
Abstract
Purpose of Review
The management of myelofibrosis is risk-adapted when considering transplant eligibility and symptom-directed, prioritizing the most burdensome symptoms for the patient. Unfortunately, myelofibrosis-anemia is common, multifactorial in its origin, and impactful regarding prognosis. While clinical trials are advised, not all patients have convenient access, and therefore, hematologists should be aware of the data supporting the use of conventional agents such as erythropoietin-stimulating agents, steroid treatments (danazol and prednisone), and immunomodulatory drugs (thalidomide and lenalidomide). This review summarizes the conventional approach to treating myelofibrosis-anemia and highlights recent data from 3 novel agents that are under phase 3 evaluation.
Recent Findings
Momelotonib is a JAK1/2 and ACVR1 inhibitor that has demonstrated not only improvements in splenomegaly and symptoms, but also amelioration of anemia on the SIMPLIFY 1 and 2 clinical trial program. This may occur through suppression of hepcidin production. Luspatercept promotes late-stage hematopoiesis, and the phase 2 study has shown promise in ameliorating anemia as a monotherapy, and especially in combination with ruxolitinib. Finally, CP-0160, a BET inhibitor, has shown efficacy as an anemia-directed agent, when used as monotherapy and in combination. This agent reduces cytokine production and promotes erythroid differentiation. Results have been presented for patients previously treated with JAK inhibitors, as well as those who were naïve to JAK inhibitor therapy.
Summary
Safety and effectiveness are reviewed for both conventional and selected novel agents used in the treatment of MF-anemia. A practical approach to treatment is presented, and data from ASH 2020 are presented.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The myeloproliferative neoplasms (MPNs) are stem cell-derived clonal disorders characterized by myeloid proliferation, with shared clinical consequences that include a significant symptom burden that impacts the quality of life, a thrombotic and/or hemorrhagic tendency, organomegaly (usually hepatomegaly or splenomegaly), and the possibility for progression to overt fibrotic or leukemic phase. Recognizing these shared features, Dr. William Dameshek first proposed that essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis. (PMF) be classified together [1]. Dr. Dameshek also suggested that these entities had a shared pathogenesis, likely due to a “myelostimulatory factor” [1]. Of course, he was correct, though it took more than 50 years before the molecular pathogenesis of the MPNs was defined. It is now clear that ET, PV, and MF harbor driver mutations (JAK2 V617F, CALR, or MPL) which lead to JAK-STAT dysregulation; additional non-JAK-STAT pathway mutations are often present and impact prognosis [2]. Recognition of JAK-STAT dysregulation in the MPNs led to the development of JAK inhibitors, which are approved for use in polycythemia vera and myelofibrosis. Two agents are commercially available, including ruxolitinib and fedratinib, and can help address inflammation-associated symptoms and splenomegaly (and phlebotomy burden in PV) but do not ameliorate cytopenias [3,4,5,6,7].
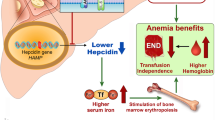
Unlike ET or PV, the course of MF is frequently complicated by anemia. Anemia in MF can be multifactorial, due to ineffective erythropoiesis, inflammatory iron sequestration, splenic sequestration or hemolysis, and, occasionally, blood loss (especially if there is portal hypertension). In addition to impacting the clinical course, anemia has an adverse impact on prognosis [8]. Management of anemia represents an unmet treatment need—unfortunately, currently available JAK inhibitors can exacerbate rather than ameliorate anemia. Conventional agents to treat MF-anemia offer modest responses, often lacking in durability. In this review, a practical approach to managing anemia is presented, with attention to conventional agents as well as three promising anemia-directed treatments on the horizon, tested in phase 3 (momelotonib, luspatercept, CPI-0610) (Fig. 1).

