
Abstract
Purpose of Review
Tauopathies represent a spectrum of incurable and progressive age-associated neurodegenerative diseases that currently are diagnosed definitively only at autopsy. Few clinical diagnoses, such as classic Richardson’s syndrome of progressive supranuclear palsy, are specific for underlying tauopathy and no clinical syndrome is fully sensitive to reliably identify all forms of clinically manifest tauopathy. Thus, a major unmet need for the development and implementation of tau-targeted therapies is precise antemortem diagnosis. This article reviews new and emerging diagnostic therapies for tauopathies including novel imaging techniques and biomarkers and also reviews recent tau therapeutics.
Recent Findings
Building evidence from animal and cell models suggests that prion-like misfolding and propagation of pathogenic tau proteins between brain cells are central to the neurodegenerative process. These rapidly growing developments build rationale and motivation for the development of therapeutics targeting this mechanism through altering phosphorylation and other post-translational modifications of the tau protein, blocking aggregation and spread using small molecular compounds or immunotherapy and reducing or silencing expression of the MAPT tau gene.
Summary
New clinical criteria, CSF, MRI, and PET biomarkers will aid in identifying tauopathies earlier and more accurately which will aid in selection for new clinical trials which focus on a variety of agents including immunotherapy and gene silencing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
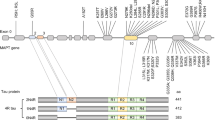
Tau is a highly soluble microtubule-associated protein which modulates stability of axonal cytoskeleton and is encoded by the MAPT gene on chromosome 17q21.3 consisting of 16 exons. Due to alternate splicing of E2, E3, and E10, six tau isoforms exist in human brain tissue that are defined by the presence or absence of E10 (the second microtubule-binding domain): three tau isoforms that contain three repeated binding domains (i.e., 3R tau) and the three tau isoforms containing E10 with four repeated binding domains (i.e., 4R tau). In the normal human brain, there exists a relative equal balance in the ratio of 3R:4R tau isoforms [1]. Tauopathies are a class of age-associated neurodegenerative diseases that are characterized by the presence of abnormal accumulations of pathogenic tau in neurons and/or glia. These disorders can be further classified by the relative balance of 3R and 4R tau isoforms found in pathological inclusions and morphological/ultrastructural features of inclusions.
Alzheimer’s disease (AD) is defined by the presence of both amyloid-beta plaques and tau neurofibrillary tangles (NFTs) [2], which consist of relatively equal proportions of 3R and 4R tau isoforms in paired helical filaments [3••]. While NFTs in AD correlate most closely with clinical symptoms [4], the precise relationship between amyloidosis, NFTs, and cognitive dysfunction are currently unclear. As such, AD can be considered a mixed tauopathy due to the consistent findings of both tau NFTs and amyloid plaques. A distinct neuropathological entity, primary age-related tauopathy (PART), has been recently proposed to distinguish the pathological findings of NFT pathology found in relative or absolute absence of amyloid plaque pathology [5]. These individuals are usually older and may have mild or no clear cognitive impairment during life, with corresponding tau pathology found restricted to the medial-temporal lobe. Others claim that PART is within the spectrum of the AD due to the lack of biochemical differences between AD and PART NFTs and universal findings of medial temporal lobe NFTs in AD [6]. Further research is needed to help support or refute the distinction of PART from AD. Finally, moderate to severe comorbid AD NFT tau and amyloid-beta plaque pathology is common (~50%) in Lewy body disorders (LBD) [7] and NFTs confer a strong effect on prognosis and timing of the expression of dementia [8]. Thus, tau-directed therapies may likely impact not only primary tauopathies but also potentially mixed tauopathies such as AD and LBD patients with AD copathology. This review will focus on primary tauopathies, which are considered part of the frontotemporal lobar degeneration (FTLD) spectrum (i.e., FTLD-Tau) [9••], as these patients have a monoproteinopathy which is advantageous for testing tau-directed therapeutics [10].
Three main strands of evidence suggest that the pathological process of tau accumulation within brain cells and propagation between cells is central to disease pathogenesis. First, pathological findings of tau pathology is the hallmark of these disorders and “gold-standard” for diagnosis, and regional topography of tau pathology in the CNS correlates well with clinical symptoms [4, 11, 12]. Second, patients with familial forms of tauopathy possess pathogenic mutations in the MAPT tau gene (FTDP-17); many of which correspond to accelerated fibrillization of tau and/or loss of microtubule binding function in vitro [13] demonstrating that altered tau function can contribute to disease pathogenesis. Finally, many recent animal- and cell-model studies find transmission of both recombinant tau and pathogenic tau-derived from brain homogenates of human tauopathy patients which can propagate from cell-to-cell in anatomically connected networks [14•, 15, 16]. These studies parallel the landmark human staging studies by Braak and Braak, which find sequential patterns of progressive cortical NFT pathology from serial cross-sectional AD autopsies [17] and provide compelling evidence that alteration of the tau protein alone is sufficient to recapitulate human disease. Further, studies using injections of brain extracts from various human tauopathies give rise to distinct morphologies of tau pathology in murine models that are similar to the features of tau pathology from human source tissue [18, 19]. In addition, inoculation with recombinant tau protein can cause distinct morphologies of endogenous tau aggregations in cell models of disease and these specific aggregation types, when injected into transgenic mice, developed different regional patterns of tau pathology [20, 21]. These innovative studies suggest that there may be distinct strains of pathogenic tau that correspond to the various clinical and pathological forms of tauopathies. These strain-like properties are similar to those seen in spongiform encephalopathies; however, a clear distinction remains in that prions are infectious proteinaceous particles [22] and there is currently no evidence to suggest that tauopathies can be spread between humans or non-human primates [23]. These distinctions aside, the prion-like mechanism of tauopathy aggregation and spread is an attractive target for therapeutic development as it is likely the most proximal cause of neurodegeneration. Transmission models show minimal neuronal toxicity associated with exogenously induced tangles [14•, 15, 16, 18], and transgenic animals may show signs of degeneration prior to tau inclusion formation [24], suggesting that the toxic species of tau may be prefibrillar tau (i.e., soluble monomers, oligomers) rather than tangles themselves [25•]. It is likely that loss of tau microtubule stabilizing function contributes as well through compromised axonal transport and resultant altered cellular metabolism [26]. Other downstream mechanisms including impaired protein degradation pathways, oxidative stress, and inflammation likely contribute in the neurodegenerative process, and targeting these systems alone or in combination with tau-directed therapies may be advantageous as well.
This review highlights the clinicopathological heterogeneity of tauopathies, followed by an overview of the state of the science in diagnostic biomarkers and emerging therapeutic strategies to slow or halt tau-mediated neurodegeneration.
Clinicopathological Complexity of Tauopathies
Primary tauopathies (FTLD-Tau) are both clinically and pathologically diverse. Figure 1 depicts the main clinicopathological associations of FTLD-Tau within the clinical spectrum of frontotemporal dementia (FTD). One major challenge to accurate diagnosis is that patients may present with either cognitive and/or motor symptoms that may be encountered at either memory or movement disorder clinic. Cognitive and motor impairment can cause additive disability and many patients require coordinated care across neurological disciplines. The main diagnostic considerations are other age-associated neurodegenerative diseases including forms of FTLD with TDP-43 or fused-in-sarcoma proteinopathy (i.e., FTLD-TDP, FTLD-FUS), AD, or LBD. Since there is no clinically available test to diagnose FTLD-Tau antemortem, it is important to exclude potentially treatable causes of “rapid-progressive dementia” in those patients with “red-flag” symptoms of acute onset, rapid progression, or atypical features such as seizure [27]. Below, we characterize the main classes of FTLD-Tau.

