramos F, Monforte C, Luengo Neuronopatia aguda sensitiva asociada a infeccion por virus varicela-zoster. Rev. Neurol. 1999;28:1067–9.
Shimazaki R, Ueyama H, Mori T, Mori M, Fujimoto S, Kumamoto T, et al. Chronic sensory neuronopathy associated with human T-cell lymphotropic virus type I infection. J Neurol Sci. 2002;194:55–8.
Mansukhani KA, Khadilkar SV. Consider leprosy as an etiology of sensory neuronopathy. Muscle Nerve. 2017;55:928.
Chiu C-C, Yang C-Y, Yang T-F, Lin K-P, Huang S-H, Wang J-C. Acute sensory neuronopathy following enterovirus infection in a 3-year-old girl. Neuropediatrics. 2017;48:190–3.
Dalakas MC. Chronic idiopathic ataxic neuropathy. Ann Neurol. 1986;19:545–54.
van Dijk GW, Wokke JH, Notermans NC, van den Berg LH, Bär PR. Indications for an immune-mediated etiology of idiopathic sensory neuronopathy. J Neuroimmunol. 1997;74:165–72.
Camdessanché J-P, Jousserand G, Ferraud K, Vial C, Petiot P, Honnorat J, et al. The pattern and diagnostic criteria of sensory neuronopathy: a case-control study. Brain. 2009;132:1723–33.
• Antoine J-C, Robert-Varvat F, Maisonobe T, Créange A, Franques J, Mathis S, et al. Testing the validity of a set of diagnostic criteria for sensory neuronopathies: a francophone collaborative study. J Neurol. 2014;261:2093–100. The sensory neuronopathy criteria, published in 2009, were validated in 210 patients in a multicenter French study.
Lauria G, Pareyson D, Sghirlanzoni A. Neurophysiological diagnosis of acquired sensory ganglionopathies. Eur Neurol. 2003;50:146–52.
Sterman AB, Schaumburg HH, Asbury AK. The acute sensory neuronopathy syndrome: a distinct clinical entity. Ann Neurol. 1980;7:354–8.
Windebank AJ, Blexrud MD, Dyck PJ, Daube JR, Karnes JL. The syndrome of acute sensory neuropathy: clinical features and electrophysiologic and pathologic changes. Neurology. 1990;40:584–91.
Rothwell JC, Traub MM, Day BL, Obeso JA, Thomas PK, Marsden CD. Manual motor performance in a deafferented man. Brain. 1982;105(Pt 3):515–42.
Molinuevo JL, Graus F, Serrano C, Reñe R, Guerrero A, Illa I. Utility of anti-Hu antibodies in the diagnosis of paraneoplastic sensory neuropathy. Ann Neurol. 1998;44:976–80.
Chalk CH, Windebank AJ, Kimmel DW, McManis PG. The distinctive clinical features of paraneoplastic sensory neuronopathy. Can J Neurol Sci. 1992;19:346–51.
Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain. 2000;123(Pt 7):1481–94.
Chartrand-Lefebvre C, Howarth N, Grenier P, Keime F, Orcel B, Beigelman C. Association of small cell lung cancer and the anti-Hu paraneoplastic syndrome: radiographic and CT findings. AJR Am J Roentgenol. 1998;170:1513–7.
Bannas P, Weber C, Derlin T, Lambert J, Leypoldt F, Adam G, et al. 18F-FDG-PET/CT in the diagnosis of paraneoplastic neurological syndromes: a retrospective analysis. Eur Radiol. 2010;20:923–30.
Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F, et al. Screening for tumours in paraneoplastic syndromes: report of an EFNS task force. Eur J Neurol. 2011;18:19–e3.
Lauria G, Pareyson D, Grisoli M, Sghirlanzoni A. Clinical and magnetic resonance imaging findings in chronic sensory ganglionopathies. Ann Neurol. 2000;47:104–9.
Bao Y-F, Tang W-J, Zhu D-Q, Li Y-X, Zee C-S, Chen X-J, et al. Sensory neuronopathy involves the spinal cord and brachial plexus: a quantitative study employing multiple-echo data image combination (MEDIC) and turbo inversion recovery magnitude (TIRM). Neuroradiology. 2013;55:41–8.
Casseb RF, de Paiva JLR, Branco LMT, Martinez ARM, Reis F, de Lima-Junior JC, et al. Spinal cord diffusion tensor imaging in patients with sensory neuronopathy. Neuroradiology. 2016;58:1103–8.
Gorson KC, Ropper AH. Positive salivary gland biopsy, Sjögren syndrome, and neuropathy: clinical implications. Muscle Nerve. 2003;28:553–60.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Nerve and Muscle
Rights and permissions
About this article

Cite this article
Crowell, A., Gwathmey, K.G. Sensory Neuronopathies. Curr Neurol Neurosci Rep 17, 79 (2017). https://doi.org/10.1007/s11910-017-0784-4
Published:
DOI: https://doi.org/10.1007/s11910-017-0784-4