The management of MF anemia requires the exclusion of potentially reversible causes, followed by an algorithmic approach, based on the erythropoietin level. An epo-stimulating agent is reasonable for patients with lower levels. Steroids can be used in non-responding patients or those with higher baseline Epo levels. Immunomodulatory drugs can be considered next. Because of the modest efficacy with these conventional agents, a clinical trial is an important consideration, even upfront
Standard Therapy
Prior to considering conventional agents to treat MF-anemia, potentially reversible, co-existing causes of anemia should be evaluated for and managed, if possible [9]. These causes typically include vitamin/mineral deficiencies (iron, B12, and/or folate deficiency) and blood loss. The latter is an important cause in MPN patients with portal hypertension, due to prior abdominal venous thrombosis, marked splenomegaly, or extramedullary hematopoiesis involving the liver. Hemolysis should be excluded, but direct antiglobulin test-positive hemolysis is very rarely identified [10]. With respect to supportive care, red cell transfusions are indicated to relieve symptoms from anemia, and leuko-reduced blood products are especially important for patients contemplating future stem cell transplantation [9]. While iron chelation is to be considered for patients that have received more than 20 units of packed red cells, and/or with a ferritin of 2500 ng/ml or more, the overall impact in MF remains unknown [9]. A recently published retrospective study of 45 MF patients treated with deferasirox demonstrated that 29% had a response to iron chelation (ferritin < 1000 mg/l or 50% reduction from baseline), and 43% had an erythroid response (17% with transfusion independence; 27% with a partial reduction in transfusion burden or hemoglobin increase) [11•]. Toxicity was noted in 20 (44%) of patients, and 24% discontinued because of grade 2 or more toxicity. Clearly, prospective studies are needed, but this retrospective study supports observations of improved hematopoiesis with iron chelation [11•].
Erythropoietin-Stimulating Agents
Erythropoiesis-stimulating agents (e.g., darbepoetin alfa or epoetin alfa) are considered as appropriate options when the serum erythropoietin (Epo) level is < 500 mU/ml [9]. While the NCCN guidelines emphasize the importance of the baseline Epo level, there are additional risk factors that may influence the response. The aim of a multi-center retrospective study of 163 MF patients treated with ESAs for anemia (transfusion-dependent or Hb < 10 g/dl at the start of ESA) was to report predictors of response [12]. In all but 16 patients, Epo levels were below 125 mU/ml, and 90% of patients started with standard doses of recombinant Epo (20,000–40,000 units per week) or darbepoetin-alpha (150mcg/week). Nearly half (72/147 patients) required dose escalation after 4–8 weeks of therapy. Anemia response was achieved in 86 patients (53%), including 7 of 24 patients dependent on transfusions; response rates were similar whether or not patients were taking JAK inhibitors with ESA therapy (62.5% vs 50%, p = 0.16) [12]. In the multivariate analysis, only female sex (OR 3.64, p = 0.007), leukocyte count ≥ 10 × 109/l (OR 2.99, p = 0.033), and serum ferritin level < 200 ng/ml (OR 4.35, p = 0.002) predicted response. Among the 86 responders, the median duration of response was 19.3 months. With over 373 patient-years of follow-up, 9 patients (2.41 per 100 patient-years) experienced thrombotic events (6 arterial, 3 venous)—this was felt comparable to the thrombotic rate in MF at large [12]. Finally, one-quarter of the patients experienced an increase in spleen size by 2 cm or more, though this was not always attributed to ESA treatment alone [12].
Another multicenter retrospective study evaluated the safety and efficacy of ESA therapy with concurrent ruxolitinib [13]. Among 59 patients, the anemia response was 54%, achieved by a median of 4 months. Similar response rates were noted among patients starting ESA therapy within 3 months of ruxolitinib therapy or after 3 months of ruxolitinib initiation [13]. Though not statistically significant, anemia responses appeared more likely in patients that experienced a reduction in splenomegaly with ruxolitinib. Here, the only predictor of an anemia response was a lower baseline Epo level (< 125 mU/ml); neither disease risk, ferritin, mutational profile, age, disease duration, nor transfusion dependency impacted response [13]. No thrombotic events were noted, and an increase in spleen size was only noted in 1 patient.
