
Abstract
In analogy to many tissues in which mature, terminally differentiated cells are continuously replenished by the progeny of less differentiated, long-lasting stem cells, it has been suspected that memory T lymphocytes might contain small numbers of stem cell-like cells. However, only recently have such cells been physically identified and isolated from humans, mice, and nonhuman primates. These cells, termed “T memory stem cells” (TSCM), represent approximately 2–4 % of all circulating T lymphocytes, seem to be extremely durable, and can rapidly differentiate into more mature central memory, effector memory, and effector T cells, while maintaining their own pool size through homeostatic self-renewal. Although it is becoming increasingly evident that that these cells have critical roles for T cell homeostasis and maintaining life-long cellular immunity against microbial pathogens during physiological conditions, they also seem intrinsically involved in many key aspects of HIV/SIV disease pathogenesis. Current data suggest that CD4+ TSCM cells represent a core element of the HIV-1 reservoir in patients treated with suppressive antiretroviral therapy (ART) and that relative resistance of CD4+ TSCM cells to SIV represents a distinguishing feature of non-pathogenic SIV infection in natural hosts. This article summarizes recent studies investigating the role of TSCM in HIV/SIV infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
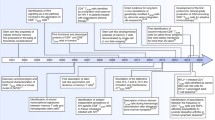
Memory T cells represent the largest lymphocyte population in the adult human body and play critical roles for maintaining life-long antimicrobial immune defense against specific pathogens. From an evolutionary perspective, memory T cells develop when antigen-specific naive CD4+ or CD8+ T cells become activated upon antigen exposure and subsequently undergo proliferative expansion and differentiation [1]. Whether naive T cells directly develop into memory T cells or first differentiate into effector cells of which a few are selected for immunological memory during a subsequent contraction phase is unclear at present [2]. Based on complementary studies in humans and mice, memory T cells can be divided into central memory (TCM) and effector memory (TEM) T cells [3–5]. TCM cells represent a long-lasting cell population that express lymphoid tissue homing markers, secrete mostly IL-2 upon TCR stimulation, and have a high proliferative capacity and a prolonged in vivo half-life. In contrast, effector memory T cells are more short-lived, express chemokine receptors and adhesion molecules that allow for extravasation into inflamed tissues, and can more rapidly execute lymphocellular effector functions. Current experimental data and theoretical considerations suggest that these memory T cell populations evolve in a hierarchical developmental process during which more immature, long-lasting T cells serve as precursors for more differentiated, mature, and short-lived memory cell subsets [3, 6, 7]. Such a linear and progressive model of memory T cell development is reminiscent of the hierarchical developmental structure of the hematological and epithelial systems, in which small populations of multipotent, tissue-specific stem cells are able to constantly replenish large populations of differentiated effector cells, while maintaining their own life-long survival through homeostatic self-renewal. Such considerations have led to the provocative hypothesis that small populations of highly undifferentiated and long-lived memory T cells with stem cell-like properties could be the basis for the continual generation of central memory, effector memory, and effector lymphocytes from the memory pool [8–11] (Fig. 1a). Recently, immense progress has been made in identifying such stem cell-like memory T cell populations in humans and other species [12, 13, 14••], and emerging data suggest that these cells play a critical role in the pathogenesis of HIV and SIV infection in humans and nonhuman primates.

