
Abstract
Purpose of the Review
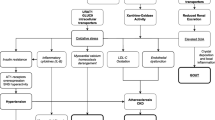
Our review explores the epidemiology, physiology, and clinical data surrounding the connection between hyperuricemia and metabolic syndrome, chronic kidney disease, and hypertension.
Recent Findings
Compelling physiologic mechanisms have been proposed to explain a causal relationship between hyperuricemia and metabolic syndrome, chronic kidney disease, and hypertension but clinical studies have given mixed results in terms of whether intervening with hyperuricemia using urate-lowering therapy has any beneficial effects for patients with these conditions.
Summary
Despite the large amount of research already put into this topic, more randomized placebo-controlled trials are needed to more firmly establish whether a cause-effect relationship exists and whether lowering uric acid levels in patients with these conditions is beneficial.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Uric acid in humans is the product of the purine degradation process in which an enzyme, xanthine oxidase, converts hypoxanthine and xanthine to poorly soluble uric acid. In contrast to humans, other mammals are further powered with an enzyme, urate oxidase, which converts uric acid to allantoin, which is water soluble, thus allowing for efficient urinary excretion. During early evolution, humans lost the activity of the urate oxidase enzyme due to selected genetic mutations, resulting in higher uric acid levels than in other mammals [1].
The kidneys eliminate approximately 70% of the daily uric acid load, while 30% is eliminated by bacterial intestinal uricolysis. In the human kidney, uric acid is freely filtered at the glomerulus; however, the proximal convoluted tubules are highly selective for uric acid. The urate transporter, URAT1, plays a key role in uric acid homeostasis resulting in < 10% of fractional excretion of filtered uric acid [2]. In most patients suffering from primary hyperuricemia and gout, the mechanisms of maintaining uric acid homeostasis are severely affected by the failing kidneys, resulting in a decline in fractional excretion of uric acid. As the diseased kidneys continue to worsen, the imbalance between daily production and ingestion of uric acid and decreased excretion can result in uric acid deposition in joints and soft tissues; these insoluble deposits are referred to as tophi [3].
An elevated plasma uric acid concentration has been described as a beneficial phenomenon acting as an antioxidant [4]. However, uric acid exerts its antioxidant properties only in the extracellular environment. Once uric acid enters the cell, including the vascular smooth muscle cells and adipocytes, it has harmful effects on cellular function [5, 6]. Moreover, uric acid has an inhibitory effect on nitric oxide production [7], induction of platelet aggregation, and pro-inflammatory activity [8, 9].
Several studies and clinical trials have proposed an intriguing relationship between hyperuricemia and the development of metabolic syndrome [10], diabetes [11], hypertension [12], chronic kidney disease, [13] and cardiovascular disorders [14]. Hyperuricemia has also been linked to both pre-eclampsia [15] and gestational diabetes [16]. In contrast, in the hemodialysis population, hyperuricemia has been associated with good nutritional status, overall lower rates of hospitalization, and lower all-cause and cardiovascular mortality [17]. However, the relationship between hyperuricemia and graft survival in kidney transplant recipients is complex and still controversial [18].
This review will focus on the literature examining the role of hyperuricemia in the progression of metabolic syndrome, diabetes mellitus, chronic kidney disease, including end-stage renal disease, the post-transplant population, and hypertension.
Hyperuricemia in the Metabolic Syndrome and Diabetes Mellitus
Metabolic syndrome is defined as a cluster of different cardiovascular risk factors, including hypertension, dyslipidemia, visceral obesity, glucose intolerance, and hyperinsulinemia. Hyperuricemia is not considered to be a part of the metabolic syndrome despite several epidemiologic studies confirming a direct relationship in children, adolescents, and adults [19,20,21].
A set of rat model experiments by Nakagawa et al. [22] answered important questions regarding a high fructose diet and the causal role of hyperuricemia in the context of insulin-resistant states. They concluded that rats fed a 60% fructose diet for 10 weeks developed features of metabolic syndrome, including hyperinsulinemia, hypertriglyceridemia, and hyperuricemia. When treated with uric acid-lowering agents such as allopurinol (a xanthine oxidase inhibitor) or benzbromarone (a uricosuric agent), these features were prevented or even reversed.
They also demonstrated that administering allopurinol prophylactically prevented hyperinsulinemia and weight gain and lowered systolic blood pressure (SBP) [22]. Other authors have shown that febuxostat (a more selective xanthine oxidase inhibitor) reversed hyperuricemia and prevented increases in BP, plasma triglycerides, and fasting plasma insulin in rats [23].
Hyperuricemia in metabolic syndrome is caused by the effects of hyperinsulinemia on the proximal tubule, which promotes an increase in uric acid absorption and a decrease in uric acid excretion [19, 24]. Moreover, hyperuricemia can cause hyperinsulinemia due to endothelial dysfunction and inhibition of nitric oxide utilization [25]. In animal studies, the causal role of hyperuricemia in the development of metabolic syndrome in fructose-fed rats could be explained by two main theories. The first, demonstrated in 1990 by Hallfrisch et al., revealed a rapid rise in uric acid when rats were given fructose [26] and activation of fructokinase, resulting in ATP consumption, intracellular phosphate depletion, and stimulation of AMP deaminase. Hyperuricemia also induces a significant increase in insulin resistance via inhibiting endothelial nitric oxide production [27, 28].
The second mechanism relating hyperuricemia to the metabolic syndrome is centered around the effects of hyperuricemia in inducing intracellular reactive oxygen species (ROS). An increase in ROS results in a decrease in nitric oxide bioavailability and further increases in insulin resistance in adipose tissues [5, 29].
Several observational studies used the uric acid level as a future marker for the development of the metabolic syndrome, obesity, and type 2 DM in humans [30,31,32]. Ryu et al. revealed an independent association between serum uric acid levels and increased risk of metabolic syndrome and proposed that uric acid can be one component of the metabolic syndrome [30].
Other investigators have discussed the association between uric acid levels and the risk of developing type 2 diabetes. The population analyzed included 4536 persons with hyperuricemia who were free of diabetes at baseline. During the 10 years of follow-up, 462 participants developed diabetes. The results indicated that one-quarter of diabetes cases can be attributed to a high serum uric acid level [31].
Niskanen et al. studied hyperuricemia and its role in predicting changes in glucose intolerance and the development of type 2 diabetes over 4.1 years. The results showed that hyperuricemia at baseline predicted a twofold increase in the likelihood of developing type 2 diabetes [32]. In a prospective follow-up of 8429 men and 1260 women aged 20–82 years, uric acid was a strong and independent predictor of developing the metabolic syndrome in both men and women [33].