Steroid Treatment
In the event of an increased baseline Epo level, or should an ESA fail to address anemia, steroid therapy is another option to treat MF-anemia. In a retrospective analysis of 30 MF (27 primary MF, 3 post-ETMF) patients treated with prednisone (0.5–1 mg daily, then tapered to minimum effective dose), 12 patients responded, typically after a median of 1.1 months, with duration for a median of 12.3 months [14]. Three of 11 patients with baseline thrombocytopenia improved their platelet count by at least 50 × 109/l. Investigators reported lower response rates in patients with constitutional symptoms or > 2% peripheral blasts, but these associations lacked statistical significance. Laboratory features suggesting hemolysis did not predict response either. There was no difference in response when considering a baseline prednisone dose of either 0.5 mg/kg or 1 mg/kg [14].
Danazol, a semi-synthetic androgen, is another option for the treatment of MF-anemia. A retrospective study has evaluated outcomes among 50 consecutively treated patients, including 23 who received first-line danazol [15]. The dose was 600 mg daily for 6 months, followed by a reduction to 400 mg daily × 6 months, and then a taper to the minimum effective dose which was usually 200 mg daily. Using accepted response criteria, anemia responses were seen in 15 patients (30%), including 5 of 27 that were transfusion-dependent. The time to response was 5 months, and the median duration of response was 14 months. An overall thrombocytopenia response of 23% was also observed (3/13 with platelet counts < 100 × 109/l) [15]. No baseline variable was considered predictive of response, though there was a trend toward worse response among transfusion-dependent patients. The most common adverse event included liver function abnormalities of grade 1 or 2 severity though 2 patients developed severe cholestatic liver injury; 1 patient developed prostate cancer and another developed liver peliosis, resulting in discontinuation [15].
Danazol has also been studied as an agent in combination. With a potential aim of offsetting treatment-induced anemia, danazol was combined with ruxolitinib in a prospective study of 14 patients [16]. Here, the most common response was that of stable disease (9 patients, 64.2%) as opposed to clinical improvement in anemia. In another study of 88 patients, when danazol was added to thalidomide/prednisone (N = 42), anemia response rates were 71% compared to 46% (p = 0.014) in those receiving thalidomide/prednisone alone [17]. In this retrospective study, authors reported a median of 2 months to response in both groups, but a longer duration of response in the triple therapy group (HR 2.18) [17].
Immunomodulatory Drugs
Thalidomide or lenalidomide are considered if ESA and/or androgen/steroid therapy fails to ameliorate anemia. A prior phase 3 study of pomalidomide versus placebo did not demonstrate benefit [18]. Modest efficacy has been noted with thalidomide and lenalidomide in prior studies [19, 20]. More recently, a single-center, retrospective study including 176 patients treated with either thalidomide (N = 79) or lenalidomide (N = 97) between 1998 and 2019 has been published [21•]. Most patients were treated concurrently with steroids. Patients treated with thalidomide were more likely to have thrombocytopenia and have higher risk of disease. Splicing mutations were identified in 41% and 61% of lenalidomide- and thalidomide-treated patients, respectively; the latter group was more likely to have SRSF2 or U2AF1 mutations [21•]. Clinical benefit was noted in 49% (16% became transfusion-independent) and 42% (11% became transfusion-independent) of lenalidomide- and thalidomide-treated patients, respectively. There was no predictive variable regarding a response or lack thereof. Adverse events were not explicitly discussed, due to the retrospective nature of the study, but the median treatment duration was 4.9 and 3.8 months for lenalidomide- and thalidomide-treated patients, respectively [21•].
Novel Agents
Momelotonib
Momelotonib is a JAK1/2 and ACVR1 inhibitor under investigation for myelofibrosis. This agent holds promise given the possibility of not only ameliorating symptoms and splenomegaly, but also addressing anemia. While JAK inhibition is typically associated with myelosuppression, this agent may address iron dysregulation. Preclinical work has shown that momelotonib inhibits the interleukin-6/JAK-STAT pathway, as well as the BMP6-ACVR1/SMAD signaling pathways, both of which upregulate hepcidin production [22•, 23]. Elevated hepcidin results in a functional iron deficiency, as seen in the anemia of inflammation. With suppression of hepcidin, the ferroportin channel opens, improving iron availability. This is the suspected mechanism by which momelotonib may actually improve anemia in some MF patients.