Schematic of CD4+ memory T cell homeostasis (a) and proposed relative contribution of CD4+ T memory subsets to the persistent reservoir of HIV/SIV (b, top panel). In b, the potential effects of drugs promoting enhanced self-renewal (middle panel) or enhanced differentiation (bottom panel) of TSCM on the overall CD4+ memory T cell reservoir are shown. Stars represent cells latently infected with HIV/SIV
Discovery of T Memory Stem Cells
A population of CD8+ T cells with stem cell-like properties, later termed “T memory stem cells” (TSCM), was first experimentally observed in a murine model of graft-versus-host disease [15]. These alloreactive CD8+ T cells demonstrated enhanced self-renewal capacity and multipotency and were able to differentiate into central memory, effector memory, and effector T cells. In this mouse model, these candidate memory stem cells were characterized by low surface expression of CD4 and high expression of CD62L, stem cell antigen-1 (Sca-1), CD122, CXCR3, and Bcl-2. Since that initial description, the properties of TSCM have been explored in detail in mice [13], humans [12], and nonhuman primates [14••], in the healthy state as well as in the setting of cancer or HIV/SIV infection [16••, 17••, 18•, 19]. In these studies, TSCM have been defined by the expression of naive T cell markers such as CD45RA and CCR7, in tandem with memory T cell markers including CD95 and CD122, among others (Table 1). Such cells were detected both within CD4+ and CD8+ T cell subsets and account for approximately 2–4 % of all cells in each compartment. As the defining functional characteristic, these long-lived TSCM exhibit a multipotent developmental program that allows them to (i) continuously regenerate their own pool size through homeostatic self-renewal and (ii) repopulate more differentiated memory T cell subsets, including central memory, transitional memory, and effector memory T cells in vitro, as well as in vivo upon serial transplantation in animal experiments [8, 12, 13, 14••, 20, 21]. Further, TSCM have been shown to be antigen-experienced T cells with a molecular signature that overlaps with but is still distinct from both naive and central memory T cells [12]. Homeostasis, proliferation, and differentiation of TSCM seem to be governed by signaling pathways that are active in hematopoietic stem cells, thereby supporting the current nomenclature terming them “stem cell-like” memory T cells or, more simply, T memory stem cells. In vitro, TSCM can be differentiated from naive T cells using TCR ligation along with co-stimulation through CD28 in the presence of IL-7 and IL-15 [22]. In this review, we will summarize recent advances in our knowledge of TSCM in HIV and SIV infection, with an emphasis on their contribution to viral persistence, and describe the role of TSCM in cellular immunity. Moreover, we will describe the signaling pathways that drive TSCM self-renewal or proliferation and postulate how these pathways may be targeted for therapeutic purposes, including for HIV cure.
Role of TSCM in Cellular Immunity
Despite the expression of several naive T cell markers, prior studies demonstrated that TSCM could rapidly execute classical lymphocellular effector functions and secrete a number of different cytokines when stimulated with cognate antigen or experimental TCR ligands. Cytokines released by TSCM upon antigenic challenge include IL-2, TNF-α, and IFN-γ [12], but the fraction of responding cells within the TSCM compartment is typically smaller when compared to more mature T cell populations, consistent with the fact that TSCM represent the least differentiated T cell subset. Other lymphocellular effector mechanisms, including cytotoxic activities and expression of lytic granules, have not yet been investigated in CD4+ or CD8+ TSCM.
So far, the presence of CMV-, Flu-, SIV-, and melanoma-specific CD8+ TSCM has been formally demonstrated [12, 14••], but CD4+ and CD8+ TSCM are likely to contribute to cellular immune responses against any microbial pathogen that challenges the host and may also be inducible by vaccines or immunogens. Yet, the extent to which these cells may represent a correlate of immune protection, either during natural infection or after vaccination, is currently unknown. In the context of HIV-1 infection, a recent study demonstrated that in untreated patients, proportions of total CD8+ TSCM were inversely correlated with levels of plasma viremia and with the degree of T cell-dependent immune activation [23], which represents an independent predictor of disease progression; moreover, there was a positive association with CD4+ T cell counts in the peripheral blood [23]. HIV-1-specific TSCM have not yet been investigated, neither in the CD4+ nor in the CD8+ T cell compartment, and their role for antiviral immune defense is uncertain at present. However, given that more immature memory HIV-1-specific CD8+ T cells were preferentially observed in persons with natural control of HIV-1 [24], it is possible that HIV-1-specific TSCM may contribute to HIV-1 restriction in this particular patient population. In the setting of SIV infection in rhesus macaques, prior studies demonstrated that SIV-specific CD8+ TSCM are generated early after infection and persist throughout the disease process, but their contribution to the total number of SIV-specific CD8+ T cells recognizing a given epitope is low in the majority of cases [14••]. Notably, long-term persistence of SIV-specific TSCM was demonstrated even after mutational escape of the targeted viral epitope, consistent with long-term antigen-independent persistence of antigen-specific CD8+ TSCM. In contrast, many antigen-specific TCM and TEM cells recognizing the original wild-type epitope disappeared relatively rapidly after the emergence of viral escape mutations, indicating that they depend on continuous antigenic challenge to maintain their long-term survival. A better characterization of antigen-specific CD4+ and CD8+ TSCM in HIV/SIV infection and other chronic infections represents an important area of future research.
