
Abstract
Uric acid is a product of purine metabolism and has been linked to gout and kidney calculi. Chronic kidney disease (CKD) and hypertension (HTN) are two major public health problems, and both are associated with increased risk of cardiovascular events. Emerging evidence suggests a pathogenic role of hyperuricemia in the development of HTN and CKD, in addition to progression of CKD, by inducing renal inflammation, endothelial dysfunction, and activation of the renin-angiotensin system. In addition, several epidemiological studies have linked hyperuricemia with an increased risk of HTN and CKD. A few clinical trials have assessed the use of uric acid-lowering therapies such as allopurinol and febuxostat in the management of HTN and delaying progression of CKD. To date, most of these trials are short-term with a small sample size; however, their results are encouraging and provide a rationale for larger randomized controlled trials to establish the role of uric acid-lowering therapies in the management of HTN, in addition to prevention of CKD progression and cardiovascular events.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Uric acid (2,6,8 trioxypurine-C5H4N4O3) is a carbon-based compound endogenously produced by animals as a purine metabolite. It is generated by the liver, as part of the normal turnover of nucleic acids, and chiefly excreted by the kidneys (65–75 %) and intestines (25–35 %) [1, 2].
Although gout has been first described by the Egyptians as early as 2640 B.C., and later on by Hippocrates in the fifth century B.C., the chemical identity of uric acid and its causal relationship to the disease and as a constituent of kidney calculi were not established until the late eighteenth century [3].
On the other hand, the possible link between hyperuricemia, hypertension (HTN), and the development and progression of chronic kidney disease (CKD) has emerged only recently [4, 5].
CKD is recognized as a worldwide public health problem with high morbidity and mortality rates due to its progression to end-stage renal disease (ESRD) and increased risk of cardiovascular events [6]. Likewise, HTN remains one of the major preventable risk factors for cardiovascular events and progression of kidney disease [7]. Any therapeutic intervention that would help decrease the burden of these two entities will most definitely have a major positive impact on public health and overall cardiovascular outcomes.
After a brief overview of uric acid homeostasis, and definition and epidemiology of hyperuricemia, our review will focus on the recent advances on the role of hyperuricemia in HTN and CKD, with an emphasis on the influence of uric acid-lowering therapies on renal and cardiovascular outcomes.
Uric Acid Homeostasis
Uric acid (UA) is produced in the liver from the breakdown of dietary and endogenously synthesized purine mononucleotides into the purine bases guanine and hypoxanthine. These latter two compounds are then metabolized to xanthine, which is irreversibly oxidized by xanthine oxidase to produce uric acid (Fig. 1). This is a continuous process resulting in UA which is considered a normal intracellular and biological fluid constituent [1, 8]. Normal humans have serum UA concentrations nearing 6.8 mg/dL [9]. In adult males, the total body urate pool is approximately 1200 mg, whereas adult females have lower UA plasma concentrations [2, 10].

Structure of uric acid and its homeostasis. Purines are oxidized by xanthine oxidase to generate uric acid which is the major end product of purine metabolism in humans. The majority of uric acid is excreted by the kidney and the rest in feces. However, a part of UA is reabsorbed at the proximal tubule. Reprinted with permission from Macmillan Publishers Ltd: [Nature Reviews Rheumatology] from reference [1]), copyright (2012)
Intestinal uricolysis, which is the degradation of UA by intestinal tract microbiota, is known to be responsible for nearly one third of total urate disposal. On the other hand, urinary UA excretion accounts for the residual two thirds of the daily UA clearance involving a complex interchange of reabsorptive and secretory pathways occurring in the renal proximal tubule [8, 9, 11].
These mechanisms work concurrently to assure net tubular reabsorption of UA filtered at the glomerulus, resulting in a normal urate excretion of about 10 % [8].
As shown in Fig. 2, a number of plasma membrane transport proteins are contributors in renal urate handling. The entry of urate into the intestine is mediated in part by Abcg2 (ATP-binding cassette subfamily G membrane 2), a high-capacity urate efflux transporter, encoded by the ABCG2 gene, which is also expressed in the proximal renal tubular epithelium. Moreover, URAT1 (urate anion exchanger 1) which is the product of the SLC22A12 gene and GLUT9 (glucose transporter type 9) which is encoded by SLC2A9 gene are considered as the main reabsorptive transporters of urate in the renal tubule. These two transporters have been proposed to be major regulators of UA homeostasis; thus, polymorphisms of these renal urate transporters are associated with the onset of hyperuricemia and gout [8, 9, 11, 12].