Momelotinib has been studied in randomized clinical trials. In SIMPLIFY-1, momelotonib 200 mg daily was compared to ruxolitinib in 432 JAK inhibitor-naïve patients [24]. The mean hemoglobin was 10.6 and 10.7 g/dl respectively in the momelotinib and ruxolitinib groups, and a similar proportion were transfusion-dependent (24.7% and 24%, respectively). The effect on anemia was analyzed as a secondary endpoint. More patients were transfusion-independent at week 24 in the momelotinib versus ruxolitinib groups (66.5% versus 49.3%, p < 0.001); fewer patients were transfusion-dependent at week 24 in the momelotinib versus ruxolitinib groups (30.2% vs. 40.1%, p = 0.019) [24]. The median rate of red cell transfusions was 0 units/month in the momelotonib group, compared to 0.4 units/month in the ruxolitinib group (p < 0.001) [24]. SIMPLIFY-2 compared momelotonib with the best available therapy (BAT) in patients previously treated with ruxolitinib [25]. Similarly, the impact on anemia was evaluated as a secondary endpoint. Here, baseline hemoglobin values were similar in momelotinib versus BAT (9.4 vs 9.5 g/dl), though more patients in the momelotonib group had hemoglobin values < 8 g/dl (26% vs. 11%). Fifty-six and 52% of patients in the momelotonib and ruxolitinib groups, respectively, were transfusion-dependent. Following treatment, more patients in the momelotinib group were transfusion-independent at week 24, compared to the BAT arm (43% vs. 21%, p = 0.0012) [25]. Forty percent of the momelotonib-treated patients did not require transfusion during the study, compared to 27% of patients treated with ruxolitinib. The median rate of RBC transfusion was 0.5 units per month compared to 1.2 units per month, in the momelotonib versus ruxolitinib groups, respectively (p = 0.39) [25].
In a subsequently published multicenter, open-label phase 2 study, momelotonib 200 mg daily was evaluated in 41 transfusion-dependent MF patients [22•]. Seventeen of 41 (41%) achieved transfusion independence for at least 12 weeks. As proof of concept, investigators measured hepcidin levels which decreased as soon as 6 h after drug administration, and downward trends were noted over the 24-week study period. Iron levels increased by week 2 in responding patients [22•]. Further, reductions in C-reactive protein were noted, consistent with the anti-inflammatory effect. Though there was missing data, total symptom score and splenomegaly responses were noted in 29% and 19% of patients, respectively. The safety profile was in keeping with prior reports, including ≥ grade 3 anemia and neutropenia in 12%; 12% (all grade 1) experienced peripheral neuropathy [22•]. The MOMENTUM study, which is currently underway, is a randomized, double-blind study comparing momelotonib with danazol (NCT04173494).
Luspatercept
Having been previously approved for the treatment of myelodysplastic syndrome with ring sideroblasts as well as transfusion-dependent beta-thalassemia, luspatercept is now under evaluation in myelofibrosis-anemia. The mechanism of action is unique—unlike recombinant erythropoietin which works at earlier stages of hematopoiesis, luspatercept promotes later stages of hematopoiesis. This occurs through the binding of TGF-β superfamily ligands to decrease signaling by SMAD2 and SMAD3, which otherwise inhibit red cell maturation.
Results from the open-label phase 2 study (ACE-536-MF-001) in myelofibrosis-associated anemia were presented at the American Society of Hematology annual meeting in 2019 and 2020. In the first update (2019), 74 patients were enrolled into 4 cohorts, depending on the use of ruxolitinib and transfusion dependence [26]. Cohorts 1 and 2 received luspatercept as monotherapy; the former were not transfusion-dependent (cohort 1, N = 20) while the latter received 2–4 units of blood in the 3 months prior to treatment (cohort 2, N = 21). Cohorts 3A and 3B received a stable dose of ruxolitinib; cohort 3A patients were not receiving transfusions, while cohort 3B received transfusions. Luspatercept was administered each 21 days, starting at 1 mg/kg, increasing to the maximal dose of 1.75 mg/kg [26]. Most patients were older (median 71 years) and classified as intermediate-2 or high risk by DIPSS classification. Among the transfusion-independent patients, 10% and 21% of cohort 1 and 3A (combination) patients experienced a Hgb increase of at least 1.5 g/dl, respectively. Of the transfusion-dependent patients, 10% and 32% of cohorts 2 (monotherapy) and 3B (combination therapy) achieved red cell transfusion independence over any 12-week period, respectively. Treatment-related adverse events included hypertension (11%), bone pain (8%), and diarrhea (4%) [26].