CD4+ TSCM as an HIV-1 Reservoir During Treatment with ART
Given that CD4+ TSCM seem to represent a very long-lasting cell population, they may represent a preferential cellular niche that supports long-term HIV-1 persistence in patients on suppressive antiretroviral therapy (ART) (Fig. 1b). To investigate this, prior studies initially analyzed the susceptibility of CD4+ TSCM to HIV-1 [25–27]. Since hematopoietic stem cells seem to be largely resistant to HIV-1, it was hypothesized that CD4+ TSCM, which appear to be regulated at least in part by stem cell-specific pathways, may possess cell-intrinsic immune defense mechanisms that protect this cell subset from retroviral infection. However, studies from a number of different laboratories demonstrated that CD4+ TSCM express CCR5, although at low levels, and are able to support productive and latent infection with R5-tropic HIV-1. In addition, CD4+ TSCM were highly permissive to ex vivo infection with a VSV-G pseudotyped HIV-1 virus and expressed comparatively low levels of cell-intrinsic viral restriction factors, such as SAMHD1, Trim5alpha, and APOBEC3G [16••]. Importantly, these investigations also demonstrated that CD4+ TSCM from untreated HIV-1-infected patients harbored high levels of HIV-1 RNA, indicating that these cells are susceptible to HIV-1 in vivo. Using CD4+ TSCM sorted from patients undergoing suppressive ART, subsequent studies demonstrated that CD4+ TSCM harbor replication-competent virus that can be reactivated in viral outgrowth assays, further suggesting that CD4+ TSCM can represent a reservoir for latent HIV-1 infection [16••, 18•]. Yet, due to their low frequency, the viral reservoir in CD4+ TSCM only accounted for a small proportion of the total reservoir in CD4+ T cells in most patients [16••].
Interestingly, the relative contribution of CD4+ TSCM to the total viral reservoir seems to critically depend on the timing and the duration of ART. For instance, in long-term treated patients who initiated ART early after infection and had a comparatively small total HIV reservoir, CD4+ TSCM appeared to make a relatively larger contribution to the total viral reservoir size, as opposed to patients who started treatment during the chronic phase of the infection and had an increased total viral reservoir size [28•]. Moreover, the contribution of CD4+ TSCM to the total viral reservoir seems to be inversely related to the total viral reservoir, an association that was exclusively observed for the CD4+ TSCM compartment, and not for alternative CD4+ T cell populations. Longitudinal evaluations spanning more than 10 continuous years of suppressive ART indicated that HIV-1 DNA in CD4+ TSCM remained largely stable, while decay of HIV-1 DNA in alternative CD4+ T cell populations was more obvious [16••, 18•]. Correspondingly, CD4+ TSCM contributed only a small proportion to the viral reservoir after short-term ART. Yet, after longer-term ART, the fraction of the viral reservoir harbored by CD4+ TSCM was significantly larger, while contributions made by other subsets, in particular short-lived CD4+ TEM cells, declined [16••]. This disproportionate increase in the contribution of CD4+ TSCM to the total viral reservoir over time is consistent with a role of CD4+ TSCM as one of the most durable and stable reservoirs for HIV-1 that becomes increasingly visible after prolonged periods of ART.
A similar observation was made in a geographically distinct population of HIV-1-infected patients treated with antiretroviral agents for varying periods of time in France [18•]. Chromosomally integrated HIV-1 DNA was readily detectable in CD4+ TSCM in all but three individuals out of a total study cohort of 38 ART-treated HIV-1-infected patients. Importantly, linear regression analyses of these data indicated that the half-life of HIV-1 DNA in CD4+ TSCM was almost twice as long as in CD4+ TCM cells, the compartment with the second-longest half-life of HIV-1 DNA, and more than three times longer than the half-life of HIV-1 DNA in CD4+ TEM cells. Interestingly, analysis of data from this study also led to the conclusion that the relative proportion of HIV-1 DNA in the CD4+ TSCM compartment progressively expands over time, while the fraction of HIV-1 DNA harbored by other CD4+ T cell subsets declined, with the net result of a contraction of the viral reservoir around a core of less differentiated, long-lasting memory CD4+ T cells. It should be mentioned, however, that these studies did not investigate whether and how HIV-1-infected CD4+ TSCM may stabilize the viral reservoir by repopulating the pool of HIV-1-infected CD4+ TEM and TCM cells through transitional proliferation. Given that TSCM can readily differentiate in more mature CD4+ T cell populations in in vitro assays and rapidly repopulate cellular immunity upon serial transplantation in animal models, it is expected that HIV-1-infected CD4+ TSCM can indeed perpetuate HIV-1 persistence by homeostatic self-renewal as well as by continuously differentiating into more mature memory CD4+ T cell populations that are also infected with HIV-1; however, this remains to be formally demonstrated.