A recent study by Rubio-Guerra et al. found a strong association between hyperuricemia and the metabolic syndrome in low-income young adults [34]. Another large study that analyzed the bidirectional relationship between metabolic syndromes in the Chinese population aged 40 or more years old demonstrated a bidirectional relationship between metabolic syndrome and serum uric acid in both sexes [35].
The intake of sugars such as high fructose corn syrup has increased significantly in the last decades, resulting in a dramatic increase in obesity, fatty liver, metabolic syndrome, and diabetes [36,37,38]. Several studies have also shown that a fructose-rich diet can raise uric acid production and induce components of the metabolic syndrome through mechanisms independent of energy intake or weight gain [39]. However, It is worth noting that these effects were not observed with a glucose-rich diet [40]. Thiazide diuretics, calcineurin inhibitors, gestational diabetes mellitus, and excessive alcohol intake have been also shown to cause hyperuricemia and the metabolic syndrome [41,42,43].
In summary, the worldwide epidemic of the metabolic syndrome and diabetes seen in the last few decades correlates strongly with an increase in total fructose intake and elevation of uric acid levels. Moreover, it is essential to recognize the pathogenesis of how a high fructose diet and hyperuricemia could lead to the development of metabolic syndrome and diabetes. The proposed mechanisms related to these findings are endothelial dysfunction, overproduction of the ROS, and insulin resistance. Administration of allopurinol has been shown to reverse the components of metabolic syndrome. However, more randomized controlled trials are needed to determine appropriate goals of therapy.
Hyperuricemia in Chronic Kidney Disease, End-Stage Kidney Disease, and Post-Kidney Transplant
In the 1890s, Haig et al. proposed that hyperuricemia played an essential role in multiple diseases including chronic kidney disease (CKD) [44]. The connection was made in the context of gouty nephropathy which occurs by the deposition of uric acid crystals into the medullary interstitial. The uric acid crystals induce a chronic inflammatory response potentially leading to interstitial fibrosis and CKD [45]. In a novel mouse model of hyperuricemia and chronic crystalluria, the authors conclude that only hyperuricemia with crystalluria induces progression of chronic kidney disease [46••]. A recent review of the role of urate-lowering therapy concludes that hyperuricemia aggravates renal dysfunction through a direct toxic effect via multiple mechanisms, including an inflammatory response to urate crystallization, activation of the renin-angiotensin-aldosterone system, and increased oxidative stress [47].
Many epidemiological studies also demonstrated strong independent links between asymptomatic hyperuricemia and the prevalence and progression of renal disease. Iseki et al. showed that asymptomatic hyperuricemia (uric acid >8 g/dl) increased CKD risk by threefold in men and tenfold in women. This higher risk was independent of age, SBP, body mass index, glucose, smoking, alcohol use, and proteinuria [48].
The large epidemiological trial, Atherosclerosis Risk in Communities (ARIC), showed that each 1 mg/dl increase in uric acid increased the risk of CKD by 7–11%. Other studies have shown a linear correlation between uric acid and the degree of CKD in the elderly [49,50,51]. Hsu et al. evaluated the most comprehensive epidemiological study linking a strong relationship between serum uric acid and CKD. In 177,570 US Renal Data System subjects that were followed over 25 years, a higher uric acid quartile unveiled a 2.14-fold increase in end-stage renal disease over time [52]. In a recently published study, Kochi et al. describe a stronger association between hypertension and proteinuria in patients with hyperuricemia compared to those without hyperuricemia and conclude that hyperuricemia is “an important contributing factor to both nephrosclerosis with ischemia and hyperfiltration” [53••]. In the commentary regarding this study, Tadashi Sofue considers hyperuricemia as the “third key player for nephrosclerosis with ischemia” [54].
Chonchol et al. evaluated 5808 patients in the Cardiovascular Health Study and found asymptomatic hyperuricemia was strongly associated with the prevalence but not the incidence of CKD [55]. In the Mild to Moderate Kidney Disease Study, Sturm et al. evaluated 277 persons with non-diabetic kidney disease and found that initial asymptomatic hyperuricemia correlated with CKD progression [56].
Other investigators have examined the relationship between serum uric acid obtained at the initiation of dialysis in patients with advanced stage 5 CKD and subsequent all-cause mortality. The major finding was that asymptomatic hyperuricemia (uric acid of 9.0 mg) had a twofold increased risk for mortality independent of potential confounders such as age, sex, eGFR, cardiovascular disease, diabetes mellitus, diuretic use, cholesterol level, CRP level, and phosphate level. They also reported an increased risk for mortality in those patients who had a uric acid level of 5.2 mg or greater [57]. In contrast, a retrospective cohort study of 200 patients on maintenance hemodialysis revealed an interesting association between longitudinal changes in uric acid, nutritional markers, and long-term survival. With each 1.0 mg/dl longitudinal increase in uric acid, there was a 13.4% slower rate of decline in geriatric nutritional risk over 3 years and lower overall mortality [58].
Asymptomatic hyperuricemia is a common finding in post-kidney transplant recipients, with the prevalence of hyperuricemia in the range of 15–52%. Other risk factors for hyperuricemia in those patients are progressive CKD and medications including hydrochlorothiazide and other thiazide-like diuretics, cyclosporine immunosuppressive therapy, and alcohol abuse [59,60,61,62]. Asymptomatic hyperuricemia and graft survival in kidney transplant recipients is an intriguing and complex topic for the transplant nephrologist. Many have investigated this relationship, and most studies suggest a negative impact of asymptomatic hyperuricemia on renal allograft function and survival [63, 64].
One study analyzed the mean uric acid level during the first 6 months post-transplant and found that higher uric acid levels were an independent predictor of long-term and short-term graft function [65]. Other authors have shown that early onset of hyperuricemia at 3 months post-kidney transplant increased risk for graft failure according to multiple Cox regression analysis, [66] and Akalin et al. found that hyperuricemia 6 months after transplantation was significantly associated with new cardiovascular events and graft dysfunction [67]. Kalil et al. used data from the randomized controlled trial, Folic Acid for Vascular Outcome Reduction in Transplantation (FAVORIT), that examined the potential association of asymptomatic hyperuricemia with cardiovascular mortality and allograft failure over a mean follow-up time of 3.9 years. The study showed that 503 patients sustained a primary cardiovascular event (P=0.5), 401 died (P=0.09), and 287 had transplant failure (P=0.1) [68]. A large retrospective cohort study divided patients into low, normal, and high uric acid groups according to mean serum uric acid level within the first year and 1–5 years after transplantation. Those with low to normal serum acid levels in the first year and 1–5 years had better renal allograft outcomes in the long-term follow-up period, compared to those with high serum uric acid [69].
