
Abstract
Purpose of review
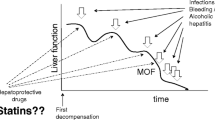
The purpose of this study is to analyze the current evidence regarding the use of statins in patients with chronic liver disease and cirrhosis.
Recent findings
Chronic liver disease (CLD), cirrhosis, and its complications, including hepatocellular carcinoma (HCC), are significant public health problems. The use of statins in patients with CLD has been a matter of concern, and physicians are often reluctant to its prescription in these patients. This mainly relates to the potential occurrence of drug-induced liver injury. However, newer evidence from pre-clinical and clinical research has shown that statins are drugs with a potentially beneficial impact on the natural history of cirrhosis, on portal hypertension, and in HCC prevention.
Summary
In this review, we summarize current evidence regarding the influence of statins in endothelial dysfunction in CLD, their ability to modulate hepatic fibrogenesis, and their vasoprotective effects in portal hypertension; we also focus on existing data about the impact of statins in cirrhosis development, progression, and complications and critically assess the current concerns about its use in patients with CLD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic liver disease (CLD), its advanced form, cirrhosis, and its complications (including variceal bleeding and hepatocellular carcinoma), are significant public health problems being among the main causes of death and disability worldwide, both in men and women [1].Some patients with CLD, particularly those with non-alcoholic fatty liver disease (NAFLD), have an increased cardiovascular risk (CVR) [2, 3]. Thus, hepatologists need to correctly assess CVR as well as be able to institute an integrated management involving lifestyle modification and drug therapy.
Statins are widely used lipid lowering agents that act through competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase impeding the conversion of HMG-CoA to mevalonic acid (a cholesterol precursor). Statins are considered one of the cornerstones of treatment as well as primary or secondary prevention of atherosclerotic cardiovascular disease [4]. They have well-known beneficial cardiovascular effects reducing cardiovascular events and mortality [5, 6], with more than 200 million users worldwide, and 30 million users in the USA [7].
Statins are often under-prescribed in patients with CLD or cirrhosis due to fear to hepatotoxicity. In light of current literature, this fear seems unjustified since statin liver toxicity is a rare event [8••]. Moreover, recent data point to potentially beneficial and previously unexpected effects of statins in CLD and cirrhosis [9]. In this review, we summarize current evidence regarding the influence of statins in endothelial dysfunction in CLD, their ability to modulate hepatic fibrogenesis, and their vasoprotective effects in portal hypertension as well as on existing data about the impact of statins in cirrhosis development, progression, and complications.
Statin Subclasses and Their Pleiotropic Effects
Pharmacological properties of subclasses of statins are different depending on their activity on HMG-CoA reductase, oral absorption, bioavailability, liver extraction, and protein binding. Whereas atorvastatin, lovastatin, simvastatin, and fluvastatin are lipophilic and metabolized by the cytochrome P-450 system; pravastatin and pitavastatin are hydrophilic and undergo minimal hepatic metabolization, while rosuvastatin has an intermediate behavior [10].
In vitro and in vivo have shown that the hydrophobic statins lovastatin and simvastatin reach a higher concentration in the liver compared to hydrophilic pravastatin [11]. Also a potential structure-related effect in prevention of cardiovascular disease and antiinflammatory properties has been described for different types of statins [12]; the exact clinical relevance of this differences is uncertain.
Numerous reports have described additional beneficial effects of statins beyond the quantitative decrease in serum lipid levels, known as pleiotropic effects. The pleiotropic effects of statins have arisen from epidemiological studies showing a beneficial impact in conditions other than cardiovascular diseases, including contrast-induced nephropathy, acute kidney injury, pancreatitis, chronic obstructive pulmonary disease, venous thromboembolism, dementia, cognition, and erectile dysfunction. However, statins have also been linked to potentially harmful effects: a modest increase in the risk of myositis and rhabdomyolysis, a modest increase in the risk of new onset diabetes with higher intensity regimens, an epidemiological association with incidence of cataracts, and a dose-dependent effect in liver enzyme alterations [13].
Several hypotheses have been proposed to explain these pleiotropic effects probably mediated by the reduced formation of isoprenoids [14]: through the reduction in overall lipoprotein load, improvement in endothelial function, upregulation of nitric oxide synthase, reduction in systemic subclinical inflammation, and modulation in components of the atherosclerotic plaque [7]. Moreover, statins have been shown to reduce the potential of mesenchymal stem cell to differentiate into macrophages, therefore reducing inflammation [15]. Indeed, the abovementioned effects can have an impact in the pathophysiological abnormalities at play in CLD and can have a meaningful clinical impact. Existing evidence on these topics is summarized as follows.