Role of TSCM in SIV Pathogenesis
Asian rhesus macaques infected with SIV represent the best animal model of HIV infection due to many similarities in terms of early transmission events, viral and CD4+ T cell dynamics, establishment of reservoirs, and disease progression [29–33]. Similar to HIV-infected patients, SIV-infected rhesus macaques display a high viral load peak that falls to a set point level during the period of primary/acute infection. Viral loads during the chronic phase of infection are typically in the range of 104–107 and are accompanied by CD4+ T cell decline that progresses to AIDS and death over 1–2 years. African monkey species, such as sooty mangabeys and African green monkeys, are naturally infected with SIV both in the wild and in captivity. Despite peak and set point viral loads that are comparable to those of HIV-infected humans and SIV-infected macaques, these “natural hosts” do not progress to AIDS and manifest only a minimal decline in peripheral CD4+ T cell counts [34]. One of the key features that differ between natural hosts and non-natural hosts (i.e., human and macaques) is the level of cell surface expression of the HIV/SIV co-receptor CCR5 [35]. In natural hosts, high-level expression of CCR5 is restricted to the effector memory subset of CD4+ T cells whereas central memory CD4+ T cells maintain low CCR5 levels even after exposure to activating stimuli in vitro or in vivo [36]. In fact, the level of CCR5 expression has been shown to correlate with the ability of SIV to infect specific cell subsets as well as with age at SIV acquisition [36–39]. These observations led to the hypothesis that differential SIV infection of CD4+ TSCM (as a consequence of the level of cell surface CCR5) may distinguish pathogenic SIV infection of rhesus macaques from non-pathogenic SIV infection of sooty mangabeys (described further below).
The phenotypic and functional properties of TSCM, both CD8+ and CD4+, in healthy and SIV-infected macaques were first described by the group of Mario Roederer [14••, 19]. Similar to their description in humans, TSCM were identified using markers of naive T cells with further characterization as CD27 + CD28 + IL-7Ralpha + CD95+ in rhesus and pigtailed macaques. These cells represented about 2–3 % of circulating CD8+ and CD4+ T cells and were slightly more abundant in lymph nodes (for CD8+ TSCM only) and less so at mucosal sites. Notably, during pathogenic HIV and SIV infection, a high level of direct infection of CD4+ TCM cells is associated with depletion of CD4+ T cells as well as chronic immune activation [36, 40–44]. Although potentially less so than TSCM, TCM cells are thought to contribute to overall CD4+ T cell homeostasis due to their ability to undergo rounds of proliferation that maintain their pool as well as differentiate into the shorter-lived effector memory subset. Direct virus infection and killing of CD4+ TCM cells therefore have implications for the maintenance of long-lived immunity as well as overall CD4+ T cell homeostasis. As the non-pathogenic SIV infection of sooty mangabeys is characterized by preservation of CD4+ TCM cells [36], we postulated that CD4+ TSCM would also be maintained and that this state of memory stem cell immunocompetence represents a hallmark of non-pathogenic SIV infection in natural hosts.
When directly compared, the levels of circulating CD4+ TSCM in healthy rhesus macaques were slightly higher than in sooty mangabeys (1–8 vs. 0.5–3 %), although CD4+ TSCM from mangabeys expressed higher levels of the proliferation marker Ki-67 [17••]. In addition, CD4+ TSCM from sooty mangabeys demonstrated an almost complete absence of cell surface CCR5, unlike CD4+ TSCM from macaques, suggesting a potential for differential levels of SIV infection. Indeed, a low level of SIV DNA in CD4+ TSCM could only be detected in 2/10 SIV-infected sooty mangabeys, in contrast to 9/9 SIV-infected rhesus macaques with SIV DNA levels in CD4+ TSCM that were comparable to those of other CD4+ T memory subsets [17••]. Furthermore, the pathogenic SIV infection of rhesus macaques was associated with a selective depletion of CCR5 + CD4+ TSCM whereas the percentage and absolute number of CD4+ TSCM along with the percentage of CD4+ TSCM expressing CCR5 did not differ in SIV-uninfected and SIV-infected mangabeys. Similar observations were also made in human viremic non-progressors, an exquisitely rare group of HIV-1-infected patients who maintain sufficient CD4+ T cell counts for prolonged periods of time in the setting of ongoing high-level HIV-1 replication, and in this way closely resemble the clinical picture of SIV infection in natural hosts. In these patients, the levels of HIV-1 DNA in CD4+ TSCM were significantly lower than in normally progressing, untreated HIV-1-infected patients [45•], suggesting that the CD4+ TSCM compartment of these specific patients does not support effective HIV-1 replication. Collectively, this work suggested that the relative resistance of CD4+ TSCM to lentiviral infection in natural hosts of SIV infection and in HIV-1-infected viremic non-progressors is associated with protection against HIV/SIV disease progression. Vice versa, perturbed homeostasis of CD4+ TSCM in SIV-infected rhesus macaques (manifested by both increased infection and depletion) likely promotes lentiviral pathogenesis. Based on these results in nonhuman primates as well as the recognized role of CD4+ TSCM as an HIV-1 reservoir in ART-treated humans, experiments are underway in our laboratories to fully understand the contribution of CD4+ TSCM to the total body SIV reservoir in ART-suppressed rhesus macaques and sooty mangabeys.