The association between hyperuricemia and chronic kidney disease is well established in observational studies. However, debate remains as to what effect urate-lowering therapies may have on CKD outcomes in the setting of asymptomatic hyperuricemia. Several single-center studies have demonstrated that the use of allopurinol or febuxostat slows the progression of CKD [70,71,72]. A meta-analysis performed by Bose [73] that included eight randomized controlled trials and a total of 476 patients showed a trend toward a higher eGFR by 3.1 ml/min/1.73 m2 [2] BSA in the allopurinol group with a median follow-up time of 11 months (95% confidence interval 0.9–7.0). However, this review was limited by its small size and that only two of the eight studies were placebo controlled.
A more recent study, the Controlled Trial of Slowing of Kidney Disease Progression from the Inhibition of Xanthine Oxidase (CKD-FIX), specifically looked at adults with baseline stage 3 or 4 CKD, albuminuria, or an eGFR decline of at least 3.0 ml/min/1.72 m2 [2] BSA in the preceding year and no history of gout [74]. A total of 369 patients from 31 centers in Australia and New Zealand were randomized to receive allopurinol or placebo. For the primary outcome, change in eGFR over 104 weeks, this study did not find a significant difference in the allopurinol group versus the placebo group. There was also no difference between the two groups with regard to secondary outcomes of change in the amount of proteinuria, change in BP, progression to ESRD, or death [74]. Other randomized placebo-controlled studies have also failed to demonstrate a statistically significant effect of febuxostat or allopurinol on the progression of CKD in patients with already established CKD [75, 76].
In summary, asymptomatic hyperuricemia seems to correlate strongly with the prevalence of CKD. In the ESRD population on hemodialysis, asymptomatic hyperuricemia has been associated with a decrease in mortality. In post-kidney transplant patients, high uric acid has been associated with poor long-term renal allograft function. However, once again, data to support the use of urate-lowering drugs to alter the outcomes of patients with CKD is not consistent. The recent studies mentioned above looked at patients with established CKD. Randomized placebo-controlled studies focusing on the initiation of urate-lowering drugs at earlier stages of CKD or in patients at elevated risk for CKD may shed more light on what potential benefits these drugs may have in preventing or slowing the progression of kidney disease.
Hyperuricemia and Hypertension
An association between uric acid levels and hypertension has been suggested since the 1870s, and studies as early as the 1920s observed this link [77]. However, this has typically been thought to be reflective of hyperuricemia being a consequence rather than a cause of hypertension. One proposed mechanism considers impaired urate excretion by the kidney in the setting of hypertension. The decreased eGFR often seen in those with chronic hypertension leads to retained uric acid. The increase in renal vascular resistance leads to decreased renal blood flow, resulting in increased sodium and uric acid resorption in the proximal tubule [77]. Furthermore, hyperuricemia may be a result of competition for the organic anion transporter in the proximal tubule by thiazide diuretics or elevated lactate levels, which can be seen in tissue ischemia in the setting of severe hypertension. Another hypothesis is that oxidative stress, common in patients with hypertension, sets off a cascade that increases the degradation of hypoxanthine and xanthine to uric acid [77].
The supposition that hyperuricemia is a result of hypertension is challenged by many studies demonstrating elevated uric acid levels prior to the development of hypertension. Hyperuricemia, independent of most other risk factors, is a predictor of hypertension development [78,79,80,81]. One study by Krishnan et al. noted baseline normotensive men with hyperuricemia to have an 80% excess risk for hypertension [79]. Another author suggests that this association is more robust in blacks compared to whites [81].
A publication by Mazzali et al. took this debate to the next level and found that raising serum uric acid levels in rats was associated with an increase in systemic BP after several weeks [82]. Further, elevation in BP was prevented if the uric acid was lowered by a xanthine oxidase inhibitor or with a uricosuric agent [23]. A similar effect was seen in humans. Segal et al. performed a double-blind randomized controlled trial of African Americans with hypertension treated with chlorthalidone and additionally given allopurinol vs placebo. While the study did not achieve statistical significance, those given allopurinol tended to have BP compared to those in the placebo group [83]. Another randomized placebo-controlled trial by Gunawardhana et al. examining the effects of febuxostat on BP found that treatment with febuxostat resulted in a significant decrease in SBP in a subgroup analysis of patients with hypertension and normal renal function [84].
While uric acid is an antioxidant in the extracellular environment, studies have demonstrated that it is pro-oxidative intracellularly, stimulating NADPH oxidase. The mechanism of this is not entirely clear [5, 77]. When introduced into cultured endothelial cells, induction of oxidative stress was associated with increased secretion of C reactive protein and other inflammatory mediators, reduced nitric oxide, and increased angiotensin II and angiotensin I receptor expression [7, 27].
It has also been suggested that the strongest association of uric acid with hypertension may be seen in the earlier stages of hypertension. Many studies have demonstrated elevated uric acid levels as a predictor of hypertension in the child and adolescent population [85, 86].
Some studies also support allopurinol as lowering BP in adolescents. In a randomized double-blinded placebo-controlled trial by Feig et al., adolescents with newly diagnosed hypertension assigned to the allopurinol group had a mean decrease in 24 h mean ambulatory systolic BP of 6.3 mmHg and a mean decrease in diastolic BP (DBP) of 4.6 mmHg. Two-thirds of the participants in the allopurinol group saw their BP reach the targeted range during the 4 weeks of the study [87]. Another particularly interesting phenomenon previously published is that elevated uric acid is associated with a non-dipping circadian rhythm pattern [88,89,90].
The physiology that explains the link between hyperuricemia and hypertension may lie within the renin-angiotensin system. A review by Soltani et al. noted several studies in animals and humans demonstrating an increase in renin aldosterone angiotensin system activation in the setting of increased uric acid levels. Their review proposed that it was time for a reappraisal of how consideration of uric acid levels is approached in the management of hypertension [91].
The mechanism by which uric acid causes hypertension has two phases. Based on animal models, uric acid causes increased renal production of renin and reduction of nitrates in the systemic circulation, resulting in increased vasoconstriction [5, 82, 92]. Rats treated with oxonic acid, a uric acid oxidase inhibitor, showed increased uric acid levels and a gradual rise in BP over 2–3 weeks. This increase in BP can be reversed by the withdrawal of oxonic acid, the addition of uric acid-lowering drugs, or renin-angiotensin system blockade [82, 93]. However, prolonged elevation of uric acid levels leads to vascular smooth muscle cell proliferation, decreased compliance of renal afferent arterioles, and a shift in the pressure natriuresis curve, resulting in an increase in salt-sensitive hypertension. In the rat model, these changes can persist for years and are not reversed by urate-lowering therapy [85].
With this biphasic model of uric acid causing hypertension, it would make sense then that xanthine oxidase inhibitors would result in improvements in BPs in hypertensive human subjects if administered in the first phase of the proposed mechanism. Some studies do support that administration of xanthine oxidase inhibitors results in improved BPs [94, 95].