The importance of autopsy confirmation in improvement of diagnosis and treatment of tauopathies. Figure depicts clinicopathological associations of the three main FTLD-Tau neuropathologies found at autopsy with clinical syndromes. Solid lines represent the strongest associations (i.e., PiD with bvFTD, CBD with CBS, and PSP with PSPS) and dashed lines represent less frequent associations. Color shading of clinical phenotype boxes depict the relative frequencies of neuropathologies found at autopsy in each syndrome (red FTLD-Tau, blue FTLD-TDP, yellow AD) and photomicrographs in each neuropathology box depict characteristic inclusion morphologies (PiD Pick bodies, CBD astrocytic plaque, PSP tufted astrocyte). Schematic illustrates how detailed multimodal evaluations of patients with longitudinal clinical, biofluid, and neuroimaging assessments followed to autopsy can improve existing clinical criteria for detection of FTLD-Tau and differentiation from other neurodegenerative diseases and provide tissue validation for biomarkers obtained during life. Autopsy tissues also provide critical source of human-derived pathogenic tau species for use in animal/cell models of disease and therapeutic response to accelerate the development of disease modifying therapies. naPPA non-fluent agrammatic variant of primary progressive aphasia, svPPA semantic variant of primary progressive aphasia
Picks Disease (3R Tauopathy)
Pick’s disease (PiD) is the sole 3R predominant tauopathy [9••]. Neuropathological findings often include severe gross atrophy of the frontotemporal lobes and corresponding tau-positive intracellular inclusions. The morphological features include prominent round tau-positive “Pick bodies” in neurons with often severe neuron loss and diffuse neuropil threads and variable amounts of glial tau pathology in ramified astrocytes and oligodendrocytes [11]. Reactivity to C-terminal truncation epitopes [28] and the amyloid-binding dye, thioflavin-S [29], thought to be markers of mature tau inclusions of AD [30], is present in a subset of PiD tau pathology [11].
Clinically, PiD is most commonly associated with behavioral-variant FTD [31] (bvFTD), a disorder of social cognition previously referred to as Pick’s disease, but can be also seen in patients with clinical corticobasal syndrome [32] (CBS) or variants of primary progressive aphasia [33] (PPA) [34•]. Due to this clinical heterogeneity and high frequency of FTLD-TDP (~50–60%) in bvFTD [34•], current nomenclature reserves the term Pick’s disease for the pathological findings above [9••].
Progressive Supranuclear Palsy (4R Tauopathy)
Progressive supranuclear palsy (PSP) has pathological features of tau-positive glial inclusions in the form of “tufted astrocytes” in gray matter and “coiled-bodies” in oligodendrocytes in white matter, along with neuronal tangles [35]. The most severe pathology is usually seen in subcortical regions including the midbrain, pons, dentate nucleus of the cerebellum, and subthalamic nucleus, where large tau-reactive “globose” tangles may be found. Tau pathology in PSP is near exclusively of the 4R tau isoform type [35] and is reactive to acetylation at K280 [30] but lacks reactivity to mature tau markers including C-terminal truncation epitopes [28] and thioflavin-S [29]. A 900-kb inversion in MAPT has led to two haplotypes of polymorphisms in high linkage disequilibrium, H1 and H2 [36]. The H1 haplotype is a risk factor for PSP, and a recent genome-wide association study (GWAS) of autopsy-confirmed PSP identified several other polymorphisms that may increase risk of PSP tauopathy [37].
While the Steele Richardson Olszewski syndrome is the most recognized PSP clinical syndrome (i.e., PSPS) [38], PSP can present initially as pure parkinsonism (PSP-P), CBS, bvFTD, a non-fluent-agrammatic form of PPA (naPPA), pure akinesia with freezing of gait, and other more rare presentations such as cerebellar disorder [39, 40]. PSP-P in particular may be mistaken for idiopathic Parkinson’s disease early on in the course as there can be no clear clinical distinguishing features and at least 20–40% have been reported in certain series to be levodopa responsive [41,42,43]. The variety of clinical presentations in part reflect different distribution of the tau pathology within the brain [12, 44]. Finally, it is not uncommon for these clinical syndromes to overlap during the course of illness, where patients with naPPA language disorder eventually develop cardinal features of PSPS (oculomotor dysfunction and axial rigidity) or PSPS patients developing slow hesitant speech consistent with naPPA.
The NINDS/SPSP clinical criteria [38] requires a progressive a syndrome of supranuclear gaze palsy and slowed vertical saccades with falls within the first year to make a diagnosis of probable PSPS. These criteria are highly specific for PSP tauopathy but often lack sensitivity and over-represent the Richardson phenotype [45]. As such, updated clinical criteria for PSPS were developed in 2016 to expand the detection of PSP pathology in the context of these other clinical presentations and improve sensitivity [46]. These resultant criteria provide three levels of certainty based on the strength of association of four main classes of clinical features predictive of PSP tauopathy from large autopsy series [47], which allow for identifying patients with high specificity for clinical trials or increased sensitivity for use in epidemiological studies or efforts for early detection [46].
Corticobasal Degeneration (4R Tauopathy)
The main neuropathological findings of corticobasal degeneration (CBD) include diffuse tau-positive threads that are glial in origin and resemble plaques (i.e., astrocytic plaques) along with often severe white matter coiled bodies and threads, tau-positive ballooned neurons, and neuronal tangles [35]. Severe pathology is often in peri-rolandic cortical regions and subcortical structures in the basal ganglia and brainstem [30]. CBD tauopathy does not react to thioflavin-S [29] or C-terminal truncation antibodies [28] but is acetylated at lysine 280 [30]. Interestingly, CBD shares several genetic risk factors, including the H1 MAPT haplotype, with PSP [48] suggesting shared mechanisms of disease.
CBD is most commonly associated with an asymmetric frontoparietal syndrome often with lateralized extrapyramidal symptoms (i.e., CBS); however, clinical CBS is only shown to have underlying CBD tauopathy in about 50% of cases, while other neurodegenerative diseases associated with this syndrome include AD, PSP, and FTLD-TDP [49,50,51]. As such, the term CBD is now used to refer to this specific 4R tauopathy, while CBS distinguishes the clinical syndrome associated with this varied pathology. Clinical criteria for CBS have been developed to improve the diagnostic accuracy for CBD tauopathy [32], but initial replication suggests poor specify and sensitivity [52]. Ongoing replication and refinement of criteria together with emerging biomarkers of tauopathy will improve diagnostic accuracy for CBD and other tauopathies (Fig. 1).
Other Tauopathies
Less common tauopathies include other 4R tau predominant findings of argyrophilic grain-like inclusions largely constrained to limbic regions (i.e., argyrophilic grain disease, AGD) [53], globular glial tau inclusions (GGT) [54], and aging-related tau astrogliopathy (ARTAG) [55]. GGT has been described in rare cases of clinical FTD, sometimes with concurrent motor neuron disease, while AGD and ARTAG may be found in cognitively normal aged individuals and the clinical significance is currently unclear. AGD with neocortical involvement can be associated with neuropsychiatric or FTD symptoms.
Biomarkers for Tauopathies
There is currently no established clinical test that can reliably identify FTLD-Tau antemortem and autopsy-confirmed studies are rare. Due to the complex clinicopathological associations of FTLD-Tau pathology, study of living patients with PSPS provides an opportunity for biomarker development due to the high predictive value for underlying tauopathy which can be further validated in other forms of tauopathy confirmed at autopsy (Fig. 1).