Signaling Pathways Active in TSCM
The provocative observation that TSCM seem to be able to imitate many of the functional characteristics conventionally ascribed to tissue-specific stem cells has raised considerable interest in the mechanisms controlling TSCM behavior. Would it be possible that transcriptional programs and regulatory mechanisms of classical tissue-specific stem cells can be activated in TSCM, despite the fact TSCM represent committed lymphocytes? Recent data suggest that this is at least partially true, specifically for the molecular control of TSCM fate decisions. In hematopoietic and epithelial stem cells, checkpoint decisions between self-renewal vs. differentiation into more mature cells are in part regulated by the phylogenetically conserved Wnt/β-catenin signaling pathway [46]. In the canonical Wnt/β-catenin signaling cascade, extracellular Wnt glycoproteins bind to 7-transmembrane Frizzled receptors and co-receptors LRP5 and LRP6, leading to phosphorylation of LRP5/6 and subsequent binding by Axin and other members of the β-catenin destruction complex [47]. In the absence of Wnt binding, this destruction complex (made up of Axin, Dishevelled, GSK3, CK1, and APC) binds and phosphorylates β-catenin in the cytoplasm leading to ubiquitination and degradation of β-catenin by the proteasome. In the presence of Wnt signaling, however, β-catenin is released from the destruction complex and can move freely into the nucleus where it binds to the TCF/LEF family of transcription factors, resulting in transcription of target genes [47, 48]. Hypothesizing that Wnt/β-catenin is also involved in regulating cell fate decision of TSCM, Gattinoni et al. demonstrated that induction of the Wnt signaling pathway using a pharmacological inhibitor of GSK-3β in the presence of antigen in CD8+ T cells led to an accumulation of β-catenin, blocked T cell differentiation, and promoted the generation of self-renewing multipotent TSCM in vitro [13]. These observations are in line with prior studies showing that mice lacking the TCF7 gene, which encodes for a downstream effector of the Wnt/β-catenin pathway, exhibited a more differentiated T cell phenotype [49] and that decreasing expression of Lef1 and TCF7 was associated with progressive differentiation of T cells in humans and mice [50]. Moreover, high-level expression of β-catenin was associated with an increased ability to form functional memory cell responses in vivo [51]. Together, these data suggest stem cell physiology and regulatory pathways involved in stem cell fate decisions can at least transiently be activated in non-stem cells such as lymphocytes and allow for a stem cell-specific functional profile in committed lymphocytes that is otherwise exclusively encountered in traditional stem cells. Whether other stem cell-specific signaling pathways, such as the Notch or sonic hedgehog signaling cascade, are also involved in regulating TSCM behavior represents an important aspect of future investigations.