Transport proteins involved in urate handling in the human proximal tubule. Reprinted from Bobulescu and Moe [11], with permission from Elsevier
Currently available pharmacological strategies to reduce serum uric acid act by reducing uric acid production by inhibiting xanthine oxidase (allopurinol and febuxostat), inhibiting uric acid reabsorption by targeting URAT1 (such as probenecid), or by metabolizing circulating uric acid to allantoin by uricase (rasburicase) [13]. In addition, the angiotensin receptor blocker losartan has been shown to lower uric acid level by inhibiting URAT1, and the anti-diabetic SGLT2 inhibitors (such as canagliflozin and empagliflozin) lower uric acid by interfering with GLUT9 [5].
Hyperuricemia: Definition and Epidemiology
There is no universally accepted definition of hyperuricemia; a practical one is a serum UA concentration greater than its solubility point (6.8 mg/dL), as measured by automated enzymatic (uricase) methods in a routine clinical laboratory [14]. In clinical trials, hyperuricemia has been defined as UA level >7 or 7.5 mg/dL in males, and UA level >6 or 6.5 mg/dL in females [15].
Risk factors for hyperuricemia include alcohol consumption, particularly beer and distilled spirits, high dietary intake of fructose, meat, and seafood, the use of diuretics, angiotensin converting enzyme inhibitors, and non-losartan angiotensin II receptor blockers, in addition to hypertension, and obesity [16, 17]. Therefore, recently observed increase in serum levels of UA may be related to the growing prevalence of overweight and obesity, as well as the rise in consumption of sugar-sweetened beverages, foods rich in purines, and alcohol [18].
In the USA, prevalence of hyperuricemia reached 13 % of the general population as defined by a serum UA of >7 mg/dL, and up to 21 % if hyperuricemia was defined by a serum UA of >7 mg/dL in males and >5.7 mg/dL in females [19].
Hyperuricemia as defined by a serum UA of >7 mg/dL is more common in men than in women; UA levels in women of reproductive age are lower than their male counterparts due to the inhibition of renal urate reabsorption with an increased renal urate clearance by estrogenic compounds [20]. Therefore, hyperuricemia in women is more prevalent after menopause. Epidemiologic research showed a steady increase of hyperuricemia in women after 50 years of age [18, 21]. In addition, lower UA levels are observed in postmenopausal women treated with hormone replacement therapy [22].
Hyperuricemia and Hypertension
The correlation between serum UA and HTN has been demonstrated in several studies. In a large meta-analysis of 18 prospective cohort studies representing data from 55,607 subjects, incident HTN increased by 13 % per 1 mg/dL increase of serum UA [23]. In a prospective, longitudinal epidemiological investigation, involving patients with no cardiovascular disease at baseline, higher serum UA was strongly associated with the development of HTN and subclinical atherosclerosis as assessed by intima-media thickness [24]. In another longitudinal cohort of healthy adults, patients with a baseline serum UA of more than 6.5 mg/dL had a 25 % increased risk of HTN after adjusting for several parameters, including kidney function [25].
Several potential pathophysiological pathways have been explored in an attempt to explain the link between UA and HTN.
In an experimental study involving human endothelial cells, UA significantly increased the production of reactive oxygen species and angiotensin II, and at concentration of more than 6 and 9 mg/dL induced senescence and apoptosis of endothelial cells, respectively. Oxidative stress is known to be a major mechanism of endothelial dysfunction, which plays an important role in the development of cardiovascular disease, hypertension, and renal disease [26].
In another experimental study, mild hyperuricemia (1.5- to 2-fold increase in serum UA) was induced in rats by providing a uricase inhibitor (oxonic acid) in the diet. Hyperuricemic rats developed significant increase in blood pressure, as compared to the control group, starting 3 weeks. This effect was independent of oxonic acid administration, since HTN could be prevented if uric acid was lowered by either a xanthine oxidase inhibitor or a uricosuric agent. Hyperuricemic rats exhibited an increase in juxtaglomerular renin and a decrease in macula densa neuronal nitric oxide synthase, in addition to renal tubulo-interstitial injury, providing another mechanistic rationale for HTN and renal disease in the setting of hyperuricemia [27].