Structural Neuroimaging
Neuroimaging techniques using structural magnetic resonance imaging (MRI) of gray matter and diffusion tensor imaging (DTI) of white matter within the context of autopsy-confirmed clinical FTD find some regional differences between subtypes of FTLD-Tau and FTLD-TDP [56]. In one study, diagnostic accuracy to differentiate FTLD-Tau from FTLD-TDP using DTI measurements of cortical white matter degeneration showed high diagnostic accuracy validated by post-mortem measure of white matter degeneration in these patients [57]. In a series of clinical CBS, anatomic dissociation of gray and white matter pathology was seen between patients with AD and CBD pathology [58], suggesting that MRI/DTI measurements may also be useful to distinguish FTLD-Tau from atypical forms of AD. PSP has been well-described to be associated with midbrain atrophy that can be appreciated on standard structural MRI as the “hummingbird sign” [59], “morning glory sign” [60], or “Mickey Mouse sign” [61]. In one study of 48 pathologically confirmed cases of PSP or synucleinopathy, 16/22 (72.7%) of PSP cases were able to be correctly identified by radiologist reviewing conventional MRI, and the presence of a hummingbird sign or morning glory sign was 100% specific but was 68.4% sensitive [62]. A variety of ratios of brainstem structures have been reported to aid in distinguishing PSP from other forms of parkinsonism and from controls; these measures have been associated with a range of sensitivity and specificity [63–68].
Molecular Imaging
Several radioligands specific for tau pathology have been recently developed [69,64,71] to detect and track progression of tau pathology in living patients. [18F]AV1451 has most extensively studied and there is a strong signal associated with AD tauopathy that recapitulates Braak tangle staging [72]; however, autoradiographic studies suggest that there may be mild or negligible binding to FTLD-Tau [73, 74]. As aforementioned, tau pathology in AD and FTLD-Tau have different biochemical and conformational properties which could contribute. Some studies have shown the ability to discriminate PSPS patients from controls and from patients with AD [26, 75, 76]; however, evidence for potential off-target binding in melanin-containing cells has been described in regions susceptible to PSP tauopathy [74] (i.e., substantia nigra, basal ganglia) which could influence interpretation. Emerging autopsy studies provide good correlation with topography of FTLD-Tau pathology post-mortem and antemortem [18F]AV1451 signal [26, 77], suggesting potential utility in FTLD-Tau but further study with tissue validation for this and other tracers is needed.
Biofluid
Cerebrospinal fluid (CSF) analysis may be another avenue for biomarker development in tauopathies. The largest body of data for CSF biomarkers exists for AD-related measures of total and phosphorylated forms of tau (t-tau, p-tau) and amyloid-beta (Aβ1–42) protein. The AD CSF signature of elevated CSF tau and decreased Aβ1–42 can differentiate AD from controls [78] and may help distinguish atypical forms of AD pathology associated with clinical FTD from those with underlying FTLD-Tau pathology [79]. Further, CSF p-tau levels directly correlate with the burden of post-mortem tau pathology in FTLD [80], and low CSF p-tau levels or the ratio of p-tau to t-tau may accurately distinguish FTLD-TDP from FTLD-Tau [81,76,83]. Measurements of other forms of tau, including specific isoforms or modifications [84,79,86], and novel analytes are an area of study needed to help provide FTLD-Tau specific markers for use in diagnostics and trial endpoints.
Therapeutic Strategies Targeting Pathological Tau
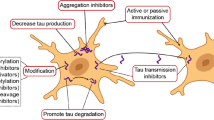
At this time, treatment of tauopathies is largely supportive [87,82,83,84,85,92] and disease modification remains a primary and unmet goal. Symptomatic therapies often consist of off-label uses of medicines focused on specific clinical features (e.g., psychiatric medications for behavioral changes in clinical FTD) but data is lacking [93]. Due to the poor specificity of most clinical diagnoses associated with FTLD-Tau (Fig. 1), current clinical trials focus on PSPS or AD. Previous disease-modulating trials using riluzole and coenzyme q10 in PSP failed to show long-term benefit [94,89,96]. Drug development efforts targeting tau currently focus on several broad strategies including inhibiting tau post-translational modifications and aggregation, immunotherapy, stabilizing microtubules, or reducing overall levels of tau protein synthesis (Table 1).
Tau Phosphorylation, Acetylation, and Aggregation
Under normal physiological conditions tau is phosphorylated at multiple residues [131], but in tauopathies, tau is hyperphosphorylated and phosphorylation at specific residues may contribute to loss of microtubule binding and promotion of aggregation [132]. Glycogen synthase kinase (GSK)-3β and CKD5 have kinase activity for tau and have been studied as potential targets for inhibition [133, 134]. Valproic acid is known to be GSK-3β inhibitor [135, 136], but a trial in PSP showed poor tolerability and failed to meet the primary endpoint [98]. Similarly, lithium is also a GSK-3β inhibitor that decreased tau accumulations in mouse models [137, 138], but a trial in humans was halted because of poor tolerability (NCT00703677) and another in AD trial failed to reduce CSF p-tau in patients after a 10-week course [97]. CDK5 activity can be inhibited by the use of siRNA or thiazolidinediones [102, 129]; however, it is not clear if aberrant CDK5 activity can be selectively reduced without affecting normal activity [139]. Tideglusib is a thiazolidinedione class small molecule with GSK-3β inhibition activity that failed to show a significant change in clinical rating scales in a phase II trial in PSP; however, MRI measurements performed during the trial showed decreased occipital lobe atrophy in patients who received the drug [99, 100]. Tideglusib also failed a phase II trial in AD as well [101].
The tau protein also undergoes several post-translational modifications including acetylation, nitration, O-glc-NAC, and caspase-mediated cleavage which all are potential therapeutic targets [140, 141]. Acetylation at specific residues of tau at lysine 174 has shown to inhibit its degradation [142] and at lysine 280 accelerate fibrillization [143]. The non-steroidal anti-inflammatory compound, salsalate, has inhibitory activity on acetyl-transferase and ameliorated tau pathology in a murine model [144]. A phase I clinical trial for salsalate is in progress for PSPS.
Methylene blue is a compound shown to have anti-aggregant properties for not only tau [145] but also TDP-43 [146], making it an attractive candidate for clinical bvFTD, which has mixed underlying pathology (Fig. 1). The mechanism of action is currently unclear but some evidence suggests that it can oxidize cysteine residues of tau to maintain a monomeric state [147]. Methylene blue-derived compounds have been tested in a phase III trials for both AD and bvFTD but clinical endpoints were not reached.
Microtubule-Stabilizing Agents
Microtubule stabilizing agents have been used in oncology to prevent aberrant cell division in solid tumors and have the potential to abrogate loss of microtubule-binding function in tauopathies. Initial studies in paclitaxel were affective in a tau murine model [148•] but may be limited by side effects from exposure to the peripheral nervous system at dosages that reach the CNS in humans; however, several later studies find related compounds with high blood brain barrier (BBB) permeability can ameliorate tau pathology and restore axonal transport in transgenic mouse models [103, 149,144,151], including the taxane derivative, TPI-287 [105], which is currently in a phase I trial for CBS/PSPS. The protective neuropeptide fragment, davunetide, has microtubule-stabilizing properties, among other potential mechanisms of action, and was recently studied in a large multicenter randomized placebo controlled stage IIb/III trial in over 300 patients with PSP but unfortunately did improve symptoms [104]. Another trial is underway in CBS/PSPS (NCT01056965).