Opportunities to Target TSCM to Reduce the HIV/SIV Reservoir
Although once regarded as an elusive goal, the development of clinical strategies that can lead to a long-term drug-free remission of HIV-1 infection has become a more and more realistic objective. This is in part related to the recent identification of patients with a sterilizing or functional cure of HIV-1 infection, which provides living evidence that at least in principle, a complete or near-complete eradication of residual HIV-1 reservoirs is possible [44, 52, 53]. Most clinical approaches that are currently evaluated as strategies to reduce HIV-1 persistence despite ART focus on the “shock and kill” strategy, which is based on the use of pharmaceutical agents that can reverse viral latency, followed by immune-based interventions that may kill cells in which viral reactivation has been successfully induced. Although this concept is currently being tested in a number of pre-clinical and clinical studies, it is uncertain whether this strategy would be effective in targeting the latent viral reservoir in CD4+ TSCM and TCM cells, which arguably represents the most durable and long-lived site for long-term viral persistence and the most critical barrier to HIV-1 cure. As an alternative to the shock and kill approach, strategies that specifically destabilize the viral reservoir in these long-lasting CD4+ TSCM and TCM cells may therefore represent promising and possibly more effective avenues for future clinical interventions to reduce HIV-1 persistence. Such approaches will likely have to specifically target molecular pathways that are responsible for self-renewal, survival, and proliferation of CD4+ TSCM and TCM cells. As described above, homeostasis of the CD4+ TSCM and TCM cell pool seems to be maintained at least in part by molecular mechanisms that are similar or identical to stem cell-specific, phylogenetically conserved signaling cascades regulating the “stemness” (i.e., multipotency, self-renewal, and long-term persistence) of classical hematopoietic or epithelial stem cells. These pathways are currently also under active investigation for targeting cancer stem cells, a small subset of long-lived cancer cells with high oncogenic potential that in many cases are responsible for persistence and recurrence of malignant diseases despite treatment [54–57], and in that sense may represent the functional analog to the reservoir of HIV-1-infected CD4+ TSCM and TCM cells that persist despite antiretroviral therapy in patients. Therefore, drugs designed to manipulate cancer stem cells through interference with stem cell-specific signaling pathways may offer novel opportunities to specifically target the long-lived core components of the HIV-1 reservoir and reduce long-term viral persistence in HIV-1-infected CD4+ TSCM and TCM cells. This strategy, whereby long-lived, latently HIV-1-infected TSCM and TCM cells are forced to differentiate into TEM and effector T cells with a much shorter half-life, could be termed “push and vanish” (Fig. 1b, bottom panel). Such approaches recognize the structural and developmental heterogeneity of HIV-1-infected CD4+ T cells in ART-suppressed patients and provide a more specific molecular strategy for selectively eliminating the cells that arguably seem most relevant for maintaining and perpetuating HIV-1 persistence.
Conclusions
The discovery of TSCM as the stem cells of cellular immune memory may have critical implications for understanding the ontogeny and the evolution of cellular immune responses and for designing immunological interventions, adoptive immunotherapy, and vaccination strategies. In the context of HIV-1 infection, the idea that HIV-1 can infect stem cells, the most long-lasting cells in the human body, to establish life-long viral persistence has remained conceptually appealing for many years [58, 59]; however, data to support HIV-1 infection of classical tissue-specific stem cells, in particular hematopoietic stem cells, remain controversial [60, 61], since these cells appear to possess defined cell-intrinsic resistance mechanisms to inhibit retroviral infection [62]. Infection of the readily susceptible CD4+ TSCM as a stem cell-like cell compartment may allow HIV-1 to exploit the stem cell characteristics of cellular immune memory to propagate viral persistence indefinitely. As evidence to support this hypothesis increases, HIV-1 CD4+ TSCM may move into the center of current efforts to reduce persistence of replication-competent HIV-1 during ART and offer novel opportunities for targeted destabilization of long-lived viral reservoirs. For instance, genetic manipulation of CD4+ TSCM through gene therapy-induced reduction of CCR5 surface expression may generate a long-lasting population of CD4+ T cells that does not support HIV-1 replication and may increase a patient’s ability to maintain at least a transient drug-free remission of his or her disease. In addition, rapid introduction of ART during the hyperacute stage of HIV-1 infection may spare infection of CD4+ TSCM, in which case a finite course of ART may be curative, or at least allow for longer periods of viral control after treatment discontinuation. A finite course of ART may also be sufficient to destabilize persistent viral reservoirs in natural hosts of SIV infection, in whom CD4+ TSCM seem to be at least partially resistant to SIV infection. Finally, pharmaceutical interventions that transform long-lived HIV-1-infected CD4+ TSCM into short-lived effector cells through interference with molecular programs governing CD4+ TSCM survival and homeostasis may represent a promising approach for reducing long-term HIV-1 persistence that would be applicable to larger populations of ART-treated HIV-1-infected patients.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Crotty S, Ahmed R. Immunological memory in humans. Semin Immunol. 2004;16:197–203.
Restifo NP, Gattinoni L. Lineage relationship of effector and memory T cells. Curr Opin Immunol. 2013;25:556–63.
Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–63.
Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291:2413–7.
Reinhardt RL, Khoruts A, Merica R, Zell T, Jenkins MK. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 2001;410:101–5.
Farber DL, Yudanin NA, Restifo NP. Human memory T cells: generation, compartmentalization and homeostasis. Nat Rev Immunol. 2014;14:24–35.
Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–84.
Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR. A distinct subset of self-renewing human memory CD8+ T cells survives cytotoxic chemotherapy. Immunity. 2009;31:834–44.
Stemberger C et al. Stem cell-like plasticity of naive and distinct memory CD8+ T cell subsets. Semin Immunol. 2009;21:62–8.
Papatriantafyllou M. T cell memory: the stem of T cell memory. Nat Rev Immunol. 2011;11:716.
Ahmed R, Bevan MJ, Reiner SL, Fearon DT. The precursors of memory: models and controversies. Nat Rev Immunol. 2009;9:662–8.
Gattinoni L et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–7.
Gattinoni L et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15:808–13.
Lugli E, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest. 2013;123:594–9. This is the first article to describe CD8+ and CD4+ T SCM cells in nonhuman primates.
Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8+ memory stem cells in graft-versus-host disease. Nat Med. 2005;11:1299–305.
Buzon MJ, et al. HIV-1 persistence in CD4 T cells with stem cell-like properties. Nat Med. 2014;20:139–42. This study first described the importance of CD4+ T SCM cells for long-term HIV-1 persistence in HIV-1-infected individuals treated with suppressive ART.
Cartwright EK, et al. Divergent CD4+ T memory stem cell dynamics in pathogenic and nonpathogenic simian immunodeficiency virus infections. J Immunol. 2014;192:4666–73. Here the authors provide the first evidence for a role of CD4+ T SCM in the pathogenesis of SIV infection. The main conclusion is that CD4+ T SCM cells are preferentially spared from SIV infection in the natural host sooty mangabeys.
Jaafoura S, et al. Progressive contraction of the latent HIV reservoir around a core of less-differentiated CD4(+) memory T cells. Nat Commun. 2014;5:5407. This work independently confirmed the contribution of CD4+ T SCM cells to the long-term HIV-1 reservoir.
Lugli E et al. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc. 2012;8:33–42.
Muranski P et al. Th17 cells are long lived and retain a stem cell-like molecular signature. Immunity. 2011;35:972–85.
Graef P et al. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8(+) central memory T cells. Immunity. 2014;41:116–26.
Cieri N et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573–84.
Ribeiro SP et al. The CD8+ memory stem T cell (TSCM) subset is associated with improved prognosis in chronic HIV-1 infection. J Virol. 2014;88:13836–44.
Ndhlovu ZM et al. Elite controllers with low to absent effector CD8+ T cell responses maintain highly functional, broadly directed central memory responses. J Virol. 2012;86:6959–69.
Cashin K et al. Differences in coreceptor specificity contribute to alternative tropism of HIV-1 subtype C for CD4+ T-cell subsets, including stem cell memory T-cells. Retrovirology. 2014;11:97.
Flynn JK et al. Quantifying susceptibility of CD4+ stem memory T-cells to infection by laboratory adapted and clinical HIV-1 strains. Viruses. 2014;6:709–26.
Tabler CO et al. CD4+ memory stem cells are infected by HIV-1 in a manner regulated in part by SAMHD1 expression. J Virol. 2014;88:4976–86.
Buzon MJ, et al. Long-term antiretroviral treatment initiated in primary HIV-1 infection affects the size, composition and decay kinetics of the reservoir of HIV-1 infected CD4 T cells. J Virol. 2014;88:10056–65. These authors show that early initiation of ART results in a smaller overall HIV-1 reservoir in CD4+ T SCM cells.
Haase AT. Early events in sexual transmission of HIV and SIV and opportunities for interventions. Annu Rev Med. 2011;62:127–39.
Valentine LE, Watkins DI. Relevance of studying T cell responses in SIV-infected rhesus macaques. Trends Microbiol. 2008;16:605–11.
Brenchley JM, Paiardini M. Immunodeficiency lentiviral infections in natural and non-natural hosts. Blood. 2011;118:847–54.
Pandrea IV et al. Acute loss of intestinal CD4+ T cells is not predictive of simian immunodeficiency virus virulence. J Immunol. 2007;179:3035–46.
Whitney JB et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature. 2014;512:74–7.
Chahroudi A, Bosinger SE, Vanderford TH, Paiardini M, Silvestri G. Natural SIV hosts: showing AIDS the door. Science. 2012;335:1188–93.
Pandrea I et al. Paucity of CD4+ CCR5+ T cells is a typical feature of natural SIV hosts. Blood. 2007;109:1069–76.
Paiardini M et al. Low levels of SIV infection in sooty mangabey central memory CD(4)(+) T cells are associated with limited CCR5 expression. Nat Med. 2011;17:830–6.