Pre-Clinical Studies of Statins in Liver Disease
Portal Hypertension and Fibrosis
Several pre-clinical studies with in vitro and in vivo animal models have described a favorable influence of statins in endothelial function, angiogenesis, portal hypertension (PHT), and fibrosis. A growing body of data regarding the effects of statins in Kruppel-like factor 2 (KLF-2) and derived transcriptional programs (endothelial NO synthase (eNOS), thrombomodulin, and C-natriuretic peptide) has been described. KFL-2 is characterized as a key component of the hepatic endothelium [16], with an important role in fibrosis and endothelial dysfunction [14]. Endothelial dysfunction, measured by flow-mediated dilatation (FMD) of the brachial artery, is frequently observed in cirrhotic patients [2]. In the liver, KLF-2 is induced early during progression of cirrhosis to ameliorate the development of vascular dysfunction acting as a defense mechanism in response to damage that occurs during the progression of the disease [16]. However, its endogenous expression results insufficient to attenuate the occurrence of portal hypertension and aggravation of cirrhosis [17•]. Simvastatin administration to sinusoidal endothelial cells induces upregulation of KLF-2 expression, deriving in vasoprotective effects on vascular dysfunction in the cirrhotic liver [16, 18]. Additionally, upregulation of hepatic endothelial KLF-2 induces hepatic stellate cells (HSC) deactivation in the presence of statins with a reduction in liver fibrosis [18], and induces attenuations in the increased portal blood flow and hepatic vascular resistance [17•]. Similarly, the addition of simvastatin to explanted rat livers also ameliorates endothelial dysfunction, preventing the decreased in KLF-2-dependent programs in cold storage conditions improving endothelial dysfunction, oxidative stress, and preventing liver damage [19] (Fig. 1).

Schematic representation of the evidence from translational research about the influence of statins in endothelial dysfunction in CLD, modulation of mechanisms of activation of fibrogenesis, and effects in portal hypertension. eNOS nitric oxide synthase, NO nitric oxide, CNP C-type natriuretic peptide, ET-1 endothelin-1, NASH non-alcoholic steatohepatitis, PPAR-γ peroxisome proliferator-activated receptor gamma
The impact of statins in HSC activation has been further characterized. Fluvastatin suppresses paracrine activation of HSCs by hepatocytes in primary rat hepatocytes and human hepatoma cell line [20]. Besides, atorvastatin inhibits the activation of HSCs to myofibroblasts in vitro, and decreases the cytokine and collagen production and proliferation of myofibroblasts, leading to decreased turnover and fibrosis in primary rat HSCs [21]. A similar effect was described with lovastatin and simvastatin [22]. In bile duct ligation (BDL) cirrhosis animal model, early atorvastatin treatment inhibits HSCs activation and fibrosis, while late treatment reduces HSC turnover and activity [23]. However, Shirin H. et al. showed that neither atorvastatin nor rosuvastatin influenced liver fibrosis, assessed by hydroxyproline content, oxidative stress assessed by hepatic malondialdehyde levels, or HSC proliferation in a rat model of thioacetamide-induced liver injury [24].
Interestingly, atorvastatin has shown different effects in cirrhotic and non-cirrhotic portal hypertension models, decreasing portal pressure, shunt flow, and angiogenesis in cirrhotic PHT; and augmenting them in non-cirrhotic portal hypertension animal models [25], by blocking the non-canonical Hedgehog (Hh) pathway Ras homolog gene family member A (RhoA) dependently in activated HSCs in cirrhosis [26] and enhancing the canonical Hh pathway in non-cirrhotic portal hypertension leading to aggravate PHT and angiogenesis [27].
Simvastatin has also shown to modulate the response to perfusion changes induced by endothelin-1 in non-cirrhotic PHT model, inducing nitric oxide (NO)-mediated vascular hypo-responsiveness in the portal-systemic collateral vascular resistance [28] and enhancing expression of eNOS, COX-2, and thromboxane A2 synthase in splenorenal shunt [29]. In a similar animal model of partial portal vein ligation (PPVL), administration of pravastatin, however, did not modify systemic and portal hemodynamics and collateral vascular response to endothelin-1 [30].The role of NO was recently highlighted when a nitric oxide-donating statin (NCX 6560) showed similar effects on lowering portal pressure, but with higher vasoprotective properties and lower toxicity in liver and muscle [31].
Complementarily to the use of angiotensin II receptor blockers, atorvastatin, and pitavastatin, have shown additive effects to losartan and candesartan, respectively, in the inhibition and attenuation of liver fibrosis in cirrhotic rat model [32, 33].
Non-Alcoholic Fatty Liver Disease
NAFLD is a significant independent risk factor for subclinical and clinical cardiovascular disease [3]. Due to the relation of NAFLD with metabolic syndrome and dyslipidemia [34], the impact of statins in NAFLD progression has been evaluated. Simvastatin decreased hepatic inflammation and fibrosis through the inhibition of Ras and RhoA pathway, but without significant changes in steatosis or intrahepatic cholesterol content in a non-alcoholic steatohepatitis (NASH) mouse model [35]. In methionine-choline-deficient diet (MCDD) fed mice, statins have shown to prevent MCDD-induced NASH, restoring mitochondrial and peroxisomal fatty acid oxidation through attenuation of the decreased expression of peroxisomal proliferator-activated receptor α (PPARα) induced by NASH, independent of cholesterol levels [36, 37]. Rosuvastatin decreases free fatty acid liver content in high-fat high-cholesterol diet-induced NASH in rats, and reduces inflammatory markers (TNF-a, IL-6, ALT) and inflammation in histology [37].