However, the second phase of this model may be less amenable to urate-lowering therapy but be mitigated by agents like the angiotensin receptor blockers (ARBs) that can slow vascular smooth muscle cell proliferation. Evidence also shows that some ARBs have a uric acid-lowering effect as well. This does not appear to be a class effect of all ARBs but losartan in particular seems to have the most consistent exposure of this lowering effect. The mechanism appears to be inhibition of URAT-1, the transporter which facilitates reabsorption of uric acid in the proximal tubule [96, 97
Despite physiologic plausibility and some studies to support it, there remains an ongoing debate as to whether there is a causal relationship between hyperuricemia and hypertension and, in the clinical setting, whether decreasing uric acid levels has a positive impact on BP control. A recent randomized double-blind crossover clinical trial by Gaffo et al. looking at young adults (age 18–40) with hypertension and serum urate levels >5.0 mg/dl for men and >4.0mg/dl for women did not show a statistically significant change in either the allopurinol treatment phase or the placebo treatment phase during the 1-month follow-up period of the study [98]. A prior meta-analysis of ten studies examining the effect of allopurinol on BP, which included 738 participants, did show decreases in SBP (3.3 mmHg) and DBP of 1.3 mmHg) in the allopurinol group. It was noted that several studies included in this systematic review were not randomized controlled trials and were considered to be of lesser quality. However, the authors of the review did find similar changes in systolic and DBPs when the analysis was restricted to higher quality randomized controlled trials [99]. The authors noted that a shortcoming of this meta-analysis is that many of the studies had patients with relatively well-controlled BPs at baseline and that other antihypertensive therapies may have changed during the course of the study.
Critics of this meta-analysis noted that another review published around the same time and that had more stringent quality inclusion criteria found only one study that met their criteria. This study showed a positive effect of allopurinol on BP lowering in the patient population studied, but the Feig study on adolescents noted above [100] further cautions that allopurinol is not without potential, albeit rare, adverse effects, including potentially fatal Stevens-Johnson syndrome and chronic interstitial nephritis. Therefore, clinicians should be cautious in using these agents because the benefit has yet to be clearly demonstrated [101].
While over the years, many studies have looked at the association between hyperuricemia and metabolic syndrome, chronic kidney disease, and hypertension, many questions about these relationships remain unanswered. That this association exists and can be explained physiologically is well established. However, association does not prove causality. It is still unclear if urate-lowering therapy is of any real benefit and if so, in what specific populations and what should therapeutic goals be.
This ongoing debate highlights the need for more high-quality randomized controlled trials to solidify the role of hyperuricemia and xanthine oxidase inhibitors in the management of metabolic syndrome, chronic kidney disease, and hypertension.
Availability of Data and Materials
No raw data or materials were used for this literature review.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Xiangwei WU, Muzny DM, Cheng Chi Lee, Caskey CT. Two independent mutational events in the loss of urate oxidase during hominoid evolution. J Mol Evol. 1992;34(1):78-84. https://www.ncbi.nlm.nih.gov/pubmed/1556746. https://doi.org/10.1007/BF00163854.
Endou H, Enomoto A, Kimura H, et al. Molecular identification of a renal urate-anion exchanger that regulates blood urate levels. Nature (London). 2002;417(6887):447-452. https://www.ncbi.nlm.nih.gov/pubmed/12024214. https://doi.org/10.1038/nature742.
Edwards NL. The role of hyperuricemia and gout in kidney and cardiovascular disease. Cleve Clin J Med. 2008;75(Suppl 5):S13-S16. http://www.ccjm.org/content/75/Suppl_5/S13.abstract. https://doi.org/10.3949/ccjm.75.Suppl_5.S13.
Johnson RJ, Kang D, Feig D, et al. Is there a pathogenetic role for uric acid in hypertension and cardiovascular and renal disease? Hypertension. 2003;41(6):1183-1190. http://hyper.ahajournals.org/cgi/content/abstract/41/6/1183. https://doi.org/10.1161/01.HYP.0000069700.62727.C5.
Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol. 2007;293(2):584-596. http://ajpcell.physiology.org/content/293/2/C584. https://doi.org/10.1152/ajpcell.00600.2006.
Corry DB, Eslami P, Yamamoto K, Nyby MD, Makino H, Tuck ML. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin–angiotensin system. J Hypertension. 2008;26(2):269-275. https://www.ncbi.nlm.nih.gov/pubmed/18192841. https://doi.org/10.1097/HJH.0b013e3282f240bf.
Kang D, Park S, Lee I, Johnson RJ. Uric acid–induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrology. 2005;16(12):3553-3562. https://doi.org/10.1681/ASN.2005050572.
Ginsberg MH, Kozin F, O'Malley M, McCarty DJ. Release of platelet constituents by monosodium urate crystals. J Clin Investig. 1977;60(5):999-1007. https://www.ncbi.nlm.nih.gov/pubmed/908764. https://doi.org/10.1172/JCI108880.
Nakagawa T, Mazzali M, Kang D, Sánchez-Lozada LG, Herrera-Acosta J, Johnson RJ. Uric acid – A uremic toxin? Blood Purif. 2005;24(1):67-70. https://www.karger.com/Article/Abstract/89440. https://doi.org/10.1159/000089440.
Masuo K, Kawaguchi H, Mikami H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension. 2003;42(4):474-480. http://hyper.ahajournals.org/cgi/content/abstract/42/4/474. https://doi.org/10.1161/01.HYP.0000091371.53502.D3.
Nakanishi N, Okamoto M, Yoshida H, Matsuo Y, Suzuki K, Tatara K. Serum uric acid and risk for development of hypertension and impaired fasting glucose or type II diabetes in Japanese male office workers. Euro J Epidemiol. 2003;18(6):523-530. https://www.jstor.org/stable/3582904. https://doi.org/10.1023/A:1024600905574.
Sundstrom J, Sullivan L, D'Agostino RB, Levy D, Kannel WB, Vasan RS. Relations of serum uric acid to longitudinal blood pressure tracking and hypertension incidence. Hypertension. 2005;45(1):28-33. http://hyper.ahajournals.org/cgi/content/abstract/45/1/28. https://doi.org/10.1161/01.HYP.0000150784.92944.9a.
De Leeuw PW, Thijs L, Rosenfeld JJ, et al. Prognostic significance of renal function in elderly patients with isolated systolic hypertension: results from the syst-eur trial. J Am Soc Nephrol. 2002;13(9):2213-2222. https://www.ncbi.nlm.nih.gov/pubmed/12191965. https://doi.org/10.1097/01.ASN.0000027871.86296.92.
Bickel C, Rupprecht HJ, Blankenberg S, et al. Serum uric acid as an independent predictor of mortality in patients with angiographically proven coronary artery disease. Am J Cardiol. 2002;89(1):12–7.