Tau Immunotherapy
Tau immunotherapy has become an interest for therapeutic development due in part to the rapid advances in transmission studies of tauopathy. These data suggest that pathogenic species of tau can be accessible in the extracellular space and thereby more accessible for antibody-mediated degeneration [152•].
Active immunization with full length tau caused an inflammatory reaction in mice [153], but immunization using different types of tau fragments and a number of different adjuvants in mouse models has shown improved safety and efficacy in reducing tau pathology in transgenic animals [106•, 107,102,103,110, 112]. A recent phase I clinical trial using active immunization was recently completed showing favorable safety profile [113]. Passive immunization studies, which circumvent activation of the innate immune system, have also been an area of intense research and find evidence for mild to moderate reduction of tau pathology and improvement in clinical phenotypes in some, but not all studies (for a recent comprehensive review please See [154•]. These studies include administration of monoclonal antibodies targeting a range of potential target epitopes including phospho-serine 396,404 [114•, 115], other phospho-epitopes [116, 117], oligomeric tau [118, 119], pathogenic conformations of tau [26, 115, 120, 121], or antibodies developed from an extracellular seeding assay [122, 123] to murine models of tauopathies.
There are several factors which could contribute to efficacy of tau immunotherapy. Only a fraction of circulating antibody can penetrate the BBB and safety and efficacy of repeated dosing of both passive and active immunization are unclear. Techniques to increase permeability such as focused ultrasound [26] or viral vector delivery [26] in murine models provide proof-of-concept for mechanisms to potentially improve CNS delivery of antibodies. The optimal epitope selection for antibody development is unclear as there are uncertainties in the pathogenic species of tau that is neurotoxic. A disease-specific epitope intuitively would be desirable to avoid degradation of normal soluble tau [26]; however, some data exists for the therapeutic potential for reducing total tau levels in tauopathies. Tau antibodies have the ability to target extracellular or intracellular tau, largely depending on the isoelectric charge of the antibody. In theory, intracellular tau targeting may result in greater efficacy, but could potentially lead to more toxicity than using an acidic, negatively charged antibody capable of only targeting extracellular tau [155]. Presumably, targeting of tau in these separate compartments would stimulate clearance by both external microglia and internal lysosomal/endosomal pathway. Lastly, the optimal affinity of antibodies to promote tau clearance is uncertain. It is possible that high-affinity antibodies could help bind smaller tau aggregates but high affinity binding could also inhibit degradation or even promote aggregation [156].
Two current human studies using an active immunization approach include one phase II trial by Axon Pharmaceuticals SE using tau fragment tau294–305 linked to keyhole limpet hemocyanin (KLH) with an alum adjuvant in patients with mild to moderate AD [111] (NCT02579252) and a phase I trial by AC Immune and Janssen using the phospho-serine 396,404 epitope with a liposomal adjuvant [110]. Passive immunization with a humanized monoclonal antibody targeting a disease specific phospho-epitope [116] was evaluated in a phase I study in 2015 but this was discontinued (NCT02281786). Passive immunization strategies currently in trials include a humanized monoclonal antibody specific for N-terminal extracellular fragments of tau in a phase II trial in PSPS patients (NCT02460094), a phase I trial in PSPS using a humanized antibody targeting extracellular tau aggregates [152•] (NCT02494024) and a phase I study of a tau-specific antibody thought to induce limited microglial activation in healthy controls (NCT02820896) (Table 1).
Gene Therapy
Reducing levels of tau may be of therapeutic benefit by reducing toxic gain-of-function. Tau knockout mice have been reported to have a largely preserved function by several groups [157, 158], but others have reported a variety of symptoms including motor deficits and weakness [159], impaired contextual and cued fear in conditioning tasks [160], parkinsonism, and cognitive impairment [159, 161, 184]. Thus, the overall safety of long-term tau suppression is currently unclear but several preclinical studies suggest that this strategy can reduce tau-mediated neurodegeneration. Reducing tau levels can potentially be accomplished by inhibiting translation through the use of small interfering RNA fragments (siRNA) or antisense oligonucleotides (ASOs). Indeed, under normal conditions, microRNA species regulate tau translation through binding to the 3′ untranslated region of tau mRNA [162]. SiRNAs are being studied in vitro and in vivo in tau transgenic mice [130]. ASOs can be created that induce the destruction of the bound mRNA by recruiting RNAseH1 or that bind mRNA without causing it to be digested. Of these non-degrading ASOs, the total protein product can be decreased by preventing the 5′ cap from forming [163] or by inducing alternative splicing if directed towards the appropriate splice site [164]. Reducing the total tau protein has been beneficial in transgenic mice overexpressing amyloid-beta [124•–127]. In tauopathies, inducing alternative splicing with ASOs may be useful to decrease the amount of 4R tau in favor of 3R tau or vice versa as appropriate for specific diseases, and in vitro experiments have been carried out to this effect [128]. Drug delivery of these compounds continues to be a challenge [165]. Intrathecal injection and intraventricular injection have been used previously in other neurodegenerative diseases [166,161,162,169]. Tagging ASOs or siRNAs to lipid-based [170] and non-lipid [171, 172]-based vectors can aid in trafficking across the BBB. Viral vectors may be used as well for siRNA delivery, which have the advantage of being able to directly target the nucleus, and such an approach has been used in animal models of Huntington’s disease and amyotrophic lateral sclerosis [173,168,175]. Intraparenchymal injections have been utilized in rat and non-human primate models of Huntington’s disease to delivery these viral vectors [176, 177]. Other strategies to transiently increase BBB permeability have been investigated as well including a variety of different compounds and most recently focused ultrasound [178,173,174,175,182].
Conclusion
Tauopathies are diverse clinicopathological entities that often require coordinated effort between cognitive and movement disorder specialists for accurate diagnosis and effective supportive care. One major obstacle for therapeutic development in tauopathies is the lack of an accurate biomarker to identify tauopathy and track disease progression. Indeed, current clinical trial outcomes largely rely on subjective cognitive or motor functional scales due to the lack of a validated prognostic marker. The high specificity of clinical PSPS and AD for tauopathy makes these patient populations eligible for many emerging biomarker and clinical trials targeting tau, while most other patients cannot currently participate due to the inability to accurately differentiate FTLD-Tau from FTLD-TDP associated with clinical bvFTD, PPA, and CBS. A rapid growth in recent basic science research on the mechanisms of tauopathy provides several avenues for potential therapeutic development of disease-modifying therapies. Coordinated efforts among patients, clinicians, and basic scientists in prospective natural history studies (Fig. 1), such as those currently ongoing in the USA (NCT02365922, NCT02372773, NCT02966145) and Europe [183], along with tissue validation are needed to improve diagnostics and accelerate the development of therapeutics in tauopathies.
References
Papers of particular interest, published recently, have been highlighted as: • of importance •• of major importance
Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9(13):4225.
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11.
•• Lee VM, Balin BJ, Otvos L Jr, Trojanowski JQ. A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science. 1991;251(4994):675–8. This is the first description of tau being the major constituent of tangle pathology in Alzheimer’s disease
Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81.
Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathologica. 2014.
Duyckaerts C, Braak H, Brion JP, Buee L, Del Tredici K, Goedert M, et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015;129(5):749–56.
Irwin DJ, Lee VM, Trojanowski JQ. Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat rev Neurosc 2013.
Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, et al. Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. The Lancet Neurology. 2017;16(1):55–65.
•• Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol. 2010;119(1):1–4. Current neuropathological criteria for FTLD-Tau
Boxer AL, Gold M, Huey E, Hu WT, Rosen H, Kramer J, et al. The advantages of frontotemporal degeneration drug development (part 2 of frontotemporal degeneration: the next therapeutic frontier). Alzheimer's & Dementia: the Journal of the Alzheimer's Association. 2012.
Irwin DJ, Brettschneider J, McMillan CT, Cooper F, Olm C, Arnold SE, et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol. 2016;79(2):272–87.
Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ, et al. Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain : a journal of neurology. 2007;130(Pt 6):1566–76.
Forman M, Trojanoswki JQ, Lee VM-Y. In: Esiri M, Lee VM-Y, JQ T, editors. Hereditary tauopathies and idiopathic frontotemporal dementias. 2nd ed. Cambridge: Cambridge University Press; 2004.
• Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–13. Novel in vivo data for transmission hypothesis of tau in murine model
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2013;33(3):1024–37.
Guo JL, Lee VM. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett. 2013;587(6):717–23.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–59.
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110(23):9535–40.
Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129(2):221–37.
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82(6):1271–88.
Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. 2016;92(4):796–812.
Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218(4579):1309–11.
Irwin DJ, Abrams JY, Schonberger LB, Leschek EW, Mills JL, Lee VM, et al. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA neurology. 2013;70(4):462–8.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–51.
• Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med. 2014;20(2):130–8. Comprehensive review of transmission studies in neurodegenerative disease
Passamonti L, Vazquez Rodriguez P, Hong YT, Allinson KS, Williamson D, Borchert RJ, et al. 18F-AV-1451 positron emission tomography in Alzheimer’s disease and progressive supranuclear palsy. Brain : a journal of neurology. 2017;140(3):781–91.
Geschwind MD, Shu H, Haman A, Sejvar JJ, Miller BL. Rapidly progressive dementia. Ann Neurol. 2008;64(1):97–108.
Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer’s disease. Biochim Biophys Acta. 2005;1739(2–3):216–23.
Schmidt ML, Schuck T, Sheridan S, Kung MP, Kung H, Zhuang ZP, et al. The fluorescent Congo red derivative, (trans, trans)-1-bromo-2,5-bis-(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (BSB), labels diverse beta-pleated sheet structures in postmortem human neurodegenerative disease brains. Am J Pathol. 2001;159(3):937–43.
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, Xie SX, Lee VM, et al. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain: a Journal of Neurology. 2012;135(Pt 3):807–18.
Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain: a Journal of Neurology. 2011;134(Pt 9):2456–77.
Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503.
Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–14.
• Irwin DJ, Cairns NJ, Grossman M, McMillan CT, Lee EB, Van Deerlin VM, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2015;129(4):469–91. Comprehensive review of clinicopathological correlations in FTLD
Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-Tau). Journal of molecular neuroscience : MN. 2011;45(3):384–9.
Wolfe MS. The role of tau in neurodegenerative diseases and its potential as a therapeutic target. Scientifica. 2012;2012.
Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43(7):699–705.
Litvan I, Agid Y, Calne D, Campbell G, Dubois B, Duvoisin RC, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9.
Respondek G, Stamelou M, Kurz C, Ferguson LW, Rajput A, Chiu WZ, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Movement disorders: Official Journal of the Movement Disorder Society. 2014;29(14):1758–66.
Lopez G, Bayulkem K, Hallett M. Progressive supranuclear palsy (PSP): Richardson syndrome and other PSP variants. Acta Neurol Scand. 2016;134(4):242–9.
Hughes AJ, Ben-Shlomo Y, Daniel SE, Lees AJ. What features improve the accuracy of clinical diagnosis in Parkinson’s disease: a clinicopathologic study. Neurology. 1992;42(6):1142–6.
Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain: a Journal of Neurology. 2002;125(Pt 4):861–70.
Adler CH, Beach TG, Hentz JG, Shill HA, Caviness JN, Driver-Dunckley E, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology. 2014;83(5):406–12.
Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA. Neuropathology of variants of progressive supranuclear palsy. Curr Opin Neurol. 2010;23(4):394–400.
Respondek G, Roeber S, Kretzschmar H, Troakes C, Al-Sarraj S, Gelpi E, et al. Accuracy of the National Institute for Neurological Disorders and Stroke/Society for Progressive Supranuclear Palsy and Neuroprotection and Natural History in Parkinson Plus Syndromes criteria for the diagnosis of progressive supranuclear palsy. Movement disorders: Official Journal of the Movement Disorder Society. 2013;28(4):504–9.
Hoglinger GU, Respondek G, Stamelou M, Kurz C, Josephs KA, Lang AE, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Movement Disorders: Official Journal of the Movement Disorder Society. 2017.
Respondek G, Kurz C, Arzberger T, Compta Y, Englund E, Ferguson LW, et al. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Movement disorders: Official Journal of the Movement Disorder Society. 2017.
Kouri N, Ross OA, Dombroski B, Younkin CS, Serie DJ, Soto-Ortolaza A, et al. Genome-wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 2015;6:7247.
Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol. 2011;70(2):327–40.
Litvan I, Agid Y, Goetz C, Jankovic J, Wenning GK, Brandel JP, et al. Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology. 1997;48(1):119–25.
Boeve BF. The multiple phenotypes of corticobasal syndrome and corticobasal degeneration: implications for further study. Journal of Molecular Neuroscience: MN. 2011;45(3):350–3.
Alexander SK, Rittman T, Xuereb JH, Bak TH, Hodges JR, Rowe JB. Validation of the new consensus criteria for the diagnosis of corticobasal degeneration. J Neurol Neurosurg Psychiatry. 2014;85(8):925–9.
Ferrer I, Santpere G, van Leeuwen FW. Argyrophilic grain disease. Brain: a Journal of Neurology. 2008;131(Pt 6):1416–32.
Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol. 2013;126(4):537–44.
Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol. 2016;131(1):87–102.
Whitwell JL, Josephs KA. Neuroimaging in frontotemporal lobar degeneration—predicting molecular pathology. Nat Rev Neurol. 2011;8(3):131–42.
McMillan CT, Irwin DJ, Avants BB, Powers J, Cook PA, Toledo JB, et al. White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2013;84(9):949–55.
McMillan CT, Boyd C, Gross RG, Weinstein J, Firn K, Toledo JB, et al. Multimodal imaging evidence of pathology-mediated disease distribution in corticobasal syndrome. Neurology. 2016;87(12):1227–34.
Kato N, Arai K, Hattori T. Study of the rostral midbrain atrophy in progressive supranuclear palsy. J Neurol Sci. 2003;210(1–2):57–60.
Adachi M, KAWANAMI T, OHSHIMA H, Sugai Y, Hosoya T. Morning glory sign: a particular MR finding in progressive supranuclear palsy. Magn Reson Med Sci. 2004;3(3):125–32.
Massey LA, Micallef C, Paviour DC, O'sullivan SS, Ling H, Williams DR, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord. 2012;27(14):1754–62.
Massey LA, Micallef C, Paviour DC, O'Sullivan SS, Ling H, Williams DR, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Movement Disorders: Official Journal of the Movement Disorder Society. 2012;27(14):1754–62.
Massey LA, Jager HR, Paviour DC, O'Sullivan SS, Ling H, Williams DR, et al. The midbrain to pons ratio: a simple and specific MRI sign of progressive supranuclear palsy. Neurology. 2013;80(20):1856–61.