At the American Society of Hematology 2020 Annual Meeting, updated results from 43 transfusion-dependent patients were presented [27]. Patients in cohort 2 (monotherapy) and 3B (ruxolitinib) received a median of 8 cycles of luspatercept. With respect to the primary endpoint, 2/21 (10%) and 6/22 (27%) achieved transfusion independence during a consecutive 12-week period. The median duration of response was 49 and 42 weeks, respectively. The authors noted that 4/15 (27%) and 8/15 (57%) in cohorts 2 and 3B, respectively, experienced clinical benefit and extended therapy [27]. Luspatercept is being compared to placebo in patients on JAK2 inhibitor therapy who require transfusions in the INDEPENDENCE trial (NCT04717414).
CPI-0610 (Now Also Known as Pelabresib)
Bromodomain and extraterminal domain (BET) protein inhibition is another promising strategy to ameliorate MF-anemia. Preclinical data have implicated aberrant NF-κβ signaling in MPN disease pathogenesis, leading to increased cytokine production and bone marrow fibrosis; BET inhibition has been shown to reduce NF-κβ signaling and cytokine production in vitro and reduce cytokine production, disease burden, and marrow fibrosis when combined with a JAK inhibitor in a mouse model [28]. These preclinical data provide a rationale for use in MF patients. CPI-0610 is a BET inhibitor in clinical development, with a mechanism of action that involves the reduction in cytokine production and promotion of erythroid and megakaryocytic differentiation through BET protein and NF-κβ inhibition.
The agent is under investigation through the MANIFEST clinical trial program, which includes monotherapy and combination therapy arms (with ruxolitinib), for transfusion-dependent and transfusion-independent patients. Updated results have been presented at the ASH 2020 annual meeting. Patients in arm 1 were intolerant or refractory, lost response to, or were ineligible for JAK inhibitors and treated with monotherapy; patients were further stratified by transfusion-independent (non-TD, N = 27) or transfusion-dependent (TD, N = 16) status [29]. Among the non-TD patients, the mean age was 68.3 years, and 81.4% had DIPSS intermediate 2 or high-risk disease. Nearly 52% had high molecular risk mutations, and 63% had a Hgb < 10 g/dl. Eleven of 19 patients experienced an at least 1.5 g/dl increase in hemoglobin. Of the 16 TD patients (mean age 71.9, 93.8% DIPSS intermediate-2 or higher risk disease, 56.3% high-molecular risk mutations), 3 (21.4%) converted to non-TD status. Common adverse events included thrombocytopenia (25.6%, 14% grade 3 or 4), anemia (11.6%, 9.3% grade 3 or 4), nausea (32.6%), and diarrhea (30.2%, 4.7% grade 3 or 4) [29].
Patients in arm 2 received CPI-0610 as an add-on to ruxolitinib in patients with a suboptimal response, after at least 6 months of ruxolitinib treatment, and 8 weeks of a stable dose [30]. Here, 44 patients were treated in the TD cohort (95.5% Hgb < 10 g/dl, 97.7% with DIPSS intermediate-2 or high-risk disease, 52.3% with high-molecular risk mutations). At the time of presentation, 11/32 (34.4%) became transfusion-independent. Results were presented for spleen volume improvement (22.2%) and symptom improvement (36.8%), but not hemoglobin improvement in 26 non-TD combination therapy patients. In this cohort, the most common AEs included thrombocytopenia (47.1%; 24.3% grade 3/4), anemia (11.4%, 8.5% grade 3 or 4), nausea (35.7%), cough (24.3%), and fatigue (22.9%) [30].