Kang D, Finch J, Nakagawa T, et al. Uric acid, endothelial dysfunction and pre-eclampsia: searching for a pathogenetic link. J Hypertension. 2004;22(2):229-235. http://ovidsp.ovid.com/ovidweb.cgi?T=JS&NEWS=n&CSC=Y&PAGE=fulltext&D=ovft&AN=00004872-200402000-00001. https://doi.org/10.1097/00004872-200402000-00001.
Laughon SK, Catov J, Provins T, Roberts JM, Gandley RE. Elevated first-trimester uric acid concentrations are associated with the development of gestational diabetes. Am J Obstet Gynecol. 2009;201(4):402.e1-402.e5. https://www.clinicalkey.es/playcontent/1-s2.0-S0002937809007601. https://doi.org/10.1016/j.ajog.2009.06.065.
Beberashvili I, Sinuani I, Azar A, Shapiro G, Feldman L, Stav K, Sandbank J, Averbukh Z Serum uric acid as a clinically useful nutritional marker and predictor of outcome in maintenance hemodialysis patients. Nutrition (Burbank, Los Angeles County, Calif.). 2015;31(1):138-147. https://www.clinicalkey.es/playcontent/1-s2.0-S0899900714003335. https://doi.org/10.1016/j.nut.2014.06.012.
Bellomo G. Uric acid and chronic kidney disease: a time to act? World J Nephrol. 2013;2(2):17-25. http://lib.cqvip.com/qk/61591X/201302/83908883504849514850484849.html. https://doi.org/10.5527/wjn.v2.i2.17.
Facchini F, Chen Y-I, Hollenbeck CB, Reaven GM. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA-J Am Med Assoc. 1991;266(21):3008–11. https://doi.org/10.1001/jama.1991.03470210076036.
Tang L, Kubota M, Nagai A, Mamemoto K, Tokuda M. Hyperuricemia in obese children and adolescents: the relationship with metabolic syndrome. Pedia Rep. 2010;2(1):e12. https://www.ncbi.nlm.nih.gov/pubmed/21589837. https://doi.org/10.4081/pr.2010.e12.
Choi HK, Ford ES. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am J Med. 2007;120(5):442-447. https://www.clinicalkey.es/playcontent/1-s2.0-S0002934306008904. https://doi.org/10.1016/j.amjmed.2006.06.040.
Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2006;290(3):625-631. http://ajprenal.physiology.org/content/290/3/F625. https://doi.org/10.1152/ajprenal.00140.2005.
Sanchez-Lozada LG, Tapia E, Bautista-Garcia P, et al. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. Am J Physiol Renal Physiol. 2008;294(4):710-718. http://ajprenal.physiology.org/content/294/4/F710. https://doi.org/10.1152/ajprenal.00454.2007.
Quinones Galvan A, Natali A, Baldi S, et al. Effect of insulin on uric acid excretion in humans. Am J Physiol Endocrinol Metab. 1995;268(1):E1-E5. http://ajpendo.physiology.org/cgi/content/abstract/268/1/E1. https://doi.org/10.1152/ajpendo.1995.268.1.E1.
Ishiro M, Takaya R, Mori Y, et al. Association of uric acid with obesity and endothelial dysfunction in children and early adolescents. Ann Nutr Metab. 2013;62(2):169-176. https://www.jstor.org/stable/48507792. https://doi.org/10.1159/000346227.
Hallfrisch, J. (National Institute on Aging, Baltimore, MD). Metabolic effects of dietary fructose. The FASEB J. 1990;4(9):2652-2660. https://agris.fao.org/agris-search/search.do?recordID=US9041786. https://doi.org/10.1096/fasebj.4.9.2189777.
Khosla UM, Zharikov S, Finch JL, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005;67(5):1739–42. https://doi.org/10.1111/j.1523-1755.2005.00273.x.
Nakagawa T, Tuttle KR, Short RA, Johnson RJ. Hypothesis: fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol. 2005;1(2):80–6. https://doi.org/10.1038/ncpneph0019.
Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Investig. 2004;114(12):1752-1761. https://www.ncbi.nlm.nih.gov/pubmed/15599400. https://doi.org/10.1172/JCI200421625.
Ryu S, Song J, Choi BY, Lee SJ, Kim WS, Chang Y, Kim DI, Suh BS, Sung KC. Incidence and risk factors for metabolic syndrome in Korean male workers, ages 30 to 39. Ann Epidemiol. 2007;17(4):245-252. https://www.clinicalkey.es/playcontent/1-s2.0-S1047279706002547. https://doi.org/10.1016/j.annepidem.2006.10.001.
Dehghan A, van Hoek M, Sijbrands EJG, Hofman B, Witteman J. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care. 2008;31(2):361-362. http://care.diabetesjournals.org/content/31/2/361.abstract. https://doi.org/10.2337/dc07-1276.
Niskanen L, Laaksanen DE, Lindstrom J, et al. Serum uric acid as a harbinger of metabolic outcome in subjects with impaired glucose tolerance: the Finnish diabetes prevention study. Diabetes Care. 2006;29(3):709-711. https://www.ncbi.nlm.nih.gov/pubmed/16505534. https://doi.org/10.2337/diacare.29.03.06.dc05-1465.
Sui X, Church TS, Meriwether RA, Lobelo F, Blair SN. Uric acid and the development of metabolic syndrome in women and men. Metab Clin Exp. 2008;57(6):845-852. https://www.clinicalkey.es/playcontent/1-s2.0-S0026049508000632. https://doi.org/10.1016/j.metabol.2008.01.030.
Rubio-Guerra AF, Morales-López H, Garro-Almendaro AK, et al. Circulating levels of uric acid and risk for metabolic syndrome. Curr Diabetes Rev. 2017;13(1):87. https://www.ncbi.nlm.nih.gov/pubmed/26419665.
Chen W, Fu Y, Zhou M. The bidirectional relationship between metabolic syndrome and hyperuricemia in china: a longitudinal study from CHARLS. Endocrine. 2022;76(1):62-69. https://doi.org/10.1007/s12020-022-02979-z.
Johnson RJ, Perez-Pozo SE, Sautin YY, et al. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocrine Rev. 2009;30(1):96–116. https://doi.org/10.1210/er.2008-0033.
Sánchez-Lozada LG, Mu W, Roncal C, et al. Comparison of free fructose and glucose to sucrose in the ability to cause fatty liver. Eur J Nutr. 2009;49(1):1-9. https://doi.org/10.1007/s00394-009-0042-x.
Bocarsly ME, Powell ES, Avena NM, Hoebel BG. High-fructose corn syrup causes characteristics of obesity in rats: increased body weight, body fat and triglyceride levels. Pharmacol Biochem Behav. 2010;97(1):101–6. https://doi.org/10.1016/j.pbb.2010.02.012.