To date, only a few clinical trials have assessed the potential role of reducing serum UA levels in the prevention or control of HTN.
In a randomized controlled, double blind trial, 60 obese children aged 11 to 17 years with prehypertension were randomly assigned to receive placebo, allopurinol, or probenecid for 7 weeks. Patients in the active treatment groups experienced a marked reduction in office systolic BP of 10.2 mmHg and diastolic BP of 9 mmHg as compared with an increase of 1.7 and 1.6 mmHg in systolic and diastolic BP, respectively, in patients taking placebo [28].
In another randomized crossover trial involving 30 adolescent patients with hyperuricemia and HTN, treatment with allopurinol decreased 24-h systolic BP by 6.8 mmHg, as compared to an increase of 0.8 mmHg in the placebo group [29].
In a non-randomized, prospective trial, allopurinol 300 mg/day was administered to 48 patients with hyperuricemia and included 34 patients with HTN. At 3 months, allopurinol achieved a significant decrease in both systolic and diastolic BP as compared to baseline (mean BP of 135.4/80.2 at baseline to 131.5/78.3 at 3 months) [30].
Hyperuricemia and Chronic Kidney Disease
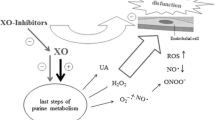
Traditionally, hyperuricemia associated with hyperuricosuria (urinary excretion of UA greater than 800 mg/day in men and greater than 750 mg/day in women) has been postulated to cause acute kidney injury by depositing intraluminal crystals in the collecting duct of the nephron causing tubular obstruction. Apart from tubular obstruction, emerging experimental data have linked hyperuricemia to induction of renal tubulo-interstial inflammation by UA crystals and renal injury by crystal-independent mechanisms such as induction of oxidative stress, activation of the renin-angiotensin system, and endothelial dysfunction, ultimately leading to development and progression of CKD [8, 31] (Fig. 3).

Mechanisms by which hyperuricemia may contribute to CKD development and progression. Reprinted from Jalal DI et al [8], with permission from Elsevier
Several studies in the general population and in patients with CKD have confirmed the association of hyperuricemia with development and progression of kidney disease.
In two community-based cohorts involving more than 13,000 healthy participants followed for up to 9 years, every 1 mg/dL increase in baseline serum UA level was associated with a 7 % increased risk for developing kidney disease (defined as an eGFR <60 mL/min) after adjusting for multiple parameters including HTN, cardiovascular disease, and baseline kidney function [32]. In a meta-analysis of 13 observational studies involving more than 190,000 participants with normal kidney function at baseline, hyperuricemia was associated with a twofold increased risk of new-onset CKD [15].
In a retrospective cohort of 803 patients with CKD, serum UA of more than 6 mg/dL was significantly associated with progression to ESRD during follow-up [33]. Furthermore, in a retrospective cohort study of 353 patients with IgA nephropathy, hyperuricemia was identified as an independent risk factor for the doubling of serum creatinine or progression to ESRD over a mean follow-up of 5 years [34].
A few small trials have assessed the effect of UA-lowering therapies in patients with CKD.
In a randomized controlled trial involving 40 hyperuricemic patients with IgA nephropathy, allopurinol (100–300 mg/day) or placebo was administered for 6 months. No difference was observed between the two groups with respect to renal function and proteinuria; however, antihypertensive drug requirement was reduced for patients in the allopurinol group [34].
In a post hoc analysis of the RENAAL trial, the risk of renal events was decreased by 6 % for every 0.5 mg/dL decrement in serum UA level during the first 6 months of treatment with losartan. Losartan is known to decrease UA levels by decreasing UA absorption in the proximal tubule [35]. In addition, in a recently published randomized controlled trial, the SGLT2 inhibitor empagliflozin slowed the progression of renal disease in patients with diabetes. Empagliflozin effectively reduced serum UA levels as compared to placebo, and a possible contribution of this effect on renal outcomes was postulated by the authors [36]. In a prospective, randomized, controlled trial, 54 hyperuricemic patients with mild to moderate CKD were randomly assigned to treatment with allopurinol, 100 to 300 mg/day, or to continue usual therapy for 12 months. There was a trend towards a lower serum creatinine level in the allopurinol group, although the result did not reach statistical significance. However, significantly more patients in the control group reached a combined endpoint of deterioration of renal function and dialysis dependence at 12 months (16 % in the allopurinol group versus 46.1 % in the control group) [37].