Chahroudi A et al. Target cell availability, rather than breast milk factors, dictates mother-to-infant transmission of SIV in sooty mangabeys and rhesus macaques. PLoS Pathog. 2014;10:e1003958.
Pandrea I et al. Paucity of CD4+ CCR5+ T cells may prevent transmission of simian immunodeficiency virus in natural nonhuman primate hosts by breast-feeding. J Virol. 2008;82:5501–9.
Pandrea I et al. Mucosal simian immunodeficiency virus transmission in African green monkeys: susceptibility to infection is proportional to target cell availability at mucosal sites. J Virol. 2012;86:4158–68.
Brenchley JM et al. T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. J Virol. 2004;78:1160–8.
Okoye A et al. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. J Exp Med. 2007;204:2171–85.
Brenchley JM et al. Differential infection patterns of CD4+ T cells and lymphoid tissue viral burden distinguish progressive and nonprogressive lentiviral infections. Blood. 2012;120:4172–81.
Descours B et al. Immune responses driven by protective human leukocyte antigen alleles from long-term nonprogressors are associated with low HIV reservoir in central memory CD4 T cells. Clin Infect Dis. 2012;54:1495–503.
Saez-Cirion A et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013;9:e1003211.
Klatt NR, et al. Limited HIV infection of central memory and stem cell memory CD4+ T cells is associated with lack of progression in viremic individuals. PLoS Pathog. 2014;10:e1004345. This study identified a fascinating subset of HIV-1-infected individuals, termed “viremic non-progressors”, that remain clinically healthy in the face of high viral loads. Interestingly, they also found low levels of HIV DNA in CD4+ T SCM cells from these patients, similar to what has been observed in sooty mangabeys.
Ring A, Kim YM, Kahn M. Wnt/catenin signaling in adult stem cell physiology and disease. Stem Cell Rev. 2014;10:512–25.
Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–205.
Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–32.
Schilham MW et al. Critical involvement of Tcf-1 in expansion of thymocytes. J Immunol. 1998;161:3984–91.
Willinger T et al. Human naive CD8 T cells down-regulate expression of the WNT pathway transcription factors lymphoid enhancer binding factor 1 and transcription factor 7 (T cell factor-1) following antigen encounter in vitro and in vivo. J Immunol. 2006;176:1439–46.
Williams MA, Ravkov EV, Bevan MJ. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity. 2008;28:533–45.
Persaud D et al. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N Engl J Med. 2013;369:1828–35.
Hutter G et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–8.
Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–62.
Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011;8:97–106.
Wang J, Sullenger BA, Rich JN. Notch signaling in cancer stem cells. Adv Exp Med Biol. 2012;727:174–85.
Lenz HJ, Kahn M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci. 2014;105:1087–92.
Carter CC et al. HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe. 2011;9:223–34.
McNamara LA, Collins KL. Hematopoietic stem/precursor cells as HIV reservoirs. Curr Opin HIV AIDS. 2011;6:43–8.
Josefsson L et al. Hematopoietic precursor cells isolated from patients on long-term suppressive HIV therapy did not contain HIV-1 DNA. J Infect Dis. 2012;206:28–34.
Durand CM et al. HIV-1 DNA is detected in bone marrow populations containing CD4+ T cells but is not found in purified CD34+ hematopoietic progenitor cells in most patients on antiretroviral therapy. J Infect Dis. 2012;205:1014–8.
Zhang J, Scadden DT, Crumpacker CS. Primitive hematopoietic cells resist HIV-1 infection via p21. J Clin Invest. 2007;117:473–81.
Acknowledgments
AC is supported by the American Foundation for AIDS Research (grant 108905-56-RGRL) and by an NICHD Child Health Research Career Development Award (K12 HD072245). ML is supported by the American Foundation for AIDS Research (grant 108302-51-RGRL), by the Doris Duke Charitable Foundation (grant 2009034), and by NIH grants AI098487 and AI106468. GS is supported by NIH grant R37AI066998.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Ann Chahroudi, Guido Silvestri, and Mathias Lichterfeld declare that they have patent pending on Wnt pathway inhibitors for treating viral infections.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on HIV Pathogenesis and Treatment
Rights and permissions
About this article

Cite this article
Chahroudi, A., Silvestri, G. & Lichterfeld, M. T Memory Stem Cells and HIV: a Long-Term Relationship. Curr HIV/AIDS Rep 12, 33–40 (2015). https://doi.org/10.1007/s11904-014-0246-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11904-014-0246-4