Finally, JAK inhibitor treatment-naïve patients in arm 3 received the combination of CPI-0610 and ruxolitinib [31]. At the time of the ASH 2020 presentation, 64 patients had been treated (mean 67.1 years, 75% DIPSS intermediate-2 or higher risk disease, 64.1% with Hgb < 10 g/dl, 53.1% with high-risk mutations) with the combination (median ruxolitinib dose 10 mg BID, CPI-0610 125 mg daily). Hemoglobin improvements in all patients and in non-TD patients were reported, but results were not quantified in the abstract presentation [31]. At the meeting, the investigators reported that 16/48 (33%) patients had an at least one grade improvement in their bone marrow fibrosis, with most (14/16) changes occurring in the first 6 months of the combination. The most common AEs included anemia (23.4%; 17.2% grade 3 or 4), thrombocytopenia (20.3%; 4.7% grade 3 or 4), diarrhea (26.6%), respiratory tract infections (18.8%, 4.7% grade 3 or 4), and nausea (18.8%) [31].
Demonstration of efficacy and safety in these 3 arms has led to the continued development of CPI-0610. In the MANIFEST-2 phase 3 study, CPI-0610 plus ruxolitinib will be compared to placebo plus ruxolitinib in JAK inhibitor–naïve MF patients with at least DIPSS intermediate-1 disease, splenomegaly by imaging, and symptomatic MF (NCT04603495).
Conclusion
The management of MF-anemia remains an unmet clinical need. In part, the challenge lies in the multifactorial set of etiologies for MF-anemia and the lack of effective agents. An algorithmic approach includes the exclusion of reversible causes, followed by treatment sequences influenced by the baseline erythropoietin level. In patients with lower Epo levels, ESA therapy is first administered, followed by steroid treatment (danazol or prednisone) in the event of treatment failure. Thalidomide or lenalidomide are typically considered thereafter.
Unfortunately, these conventional options offer only modest responses, often with a limited duration of effectiveness. Clearly, novel treatments for MF-anemia are needed, and thankfully, there are three agents in phase 3 clinical trials that hold promise in the treatment of MF anemia. Importantly, these agents appear tolerable and effective when combined with JAK inhibitor therapy, often a backbone of MF therapy, when symptoms and splenomegaly are present. Hematologists will eagerly await results from the phase 3 evaluation of these novel agents.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6(4):372–5.
Marneth AE, Mullally A. The molecular genetics of myeloproliferative neoplasms. Cold Spring Harb Perspect Med. 2020;10(2):a034876. https://doi.org/10.1101/cshperspect.a034876.
Verstovsek S, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10(1):55.
Harrison CN, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30(8):1701–7.
Vannucchi AM. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(17):1670–1.
Pardanani A, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1(5):643–51.
Harrison CN, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017;4(7):e317–24.
Guglielmelli P, et al. MIPSS70: mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J Clin Oncol. 2018;36(4):310–8.
Myeloproliferative neoplasms. NCCN Guidelines 2021 [cited 2021 2/22/21].
Cervantes F. How I treat myelofibrosis. Blood. 2014;124(17):2635–42.
• Elli EM, et al. Deferasirox in the management of iron-overload in patients with myelofibrosis: a multicentre study from the Rete Ematologica Lombarda (IRON-M study). Br J Haematol. 2019;186(5):e123–6. The role of iron chelation is unclear in Myelofibrosis. This is a multicenter study showed that 43% had an erythroid response to iron chelation.
Hernandez-Boluda JC, et al. Predictive factors for anemia response to erythropoiesis-stimulating agents in myelofibrosis. Eur J Haematol. 2017;98(4):407–14.
Crisa E, et al. The use of erythropoiesis-stimulating agents is safe and effective in the management of anaemia in myelofibrosis patients treated with ruxolitinib. Br J Haematol. 2018;182(5):701–4.
Hernandez-Boluda JC, et al. Long-term results of prednisone treatment for the anemia of myelofibrosis. Leuk Lymphoma. 2016;57(1):120–4.