Quattrone A, Nicoletti G, Messina D, Fera F, Condino F, Pugliese P, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. 2008;246(1):214–21.
Moller L, Kassubek J, Sudmeyer M, Hilker R, Hattingen E, Egger K, et al. Manual MRI morphometry in Parkinsonian syndromes. Movement Disorders: Official Journal of the Movement Disorder Society. 2017;32(5):778–82.
Nigro S, Arabia G, Antonini A, Weis L, Marcante A, Tessitore A, et al. Magnetic resonance parkinsonism index: diagnostic accuracy of a fully automated algorithm in comparison with the manual measurement in a large Italian multicentre study in patients with progressive supranuclear palsy. Eur Radiol. 2017;27(6):2665–75.
Zanigni S, Calandra-Buonaura G, Manners DN, Testa C, Gibertoni D, Evangelisti S, et al. Accuracy of MR markers for differentiating progressive supranuclear palsy from Parkinson's disease. NeuroImage Clinical. 2016;11:736–42.
Hussl A, Mahlknecht P, Scherfler C, Esterhammer R, Schocke M, Poewe W, et al. Diagnostic accuracy of the magnetic resonance parkinsonism index and the midbrain-to-pontine area ratio to differentiate progressive supranuclear palsy from Parkinson’s disease and the Parkinson variant of multiple system atrophy. Movement Disorders: Official Journal of the Movement Disorder Society. 2010;25(14):2444–9.
Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. Journal of Alzheimer’s Disease: JAD. 2013;34(2):457–68.
Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79(6):1094–108.
Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, et al. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain: a Journal of Neurology. 2011;134(Pt 4):1089–100.
Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol. 2016;79(1):110–9.
Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, et al. An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun. 2016;4(1):58.
Marquie M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78(5):787–800.
Cho H, Choi JY, Hwang MS, Lee SH, Ryu YH, Lee MS, et al. Subcortical 18 F-AV-1451 binding patterns in progressive supranuclear palsy. Movement Disorders: Official Journal of the Movement Disorder Society. 2017;32(1):134–40.
Smith R, Schain M, Nilsson C, Strandberg O, Olsson T, Hagerstrom D, et al. Increased basal ganglia binding of 18 F-AV-1451 in patients with progressive supranuclear palsy. Movement Disorders: Official Journal of the Movement Disorder Society. 2017;32(1):108–14.
McMillan CT, Irwin DJ, Nasrallah I, Phillips JS, Spindler M, Rascovsky K, et al. Multimodal evaluation demonstrates in vivo 18F-AV-1451 uptake in autopsy-confirmed corticobasal degeneration. Acta Neuropathol. 2016;132(6):935–7.
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403–13.
Irwin DJ, McMillan CT, Toledo JB, Arnold SE, Shaw LM, Wang LS, et al. Comparison of cerebrospinal fluid levels of tau and Abeta 1-42 in Alzheimer disease and frontotemporal degeneration using 2 analytical platforms. Arch Neurol. 2012;69(8):1018–25.
Irwin Dea. Ante mortem CSF tau levels correlate with post mortem tau pathology in FTLD. Ann Neurol. 2017; in press. doi:10.1002/ana.24996.
Grossman M, Elman L, McCluskey L, McMillan CT, Boller A, Powers J, et al. Phosphorylated tau as a candidate biomarker for amyotrophic lateral sclerosis. JAMA neurology. 2014;71(4):442–8.
Hu WT, Watts K, Grossman M, Glass J, Lah JJ, Hales C, et al. Reduced CSF p-Tau181 to Tau ratio is a biomarker for FTLD-TDP. Neurology. 2013;in press.
Borroni B, Benussi A, Archetti S, Galimberti D, Parnetti L, Nacmias B, et al. Csf p-tau181/tau ratio as biomarker for TDP pathology in frontotemporal dementia. Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration. 2015;16(1–2):86–91.
Borroni B, Malinverno M, Gardoni F, Alberici A, Parnetti L, Premi E, et al. Tau forms in CSF as a reliable biomarker for progressive supranuclear palsy. Neurology. 2008;71(22):1796–803.
Saijo E, Ghetti B, Zanusso G, Oblak A, Furman JL, Diamond MI, et al. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017;133(5):751–65.
Barthelemy NR, Gabelle A, Hirtz C, Fenaille F, Sergeant N, Schraen-Maschke S, et al. Differential mass spectrometry profiles of tau protein in the cerebrospinal fluid of patients with Alzheimer's disease, progressive supranuclear palsy, and dementia with Lewy bodies. Journal of Alzheimer's Disease: JAD. 2016;51(4):1033–43.
Fabbrini G, Barbanti P, Bonifati V, Colosimo C, Gasparini M, Vanacore N, et al. Donepezil in the treatment of progressive supranuclear palsy. Acta Neurol Scand. 2001;103(2):123–5.
Lepore V, Defazio G, Acquistapace D, Melpignano C, Pomes L, Lamberti P, et al. Botulinum A toxin for the so-called apraxia of lid opening. Movement Disorders: Official Journal Of the Movement Disorder Society. 1995;10(4):525–6.
Liepelt I, Gaenslen A, Godau J, Di Santo A, Schweitzer KJ, Gasser T, et al. Rivastigmine for the treatment of dementia in patients with progressive supranuclear palsy: clinical observations as a basis for power calculations and safety analysis. Alzheimer’s & Dementia: The Journal of the Alzheimer's Association. 2010;6(1):70–4.
Litvan I, Phipps M, Pharr VL, Hallett M, Grafman J, Salazar A. Randomized placebo-controlled trial of donepezil in patients with progressive supranuclear palsy. Neurology. 2001;57(3):467–73.
Nieforth KA, Golbe LI. Retrospective study of drug response in 87 patients with progressive supranuclear palsy. Clin Neuropharmacol. 1993;16(4):338–46.
Polo KB, Jabbari B. Botulinum toxin-A improves the rigidity of progressive supranuclear palsy. Ann Neurol. 1994;35(2):237–9.
Huey ED, Putnam KT, Grafman J. A systematic review of neurotransmitter deficits and treatments in frontotemporal dementia. Neurology. 2006;66(1):17–22.
Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain: a journal of Neurology. 2009;132(Pt 1):156–71.
Stamelou M, Reuss A, Pilatus U, Magerkurth J, Niklowitz P, Eggert KM, et al. Short-term effects of coenzyme Q10 in progressive supranuclear palsy: a randomized, placebo-controlled trial. Movement Disorders: Official Journal of the Movement Disorder Society. 2008;23(7):942–9.
Apetauerova D, Scala SA, Hamill RW, Simon DK, Pathak S, Ruthazer R, et al. CoQ10 in progressive supranuclear palsy: a randomized, placebo-controlled, double-blind trial. Neurology(R) neuroimmunology & neuroinflammation. 2016;3(5):e266.
Hampel H, Ewers M, Burger K, Annas P, Mortberg A, Bogstedt A, et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. The Journal of Clinical Psychiatry. 2009;70(6):922–31.
Leclair-Visonneau L, Rouaud T, Debilly B, Durif F, Houeto JL, Kreisler A, et al. Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clin Neurol Neurosurg. 2016;146:35–9.
Hoglinger GU, Huppertz HJ, Wagenpfeil S, Andres MV, Belloch V, Leon T, et al. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Movement Disorders: Official Journal of the Movement Disorder Society. 2014;29(4):479–87.