Havel PJ. Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr Rev. 2005;63(5):133–57.
Cox CL, Stanhope KL, Keim NL, et al. Consumption of fructose-sweetened beverages for 10 weeks reduces net fat oxidation and energy expenditure in overweight obese men and women. Euro J Clin Nutr. 2012;66(2):201–8. https://doi.org/10.1038/ejcn.2011.159.
Reunjui S, Roncal CA, Wei MU, et al. Thiazide diuretics exacerbate fructose-induced metabolic syndrome. J Am Soc Nephrol. 2007;18(10):2724-2731. https://www.ncbi.nlm.nih.gov/pubmed/17855639. https://doi.org/10.1681/ASN.2007040416.
Jacob S. Patient with beer belly or hypertension. check for a metabolic syndrome! MMW Fortschr Med. 2005;147(9):45. https://www.ncbi.nlm.nih.gov/pubmed/15794356.
Kappaganthu A, Sachan J, Shailaja G. Hyperuricemia in early pregnancy: a marker for gestational diabetes mellitus. IOSR J Dent Med Sci. 2014;13(12):51–4. https://doi.org/10.9790/0853-131265154.
Haig A. Uric acid as a factor in the causation of disease. 4th ed., with sixty-five illustrations. ed. London: J. & A. Churchill; 1897. http://catalog.crl.edu/record=b1040761.
Johnson RJ, Kivlighn SD, Kim Y, Suga S, Fogo AB. Reappraisal of the pathogenesis and consequences of hyperuricemia in hypertension, cardiovascular disease, and renal disease. Am J Kidney Dis. 1999;33(2):225–34. https://doi.org/10.1016/S0272-6386(99)70295-7.
•• Sellmayr M, Hernandez Petzsche MR, Ma Q, et al. Only hyperuricemia with crystalluria, but not asymptomatic hyperuricemia, drives progression of chronic kidney disease. Clin J Am Soc Nephrol. 2020;31(12):2773-2792. https://www.narcis.nl/publication/RecordID/oai:pure.eur.nl:publications%2Fce579fb7-10e7-4492-8f5b-a66c294ff1d6. https://doi.org/10.1681/ASN.2020040523. In a novel mouse model, the authors prove that the uric acid causes tubular injury, inflammation, fibrosis, and granulomatosis interstitial nephritis.
Lee TH, Chen J, Wu C, Yang C, Yang H. Hyperuricemia and progression of chronic kidney disease: a review from physiology and pathogenesis to the role of urate-lowering therapy. Diagnostics (Basel). 2021;11(9):1674. https://search.proquest.com/docview/2576393675. https://doi.org/10.3390/diagnostics11091674.
Iseki K, Oshiro S, Tozawa M, Iseki C, Ikemiya Y, Taskishita S. Significance of hyperuricemia on the early detection of renal failure in a cohort of screened subjects. Hypertension Res. 2001;24(6):691-697. https://www.jstage.jst.go.jp/article/hypres/24/6/24_6_691/_article/-char/en. https://doi.org/10.1291/hypres.24.691.
Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS. Uric acid and incident kidney disease in the community. J Am Soc Nephrol. 2008;19(6):1204-1211. https://www.ncbi.nlm.nih.gov/pubmed/18337481. https://doi.org/10.1681/ASN.2007101075.
Chen N, Wang W, Huang Y, et al. Community-based study on CKD subjects and the associated risk factors. Nephrol Dial Transplant. 2009;24(7):2117-2123. https://api.istex.fr/ark:/67375/HXZ-TDF816C3-S/fulltext.pdf. https://doi.org/10.1093/ndt/gfn767.
Chen Y, Su C, Wang S, Lee H, Lin S. A preliminary investigation of the association between serum uric acid and impaired renal function. Chang Gung Med J. 2009;32(1):66-71. https://www.ncbi.nlm.nih.gov/pubmed/19292941.
Hsu C, Iribarren C, McCulloch CE, Darbinian J, Go AS. Risk factors for end-stage renal disease: 25-year follow-up. Arch Intern Med. (1960). 2009;169(4):342-350. https://doi.org/10.1001/archinternmed.2008.605.
•• Kochi M, Kohagura K, Oshiro N, et al. Association of blood pressure and hyperuricemia with proteinuria and reduced renal function in the general population. Hypertension Res. 2023. https://www.ncbi.nlm.nih.gov/pubmed/36991065https://doi.org/10.1038/s41440-023-01250-w. The authors found a strong association between hypertension and proteinuria in patients with hyperuricemia when compared with subjects without hyperuricemia.
Sofue T. Hyperuricemia: the third key player for nephrosclerosis with ischemia. Hypertens Res. 2023. https://www.ncbi.nlm.nih.gov/pubmed/37081158. https://doi.org/10.1038/s41440-023-01294-y.
Chonchol M, Shlipak MG, Katz R, Sarnak MJ, Newman AB, Siscovick DS, Kestenbaum B, Carney JK, Fried LF. Relationship of uric acid with progression of kidney disease. Am J Kidney Dis. 2007;50(2):239-247. https://www.clinicalkey.es/playcontent/1-s2.0-S0272638607008323. https://doi.org/10.1053/j.ajkd.2007.05.013.
Sturm G, Kollerits B, Neyer U, Ritz E, Kronenberg F. Uric acid as a risk factor for progression of non-diabetic chronic kidney disease? the mild to moderate kidney disease (MMKD) study. Expe Gerontol. 2008;43(4):347–52. https://doi.org/10.1016/j.exger.2008.01.006.
Suliman ME, Johnson RJ, García-López E, et al. J-shaped mortality relationship for uric acid in CKD. Am J Kidney Dis. 2006;48(5):761–71. https://doi.org/10.1053/j.ajkd.2006.08.019.
Beberashvili I, Erlich A, Azar A, et al. Longitudinal study of serum uric acid, nutritional status, and mortality in maintenance hemodialysis patients. Clin J Am Soc Nephrol. 2016;11(6):1015-1023. https://www.ncbi.nlm.nih.gov/pubmed/27026520. https://doi.org/10.2215/CJN.10400915.
Kalantar E, Khalili N, Hossieni M-, Rostami Z, Einollahi B. Hyperuricemia after renal transplantation. Transplant Proc. 2011;43(2):584-585. https://www.clinicalkey.es/playcontent/1-s2.0-S0041134511000637. https://doi.org/10.1016/j.transproceed.2011.01.062.
Kim KM, Kim S, Han DJ, Yang WS, Park JS, Park SK. Hyperuricemia in kidney transplant recipients with intact graft function. Transplant Proc. 2010;42(9):3562-3567. https://www.clinicalkey.es/playcontent/1-s2.0-S0041134510012972.