In another prospective, randomized trial that includes 113 patients with an estimated GFR (eGFR) <60 mL/min, allopurinol therapy slowed CKD progression with an increase in eGFR of 1.3 ± 1.3 mL/min/1.73 m2 versus a decrease of 3.3 ± 1.2 mL/min per 1.73 m2 in the control group, after 24 months. More importantly, allopurinol decreased cardiovascular events by 71 %. However, the results of this trial need to be interpreted cautiously since it was not blinded, and the use of other medications such as statins and RAAS inhibitors was left to the judgment of the treating physician, therefore not necessarily balanced between the two groups [38].
And finally, the use of febuxostat was recently evaluated in a randomized, double blind, placebo-controlled study in 93 patients with advanced CKD (stages 3 and 4) and asymptomatic hyperuricemia (UA ≥7 mg/dL). After 6 months, mean changes in eGFR were +3.2 and −4.4 mL/min/1.73 m2 in the febuxostat and placebo groups, respectively. Although the results of this trial are encouraging, its limitations included a small sample size, a short follow-up time, and the fact that it was a single center trial. These results need to be confirmed in larger multicenter trials, with longer follow-up [39].
Conclusions
In summary, epidemiologic and experimental findings support the hypothesis that the increase in serum UA levels can antedate the development of hypertension and can play a major pathogenic role in the development of cardiovascular disease [40]. In addition, hyperuricemia may have a pivotal role in the development and progression of CKD. Available evidence suggests that UA-lowering therapy may have a favorable role in the management of HTN and in the slowing progression of CKD. Large randomized controlled trials with prolonged follow-up of UA-lowering therapies versus placebo are warranted and needed to evaluate safety and efficacy of such therapies in HTN and CKD. In addition, further studies are needed to clarify both the serum UA level at which intervention is warranted and the optimal target level needed to prevent the progression of CKD and cardiovascular events.
References
Rock KL, Kataoka H, Lai JJ. Uric acid as a danger signal in gout and its comorbidities. Nat Rev Rheumatol. 2013;9(1):13–23.
de Oliveira EP, Burini RC. High plasma uric acid concentration: causes and consequences. Diabetol Metab Syndr. 2012;4:12.
Nuki G, Simkin PA. A concise history of gout and hyperuricemia and their treatment. Arthritis Res Ther. 2006;8 Suppl 1:S1.
Kanbay M et al. Uric acid in metabolic syndrome: from an innocent bystander to a central player. Eur J Intern Med. 2016;29:3–8.
Mende C. Management of chronic kidney disease: the relationship between serum uric acid and development of nephropathy. Adv Ther. 2015;32(12):1177–91.
Couser WG et al. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011;80(12):1258–70.
Mallat SG, Itani HS, Tanios BY. Current perspectives on combination therapy in the management of hypertension. Integr Blood Press Control. 2013;6:69–78.
Jalal DI et al. Uric acid as a target of therapy in CKD. Am J Kidney Dis. 2013;61(1):134–46.
Maiuolo J et al. Regulation of uric acid metabolism and excretion. Int J Cardiol. 2016;213:8–14.
Moriwaki Y. Effects on uric acid metabolism of the drugs except the antihyperuricemics. J Bioequivalence Bioavailab. 2014;2014.
Bobulescu IA, Moe OW. Renal transport of uric acid: evolving concepts and uncertainties. Adv Chronic Kidney Dis. 2012;19(6):358–71.
Lioté F. Hyperuricemia and gout. Curr Rheumatol Rep. 2003;5(3):227–34.
McDonagh EM et al. PharmGKB summary: uric acid-lowering drugs pathway, pharmacodynamics. Pharmacogenet Genomics. 2014;24(9):464–76.
Terkeltaub R. Update on gout: new therapeutic strategies and options.
Li L et al. Is hyperuricemia an independent risk factor for new-onset chronic kidney disease?: a systematic review and meta-analysis based on observational cohort studies. BMC Nephrol. 2014;15:122.
Lapi F et al. Concurrent use of diuretics, angiotensin converting enzyme inhibitors, and angiotensin receptor blockers with non-steroidal anti-inflammatory drugs and risk of acute kidney injury: nested case-control study. BMJ. 2013;346:e8525.
Zhu, J., et al., Dietary factors associated with hyperuricemia and glyeolipid metabolism disorder in middle-aged and elderly people. Sichuan da xue xue bao. Yi xue ban = Journal of Sichuan University. Medical science edition, 2016. 47(1): p. 68.