Tolosa E, Litvan I, Hoglinger GU, Burn D, Lees A, Andres MV, et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Movement Disorders: Official Journal of the Movement Disorder Society. 2014;29(4):470–8.
Lovestone S, Boada M, Dubois B, Hull M, Rinne JO, Huppertz HJ, et al. A phase II trial of tideglusib in Alzheimer’s disease. Journal of Alzheimer's Disease: JAD. 2015;45(1):75–88.
Cho DH, Lee EJ, Kwon KJ, Shin CY, Song KH, Park JH, et al. Troglitazone, a thiazolidinedione, decreases tau phosphorylation through the inhibition of cyclin-dependent kinase 5 activity in SH-SY5Y neuroblastoma cells and primary neurons. J Neurochem. 2013;126(5):685–95.
Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2012;32(11):3601–11.
Boxer AL, Lang AE, Grossman M, Knopman DS, Miller BL, Schneider LS, et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. The Lancet Neurology. 2014;13(7):676–85.
Fitzgerald DP, Emerson DL, Qian Y, Anwar T, Liewehr DJ, Steinberg SM, et al. TPI-287, a new taxane family member, reduces the brain metastatic colonization of breast cancer cells. Mol Cancer Ther. 2012;11(9):1959–67.
• Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2007;27(34):9115–29. First preclinical data showing efficacy for active immunization study using tau fragments
Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010;224(2):472–85.
Bi M, Ittner A, Ke YD, Gotz J, Ittner LM. Tau-targeted immunization impedes progression of neurofibrillary histopathology in aged P301L tau transgenic mice. PLoS One. 2011;6(12):e26860.
Rozenstein-Tsalkovich L, Grigoriadis N, Lourbopoulos A, Nousiopoulou E, Kassis I, Abramsky O, et al. Repeated immunization of mice with phosphorylated-tau peptides causes neuroinflammation. Exp Neurol. 2013;248:451–6.
Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, Lopez-Deber MP, Reis P, et al. Efficacy and safety of a liposome-based vaccine against protein tau, assessed in tau.P301L mice that model tauopathy. PLoS One. 2013;8(8):e72301.
Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer's disease model. Alzheimers Res Ther. 2014;6(4):44.
Selenica ML, Davtyan H, Housley SB, Blair LJ, Gillies A, Nordhues BA, et al. Epitope analysis following active immunization with tau proteins reveals immunogens implicated in tau pathogenesis. J Neuroinflammation. 2014;11:152.
Novak P, Schmidt R, Kontsekova E, Zilka N, Kovacech B, Skrabana R, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16(2):123–34.
• Boutajangout A, Ingadottir J, Davies P, Sigurdsson EM. Passive immunization targeting pathological phospho-tau protein in a mouse model reduces functional decline and clears tau aggregates from the brain. J Neurochem. 2011;118(4):658–67. First report of preclinical data for passive tau immunotherapy in murine model of tauopathy
Chai X, Wu S, Murray TK, Kinley R, Cella CV, Sims H, et al. Passive immunization with anti-tau antibodies in two transgenic models: reduction of tau pathology and delay of disease progression. J Biol Chem. 2011;286(39):34457–67.
Collin L, Bohrmann B, Göpfert U, Oroszlan-Szovik K, Ozmen L, Grüninger F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer‘s disease. Brain : a journal of neurology. 2014;137(10):2834–46.
Walls KC, Ager RR, Vasilevko V, Cheng D, Medeiros R, LaFerla FM. p-Tau immunotherapy reduces soluble and insoluble tau in aged 3xTg-AD mice. Neurosci Lett. 2014;575:96–100.
Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700.
Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Kayed R. Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. 2014;40(s1):S97–S111.
Ittner A, Bertz J, Suh LS, Stevens CH, Gotz J, Ittner LM. Tau-targeting passive immunization modulates aspects of pathology in tau transgenic mice. J Neurochem. 2015;132(1):135–45.
Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, Cadelina G, et al. Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS One. 2015;10(5):e0125614.
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80(2):402–14.
Yanamandra K, Jiang H, Mahan TE, Maloney SE, Wozniak DF, Diamond MI, et al. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Annals of Clinical and Translational Neurology. 2015;2(3):278–88.
• Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid ß-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–4. Novel data suggesting reducing tau expression could be a therapeutic strategy for AD
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer's disease mouse models. Cell. 2010;142(3):387–97.
Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31(2):700–11.
Leroy K, Ando K, Laporte V, Dedecker R, Suain V, Authelet M, et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol. 2012;181(6):1928–40.
Peacey E, Rodriguez L, Liu Y, Wolfe MS. Targeting a pre-mRNA structure with bipartite antisense molecules modulates tau alternative splicing. Nucleic acids res. 2012:gks710.
Piedrahita D, Hernandez I, Lopez-Tobon A, Fedorov D, Obara B, Manjunath BS, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer's mice. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2010;30(42):13966–76.
Xu H, Rosler TW, Carlsson T, de Andrade A, Fiala O, Hollerhage M, et al. Tau silencing by siRNA in the P301S mouse model of tauopathy. Current Gene Therapy. 2014;14(5):343–51.
Matsuo ES, Shin RW, Billingsley ML, Van de Voorde A, O'Connor M, Trojanowski JQ, et al. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron. 1994;13(4):989–1002.
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33(1):95–130.
Ferrer I, Barrachina M, Puig B. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2002;104(6):583–91.
Patrick GN, Zukerberg L, Nikolic M, de La Monte S, Dikkes P, Tsai L-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402(6762):615–22.
Long ZM, Zhao L, Jiang R, Wang KJ, Luo SF, Zheng M, et al. Valproic acid modifies synaptic structure and accelerates Neurite outgrowth via the glycogen synthase kinase-3beta signaling pathway in an Alzheimer’s disease model. CNS Neuroscience & Therapeutics. 2015;21(11):887–97.
Xuan AG, Pan XB, Wei P, Ji WD, Zhang WJ, Liu JH, et al. Valproic acid alleviates memory deficits and attenuates amyloid-beta deposition in transgenic mouse model of Alzheimer’s disease. Mol Neurobiol. 2015;51(1):300–12.
Nakashima H, Ishihara T, Suguimoto P, Yokota O, Oshima E, Kugo A, et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 2005;110(6):547–56.
Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A. 2005;102(19):6990–5.
Sundaram JR, Poore CP, Sulaimee NH, Pareek T, Asad AB, Rajkumar R, et al. Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2013;33(1):334–43.
Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, et al. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8(4):393–9.
Wang AC, Jensen EH, Rexach JE, Vinters HV, Hsieh-Wilson LC. Loss of O-GlcNAc glycosylation in forebrain excitatory neurons induces neurodegeneration. Proc Natl Acad Sci U S A. 2016;113(52):15120–5.
Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67(6):953–66.
Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252.
Bachanova V, Burns LJ, Ahn KW, Laport GG, Akpek G, Kharfan-Dabaja MA, et al. Impact of pretransplantation (18)F-fluorodeoxy glucose-positron emission tomography status on outcomes after allogeneic hematopoietic cell transplantation for non-Hodgkin lymphoma. Biol Blood Marrow Transplant. 2015;21(9):1605–11.
Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996;93(20):11213–8.
Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, et al. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology: Official Journal of the Japanese Society of Neuropathology. 2010;30(2):170–81.
Crowe A, James MJ, Lee VM, Smith AB 3rd, Trojanowski JQ, Ballatore C, et al. Aminothienopyridazines and methylene blue affect tau fibrillization via cysteine oxidation. J Biol Chem. 2013;288(16):11024–37.
• Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, et al. Microtubule-binding drugs offset tau sequestration by stabilizing microtubules and reversing fast axonal transport deficits in a tauopathy model. Proc Natl Acad Sci U S A. 2005;102(1):227–31. Detailed preclinical study of microtubule stabalizing agents in murin model of tauopathy
Magen I, Ostritsky R, Richter F, Zhu C, Fleming SM, Lemesre V, et al. Intranasal NAP (davunetide) decreases tau hyperphosphorylation and moderately improves behavioral deficits in mice overexpressing alpha-synuclein. Pharmacol Res Perspect. 2014;2(5):e00065.
Barten DM, Fanara P, Andorfer C, Hoque N, Wong PY, Husted KH, et al. Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2012;32(21):7137–45.
Brunden KR, Gardner NM, James MJ, Yao Y, Trojanowski JQ, Lee VM, et al. MT-stabilizer, dictyostatin, exhibits prolonged brain retention and activity: potential therapeutic implications. ACS Med Chem Lett. 2013;4(9):886–9.
• Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of tau aggregation by fibrillar species. J Biol Chem. 2012;287(23):19440–51. Novel data demonstrating propogation of tau aggregations between cells which is blocked by tau-specific antibodies
Rosenmann H, Grigoriadis N, Karussis D, Boimel M, Touloumi O, Ovadia H, et al. Tauopathy-like abnormalities and neurologic deficits in mice immunized with neuronal tau protein. Arch Neurol. 2006;63(10):1459–67.
• Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer’s disease. Trends Mol Med. 2015;21(6):394–402. Comprehensive review of epitopes studied for tau directed passive immunotherapies
Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K. Engineering the variable region of therapeutic IgG antibodies. MAbs. 2011;3(3):243–52.
Sigurdsson EM. Tau immunotherapy. Neurodegener Dis. 2016;16(1–2):34–8.
Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from τ deficient mice. J Cell Sci. 2001;114(6):1179–87.
Morris M, Hamto P, Adame A, Devidze N, Masliah E, Mucke L. Age-appropriate cognition and subtle dopamine-independent motor deficits in aged tau knockout mice. Neurobiol Aging. 2013;34(6):1523–9.
Lei P, Ayton S, Moon S, Zhang Q, Volitakis I, Finkelstein DI, et al. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol Neurodegener. 2014;9:29.
Ahmed T, Van der Jeugd A, Blum D, Galas M-C, D’Hooge R, Buee L, et al. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol Aging. 2014;35(11):2474–8.
Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med. 2012;18(2):291–5.
Santa-Maria I, Alaniz ME, Renwick N, Cela C, Fulga TA, Van Vactor D, et al. Dysregulation of microRNA-219 promotes neurodegeneration through post-transcriptional regulation of tau. J Clin Invest. 2015;125(2):681–6.
DeVos SL, Miller TM. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics. 2013;10(3):486–97.
Sud R, Geller ET, Schellenberg GD. Antisense-mediated exon skipping decreases tau protein expression: a potential therapy for tauopathies. Molecular Therapy-Nucleic Acids. 2014;3:e180.
Smith RA, Miller TM, Yamanaka K, Monia BP, Condon TP, Hung G, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest. 2006;116(8):2290–6.
Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. The Lancet Neurology. 2013;12(5):435–42.
Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, et al. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–44.
Finkel RS, Chiriboga CA, Vajsar J, Day JW, Montes J, De Vivo DC, et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet (London, England). 2016;388(10063):3017–26.
DeVos SL, Miller TM. Direct intraventricular delivery of drugs to the rodent central nervous system. JoVE (Journal of Visualized Experiments). 2013(75):e50326-e.
Gomes MJ, Dreier J, Brewer J, Martins S, Brandl M, Sarmento B. A new approach for a blood-brain barrier model based on phospholipid vesicles: membrane development and siRNA-loaded nanoparticles permeability. J Membr Sci. 2016;503:8–15.
Shen H, Sun T, Ferrari M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012;19(6):367–73.
Wohlfart S, Gelperina S, Kreuter J. Transport of drugs across the blood–brain barrier by nanoparticles. J Control Release. 2012;161(2):264–73.
Morris K, Rossi J. Lentiviral-mediated delivery of siRNAs for antiviral therapy. Gene Ther. 2006;13(6):553–8.
Franich NR, Fitzsimons HL, Fong DM, Klugmann M, During MJ, Young D. AAV vector–mediated RNAi of mutant Huntingtin expression is neuroprotective in a novel genetic rat model of Huntington's disease. Mol Ther. 2008;16(5):947–56.
Raoul C, Abbas-Terki T, Bensadoun J-C, Guillot S, Haase G, Szulc J, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11(4):423–8.
Ceccarelli I, Fiengo P, Remelli R, Miragliotta V, Rossini L, Biotti I, et al. Recombinant adeno associated viral (AAV) vector type 9 delivery of Ex1-Q138-mutant Huntingtin in the rat striatum as a short-time model for in vivo studies in drug discovery. Neurobiol Dis. 2016;86:41–51.
Grondin R, Kaytor MD, Ai Y, Nelson PT, Thakker DR, Heisel J, et al. Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain: a Journal of Neurology. 2012;135(Pt 4):1197–209.
Alecou T, Giannakou M, Damianou C. Amyloid-beta plaque reduction with antibodies crossing the blood-brain barrier, which was opened in 3 sessions of focused ultrasound in a rabbit model. Journal of Ultrasound in Medicine: Official Journal of the American Institute of Ultrasound in Medicine. 2017.
Mead BP, Kim N, Miller GW, Hodges D, Mastorakos P, Klibanov AL, et al. Novel focused ultrasound gene therapy approach noninvasively restores dopaminergic neuron function in a rat Parkinson’s disease model. Nano Lett. 2017.
Yin Y, Cao L, Ge H, Duanmu W, Tan L, Yuan J, et al. L-Borneol induces transient opening of the blood-brain barrier and enhances the therapeutic effect of cisplatin. Neuroreport. 2017;28(9):506–13.
Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y, et al. Glutamate-mediated blood-brain barrier opening: implications for neuroprotection and drug delivery. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2016;36(29):7727–39.
Gao X, Qian J, Zheng S, Changyi Y, Zhang J, Ju S, et al. Overcoming the blood-brain barrier for delivering drugs into the brain by using adenosine receptor nanoagonist. ACS Nano. 2014;8(4):3678–89.
Rohrer JD, Nicholas JM, Cash DM, van Swieten J, Dopper E, Jiskoot L, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol. 2015;14(3):253–62.
Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M, et al. Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris Water Maze with aging. The Journal of Neuroscience: the Official Journal of the Society for Neuroscience. 2014;34(21):7124–36.
Acknowledgements
David Coughlin is supported by the Penn Institute for Translational Medicine and Therapeutics and David J. Irwin is supported by NIH grant K23NS088341, Brightfocus Foundation A2016244S and the Penn Institute on Aging. We thank the patients and their families who participated in brain donation and clinical research reviewed in this manuscript for without their time and effort, these advances would not be possible.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
David Coughlin declares that he has no conflict of interest.
David J. Irwin reports other from GE Healthcare.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Dementia
Rights and permissions
About this article

Cite this article
Coughlin, D., Irwin, D.J. Emerging Diagnostic and Therapeutic Strategies for Tauopathies. Curr Neurol Neurosci Rep 17, 72 (2017). https://doi.org/10.1007/s11910-017-0779-1
Published:
DOI: https://doi.org/10.1007/s11910-017-0779-1