Malheiro J, Almeida M, Fonseca I, et al. Hyperuricemia in adult renal allograft recipients: prevalence and predictors. Transplant Proc. 2012;44(8):2369-2372. https://www.clinicalkey.es/playcontent/1-s2.0-S0041134512007294. https://doi.org/10.1016/j.transproceed.2012.07.033.
Numakura K, Satoh S, Kagaya H, et al. Hyperuricemia at 1 year after renal transplantation, its prevalence, associated factors, and graft survival. Transplant. 2012;94(2):145-151. https://www.ncbi.nlm.nih.gov/pubmed/22728291. https://doi.org/10.1097/TP.0b013e318254391b.
Gerhardt U, Große Hüttmann M, Hohage H. Influence of hyperglycemia and hyperuricemia on long-term transplant survival in kidney transplant recipients. Clin Transplant. 1999;13(5):375-379. https://api.istex.fr/ark:/67375/WNG-WHGCFHNZ-8/fulltext.pdf. https://doi.org/10.1034/j.1399-0012.1999.130502.x.
Armstrong KA, Johnson DW, Campbell SB, Isbel NM, Hawley CM. Does uric acid have a pathogenetic role in graft dysfunction and hypertension in renal transplant recipients? Transplant. 2005;80(11):1565-1571. https://www.ncbi.nlm.nih.gov/pubmed/16371927. https://doi.org/10.1097/01.tp.0000183895.88572.13.
Haririan A, Noguiera JM, Zandi-Nejad K, et al. The independent association between serum uric acid and graft outcomes after kidney transplantation. Transplantation. 2010;89(5):573-579. https://www.ncbi.nlm.nih.gov/pubmed/19997058. https://doi.org/10.1097/TP.0b013e3181c73c18.
Han M, Lee JP, Park S, et al. Early onset hyperuricemia is a prognostic marker for kidney graft failure: propensity score matching analysis in a Korean multicenter cohort. PLoS ONE. 2017;12(6). https://explore.openaire.eu/search/publication?articleId=od_______267::cc09b79f1deda615ed0575ee99908162. https://doi.org/10.1371/journal.pone.0179779.
Aakalin E, Venkatesh Ganeshan S, Winston J, Muntner P. Hyperuricemia is associated with the development of the composite outcomes of new cardiovascular events and chronic allograft nephropathy. Transplantation. 2008;86(5):652-658. https://www.ncbi.nlm.nih.gov/pubmed/18791445. https://doi.org/10.1097/TP.0b013e3181814f5b.
Kalil RS, Carpenter MA, Ivanova A, et al. Impact of hyperuricemia on long-term outcomes of kidney transplantation: analysis of the FAVORIT study. Am J Kidney Dis. 2017;70(6):762–9. https://doi.org/10.1053/j.ajkd.2017.06.013.
Kim DG, Choi HY, Kim HY, et al. Association between post-transplant serum uric acid levels and kidney transplantation outcomes. PLoS ONE. 2018;13(12):e0209156. https://www.ncbi.nlm.nih.gov/pubmed/30550582. https://doi.org/10.1371/journal.pone.0209156.
Siu Y, Leung K, Tong MK, Kwan T. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47(1):51–9. https://doi.org/10.1053/j.ajkd.2005.10.006.
Goicoechea M, de Vinuesa SG, Verdalles U, et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5(8):1388-1393. http://cjasn.asnjournals.org/content/5/8/1388.abstract. https://doi.org/10.2215/CJN.01580210.
Sircar D, Chatterjee S, Waikhom R, Golay V, Raychaudhury A, Chatterjee S, Pandey R. Efficacy of febuxostat for slowing the GFR decline in patients with CKD and asymptomatic hyperuricemia: a 6-month, double-blind, randomized, placebo-controlled trial. Am J Kidney Dis. 2015;66(6):945-950. https://www.clinicalkey.es/playcontent/1-s2.0-S027263861500846X. https://doi.org/10.1053/j.ajkd.2015.05.017.
Bose B, Badve SV, Hiremath SS, et al. Effects of uric acid-lowering therapy on renal outcomes: a systematic review and meta-analysis. Nephrol Dial Transplant. 2014;29(2):406-413. https://www.ncbi.nlm.nih.gov/pubmed/24042021. https://doi.org/10.1093/ndt/gft378.
Badve SV, Pascoe EM, Tiku A, et al. Effects of allopurinol on the progression of chronic kidney disease. N Engl J Med. 2020;382(26). http://handle.unsw.edu.au/1959.4/unsworks_76546. https://doi.org/10.1056/NEJMoa1915833.
Doria A, Galecki AT, Spino C, et al. Serum urate lowering with allopurinol and kidney function in type 1 diabetes. N Engl J Med. 2020;382(26):2493-2503. https://doi.org/10.1056/NEJMoa1916624.
Kimura K, Hosoya T, Uchida S, et al. Febuxostat therapy for patients with stage 3 CKD and asymptomatic hyperuricemia: a randomized trial. Am J Kidney Dis. 2018;72(6):798–810. https://doi.org/10.1053/j.ajkd.2018.06.028.
Mazzali M, Kanbay M, Segal MS, et al. Uric acid and hypertension: cause or effect? Curr Rheumatol Rep. 2010;12(2):108-117. https://doi.org/10.1007/s11926-010-0094-1.
Forman JP, Choi H, Curhan GC. Uric acid and insulin sensitivity and risk of incident hypertension. Arch Intern Med (1960). 2009;169(2):155-162. https://doi.org/10.1001/archinternmed.2008.521.
Krishnan E, Kwoh CK, Schumacher HR, Kuller L. Hyperuricemia and incidence of hypertension among men without metabolic syndrome. Hypertension. 2007;49(2):298-303. http://hyper.ahajournals.org/cgi/content/abstract/49/2/298. https://doi.org/10.1161/01.HYP.0000254480.64564.b6.
Perlstein TS, Gumieniak O, Williams GH, et al. Uric acid and the development of hypertension: the normative aging study. Hypertension. 2006;48(6):1031-1036. http://hyper.ahajournals.org/cgi/content/abstract/48/6/1031. https://doi.org/10.1161/01.HYP.0000248752.08807.4c.
Mellen PB, Bleyer AJ, Erlinger TP, et al. Serum uric acid predicts incident hypertension in a biethnic cohort: the atherosclerosis risk in communities study. Hypertension. 2006;48(6):1037-1042. http://hyper.ahajournals.org/cgi/content/abstract/48/6/1037. https://doi.org/10.1161/01.HYP.0000249768.26560.66.
Mazzali M, Hughes J, Kim Y, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;38(5):1101-1106. http://hyper.ahajournals.org/cgi/content/abstract/38/5/1101. https://doi.org/10.1161/hy1101.092839.