Cervantes F, et al. Danazol therapy for the anemia of myelofibrosis: assessment of efficacy with current criteria of response and long-term results. Ann Hematol. 2015;94(11):1791–6.
Gowin K, et al. Multicenter phase 2 study of combination therapy with ruxolitinib and danazol in patients with myelofibrosis. Leuk Res. 2017;60:31–5.
Luo X, et al. Thalidomide plus prednisone with or without danazol therapy in myelofibrosis: a retrospective analysis of incidence and durability of anemia response. Blood Cancer J. 2018;8(1):9.
Tefferi A, et al. A randomized study of pomalidomide vs placebo in persons with myeloproliferative neoplasm-associated myelofibrosis and RBC-transfusion dependence. Leukemia. 2017;31(4):896–902.
Thapaliya P, et al. International working group for myelofibrosis research and treatment response assessment and long-term follow-up of 50 myelofibrosis patients treated with thalidomide-prednisone based regimens. Am J Hematol. 2011;86(1):96–8.
Mesa RA, et al. Lenalidomide and prednisone for myelofibrosis: Eastern Cooperative Oncology Group (ECOG) phase 2 trial E4903. Blood. 2010;116(22):4436–8.
• Castillo-Tokumori F, et al. Retrospective analysis of the clinical use and benefit of lenalidomide and thalidomide in myelofibrosis. Clin Lymphoma Myeloma Leuk. 2020;20(12):e956–60. This is a more contemporary retrospective study demonstrating response rates for immunomodulatory drugs when used to treat MF anemia.
• Oh ST, et al. ACVR1/JAK1/JAK2 inhibitor momelotinib reverses transfusion dependency and suppresses hepcidin in myelofibrosis phase 2 trial. Blood Adv. 2020;4(18):4282–91. This study provides some proof of principle regarding the ability of momelotonib to suppress hepcidin and offer anemia responses in some patients with MF and transfusion-dependent anemia.
Asshoff M, et al. Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood. 2017;129(13):1823–30.
Mesa RA, et al. SIMPLIFY-1: a phase III randomized trial of momelotinib versus ruxolitinib in Janus kinase inhibitor-naive patients with myelofibrosis. J Clin Oncol. 2017;35(34):3844–50.
Harrison CN, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018;5(2):e73–81.
Gerds AT, et al. A phase 2 study of luspatercept in patients with myelofibrosis-associated anemia. Blood. 2019;134(Supplement_1):557–557.
Gerds AT, et al. Duration of response to luspatercept in patients (Pts) requiring red blood cell (RBC) transfusions with myelofibrosis (MF) - updated data from the phase 2 ACE-536-MF-001 Study. Blood. 2020;134(Supplement_1):557 ((557)).
Kleppe M, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell. 2018;33(1):29-43 e7.
Talpaz Mea. CPI-0610, a bromodomain and extraterminal domain protein (BET) inhibitor, as monotherapy in advanced myelofibrosis patients refractory/intolerant to JAK inhibitor: update from phase 2 MANIFEST Study. Blood. 2020. 134(Paper 2163).
Verstovsek S et al. CPI-0610, bromodomain and extraterminal domain protein (BET) inhibitor, as “add-on” to ruxolitinib, in advanced myelofibrosis patients with suboptimal response: update of MANIFEST Phase 2 Study. Blood. 2020(Supplement_1): p. Abstract 56.
Mascarenhas J et al. CPI-0610, a bromodomain and extraterminal domain protein (BET) inhibitor, in combination with ruxolitinib, in JAK-inhibitor-naïve myelofibrosis patients: update of MANIFEST Phase 2 Study. Blood. 2020(Supplement_1): p. Abstract 55.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by the author.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Myeloproliferative Neoplasms
Rights and permissions
About this article

Cite this article
Stein, B.L. Management of Myelofibrosis-Associated Anemia: Focus on Standard Agents and Novel Therapeutics in Phase 3 Clinical Trials. Curr Hematol Malig Rep 16, 483–489 (2021). https://doi.org/10.1007/s11899-021-00651-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11899-021-00651-3