Guan S et al. Prevalence of hyperuricemia among Beijing post-menopausal women in 10 years. Arch Gerontol Geriatr. 2016;64:162–6.
Zhu Y, Pandya BJ, Choi HK. Prevalence of gout and hyperuricemia in the US general population: the national health and nutrition examination survey 2007–2008. Arthritis Rheum. 2011;63(10):3136–41.
Anton FM et al. Sex differences in uric acid metabolism in adults: evidence for a lack of influence of estradiol-17 beta (E2) on the renal handling of urate. Metabolism. 1986;35(4):343–8.
Liu B, et al. The prevalence of hyperuricemia in China: a meta-analysis. BMC Public Health. 2011;11(1):1–10.
Hak AE, Choi HK. Menopause, postmenopausal hormone use and serum uric acid levels in US women—the third national health and nutrition examination survey. Arthritis Res Ther. 2008;10(5):R116.
Grayson PC et al. Hyperuricemia and incident hypertension: a systematic review and meta-analysis. Arthritis Care Res. 2011;63(1):102–10.
Cicero AF et al. Association between serum uric acid, hypertension, vascular stiffness and subclinical atherosclerosis: data from the Brisighella heart study. J Hypertens. 2014;32(1):57–64.
Perlstein TS et al. Uric acid and the development of hypertension: the normative aging study. Hypertension. 2006;48(6):1031–6.
Yu M-A et al. Oxidative stress with an activation of the renin–angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens. 2010;28:1234–42.
Mazzali M, Hughes J, Kim YG, et al. Elevated uric acid increases blood pressure in the rat by a novel crystal-independent mechanism. Hypertension. 2001;5(38):1101–6.
Soletsky B, Feig DI. Uric acid reduction rectifies prehypertension in obese adolescents. Hypertension. 2012;60(5):1148–56.
Feig DI, Soletsky B, Johnson RJ. Effect of allopurinol on blood pressure of adolescents with newly diagnosed essential hypertension: a randomized trial. JAMA. 2008;300(8):924–32.
Kanbay M et al. Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearence, and proteinuria in patients with normal renal functions. Int Urol Nephrol. 2007;39(4):1227–33.
Isaka Y, et al. Hyperuricemia-induced inflammasome and kidney diseases. Nephrol Dial Transplant. 2015.
Weiner DE et al. Uric acid and incident kidney disease in the community. J Am Soc Nephrol. 2008;19(6):1204–11.
Uchida S et al. Targeting uric acid and the inhibition of progression to end-stage renal disease—a propensity score analysis. PLoS One. 2015;10(12):e0145506.
Shi Y et al. Clinical outcome of hyperuricemia in IgA nephropathy: a retrospective cohort study and randomized controlled trial. Kidney Blood Press Res. 2011;35(3):153–60.
Miao Y et al. Effect of a reduction in uric acid on renal outcomes during losartan treatment: a post hoc analysis of the reduction of endpoints in non-insulin-dependent diabetes mellitus with the Angiotensin II Antagonist Losartan Trial. Hypertension. 2011;58(1):2–7.
Wanner C et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375(4):323–34.
Siu YP et al. Use of allopurinol in slowing the progression of renal disease through its ability to lower serum uric acid level. Am J Kidney Dis. 2006;47(1):51–9.
Goicoechea M et al. Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol. 2010;5(8):1388–93.
Sircar D et al. Efficacy of febuxostat for slowing the GFR Decline in patients With CKD and asymptomatic hyperuricemia: a 6-month, double-blind, randomized, placebo-controlled trial. Am J Kidney Dis. 2015;66(6):945–50.
Mancia G, Grassi G, Borghi C. Hyperuricemia, urate deposition and the association with hypertension. Curr Med Res Opin. 2015;31 Suppl 2:15–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Drs. Mallat, Al Kattar, Tanios, and Jurjus declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Hypertension and the Kidney
Rights and permissions
About this article

Cite this article
Mallat, S.G., Al Kattar, S., Tanios, B.Y. et al. Hyperuricemia, Hypertension, and Chronic Kidney Disease: an Emerging Association. Curr Hypertens Rep 18, 74 (2016). https://doi.org/10.1007/s11906-016-0684-z
Published:
DOI: https://doi.org/10.1007/s11906-016-0684-z