Segal MS, Srinivas TR, Mohandas R, Shuster JJ, Wen X, Whidden E, Tantravahi J, Johnson RJ. The effect of the addition of allopurinol on blood pressure control in African Americans treated with a thiazide-like diuretic. Am J Hypertens. 2015;9(8):610-619.e1. https://www.clinicalkey.es/playcontent/1-s2.0-S1933171115004799. https://doi.org/10.1016/j.jash.2015.05.009.
Gunawardhana L, McLean L, Punzi HA, et al. Effect of febuxostat on ambulatory blood pressure in subjects with hyperuricemia and hypertension: a phase 2 randomized Placebo‐Controlled study. Am Heart J. 2017;6(11):n/a. https://doi.org/10.1161/JAHA.117.006683.
Yanik M, Feig D. Serum urate: a biomarker or treatment target in pediatric hypertension? Curr Opin Cardiol. 2013;28(4):433-438. http://ovidsp.ovid.com/ovidweb.cgi?T=JS&NEWS=n&CSC=Y&PAGE=fulltext&D=ovft&AN=00001573-201307000-00009. https://doi.org/10.1097/HCO.0b013e32836205ff.
Feig DI, Johnson RJ. Hyperuricemia in childhood primary hypertension. Hypertension. 2003;42(3):247-252. http://hyper.ahajournals.org/cgi/content/abstract/42/3/247. https://doi.org/10.1161/01.HYP.0000085858.66548.59.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA : J Am Med Assoc. 2008;300(8):924–32. https://doi.org/10.1001/jama.300.8.924.
Giallauria F, Predotti P, Casciello A, et al. Serum uric acid is associated with non-dipping circadian pattern in young patients (30-40 years old) with newly diagnosed essential hypertension. Clin Expe Hypertens (1993). 2016;38(2):233-237. https://doi.org/10.3109/10641963.2015.1081230.
Ahbap E, Sakaci T, Kara E, et al. Serum uric acid levels and inflammatory markers with respect to dipping status: a retrospective analysis of hypertensive patients with or without chronic kidney disease. Clin Expe Hypertens (1993). 2016;38(6):555-563. https://doi.org/10.3109/10641963.2016.1174251.
Turak O, Özcan F, Tok D, et al. Serum uric acid, inflammation, and nondipping circadian pattern in essential hypertension. J Clin Hypertens (1993) (Greenwich, Conn.). 2013;15(1):7-13. https://doi.org/10.1111/jch.12026.
Soltani Z, Rasheed K, Kapusta DR, Reisin E. Potential role of uric acid in metabolic syndrome, hypertension, kidney injury, and cardiovascular diseases: is it time for reappraisal? Curr Hypertens Rep. 2013;15(3):175-181. https://doi.org/10.1007/s11906-013-0344-5.
Laura G. Sánchez-Lozada, Edilia Tapia, Rubén López-Molina, et al. Effects of acute and chronic l-arginine treatment in experimental hyperuricemia. Am J Physiol Renal Physiol. 2007;292(4):1238-1244. http://ajprenal.physiology.org/content/292/4/F1238. https://doi.org/10.1152/ajprenal.00164.2006.
Mazzali M, Kanellis J, Han L, et al. Hyperuricemia induces a primary renal arteriolopathy in rats by a blood pressure-independent mechanism. Am J Physiol Renal Physiol. 2002;282(6):991-997. http://ajprenal.physiology.org/content/282/6/F991. https://doi.org/10.1152/ajprenal.00283.2001.
Yamagishi T. Effects of xanthine oxidase inhibitors on blood pressure, baPWV, carotid arterial IMT and elastic modulus in hypertensive patients with hyperuricemia. J Hypertens. 2019;37: e105. https://doi.org/10.1097/01.hjh.0000571356.43815.16.
Bove M, Cicero AFG, Borghi C. The effect of xanthine oxidase inhibitors on blood pressure and renal function. Curr Hypertens Rep. 2017;19(12):95-6. https://doi.org/10.1007/s11906-017-0793-3. https://doi.org/10.1007/s11906-017-0793-3.
Hamada T, Ichida K, Hosoyamada M, et al. Uricosuric action of losartan via the inhibition of urate transporter 1 (URAT 1) in hypertensive patients. Am J Hypertens. 2008;21(10):1157-1162. https://api.istex.fr/ark:/67375/HXZ-SBBS4893-C/fulltext.pdf. https://doi.org/10.1038/ajh.2008.245.
Nishida Y, Takahashi Y, Susa N, Kanou N, Nakayama T, Asai S. Comparative effect of angiotensin II type I receptor blockers on serum uric acid in hypertensive patients with type 2 diabetes mellitus: a retrospective observational study. Cardiovasc Diabetol. 2013;12(1):159. https://www.ncbi.nlm.nih.gov/pubmed/24180232. https://doi.org/10.1186/1475-2840-12-159.
•• Gaffo AL, Calhoun DA, Rahn EJ, et al. Effect of serum urate lowering with allopurinol on blood pressure in young adults: a randomized, controlled, crossover trial. Arthritis & rheumatology (Hoboken, N.J.). 2021;73(8):1514-1522. https://doi.org/10.1002/art.41749. This is a single center randomized double blind crossover clinical trial examining differences in allopurinol vs placebo on patients with hypertension. Despite seeing differences in flow mediated dilation (a marker of endothelial dysfunction), no significant improvements in BP lowering was found in the allopurinol group.
Agarwal V, Hans N, Messerli FH. Effect of allopurinol on blood pressure: a systematic review and meta‐analysis. J Clin Hypertens (Greenwich, Conn.). 2013;15(6):435-442. https://doi.org/10.1111/j.1751-7176.2012.00701.x.
Gois P SE. Pharmacotherapy for hyperuricemia in hypertensive patients. https://pubmed.ncbi.nlm.nih.gov/23440832/.
Gois PLW, Seguro A. Allopurinol on hypertension: insufficient evidence to recommend. J Clin Hypertens. 2013;15(9):700.
Acknowledgements
Current Stem Cell Reports is grateful to Dr. Suzanne Oparil for her review of this manuscript.
Author information
Authors and Affiliations
Contributions
R.V., A.P., and E.R. wrote the main manuscript text, and all three authors also reviewed the final manuscript
Corresponding author
Ethics declarations
Ethical Approval
Not applicable as this was a literature review.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vareldzis, R., Perez, A. & Reisin, E. Hyperuricemia: An Intriguing Connection to Metabolic Syndrome, Diabetes, Kidney Disease, and Hypertension. Curr Hypertens Rep 26, 237–245 (2024). https://doi.org/10.1007/s11906-024-01295-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11906-024-01295-3