Pitavastatin, a relatively novel statin subclass, also mitigates the progression of NASH in ovariectomized mice that, due to estrogen deficiency, develop a more severe histological lesion [38]. In this model, pitavastatin reduces hepatic inflammatory genes, CCR2, CCL2, TNF-α, and IFN-γ; and inflammation [39]. In contrast, in ApoE−/− mice, that have decreased serum apolipoprotein E and exhibit lipid abnormalities and atherosclerosis even on a low-cholesterol diet, atorvastatin treatment exacerbated hepatic steatosis, inflammation, and fibrosis, as well as increased hepatic oxidative stress under conditions of inflammatory stress. This effect was not observed without the presence of inflammatory stress, suggesting a pre-disposition to statin injury by inflammatory conditions in the liver [40].
In summary, in pre-clinical studies, statins have shown beneficial effects through diverse mechanisms in endothelial dysfunction, portal hypertension, and fibrosis in CLD models.
Clinical Studies
Clinical data about the use of statins in CLD have been growing in recent years, mostly arising from epidemiological cross-sectional or retrospective studies, whereas most prospective studies are focused on the use of statins in cirrhosis, portal hypertension, and its complications. The most relevant studies are summarized as follows.
Primary Biliary Cholangitis
One of the first evidence on the use of statins in CLD arose from a case report of improved cholestasis with the use of simvastatin in a patient with primary biliary cholangitis (PBC) [41]. Further, prospective studies of statins and PBC have been focused in the safety profile of this drug in these patients, showing improved lipid profile and vascular function measured by flow-mediated dilation (FMD) of the brachial artery with the use of atorvastatin, without deterioration of liver function tests or cholestasis [42]. Similar findings emanate from a randomized control trial (RCT) of simvastatin in PBC patients, where also improvements in antioxidant status compared to placebo were suggested by the evaluation of lipid hydroperoxide serum content [43].
Hepatitis C and Hepatitis B Chronic Infection
Initially, statin therapy was related to a decrease in serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in a retrospective cohort study in 20 biopsy-proven hepatitis C virus (HCV)-infected veterans matched 1:3 to patients not using statins, mostly non-cirrhotic patients with 1-year follow-up [44]. Several confounders could not be addressed in this report, including assessment of changes in metabolic factors and weight loss during the year of follow-up, focusing the conclusion of this report in the safety of the use of statins in HCV patients.
More recently, the use of statins was associated with a reduced risk of fibrosis progression in 543 HALT-C trial cohort of HCV chronic hepatitis patients followed for 42 months with compared histology along the follow-up. The adjusted hazard ratio (aHR) was 0.31 (0.1–0.97), with a significant decrease in 0.34 points in Ishak fibrosis score in continuous statin users [45] compared to non-users, adjusted by other potentially confounding factors associated with fibrosis progression.
Data derived from the ERCHIVES cohort, considering 7248 subjects, linked the use of statins to increased response to IFN-based antiviral treatment with increased sustained virological response (SVR) (odds ratio (OR) 1.44), decreased cirrhosis development and progression in 10 years of follow-up (HR 0.56) according to the fibrosis 4 (FIB-4) score, and a reduction in incidental HCC (HR 0.51) in both SVR and non-SVR patients [46]. Coincident findings were described in a population-based retrospective cohort study in Taiwan, examining database information from 226,856 HCV patients, where a dose-dependent reduction on cirrhosis development was seen in statin users, with a higher reduction in cirrhosis emergence in patients with higher statins cumulative daily doses (adjusted HR 0.13) [47]. Multiple confounders could not be completely addressed as the statin users group had increased use of other cardiovascular medications (e.g., metformin, aspirin, angiotensin-converting enzyme inhibitors) and higher comorbidity.
A similar population-based retrospective cohort study of the association of statins use with cirrhosis development and decompensation was reported in hepatitis B virus (HBV) Taiwanese patients. Statin users developed less cirrhosis (aHR 0.51) and had less decompensation episodes (aHR 0.53), after adjusting for multiple confounding factors (age, gender, comorbidity index, hypertension, diabetes, hyperlipidemia, HCC, obesity, NAFLD, aspirin use, diabetes medications, chronic hepatitis B treatment, and other lipid-lowering drugs). A dose-related beneficial effect of statins in cirrhosis was also described in this report. A positive effect of the use of statins in overall mortality in this HBV patient’s cohort was described as well. In this study, the use of triglyceride-lowering drugs (fibrates) was also associated with decreased risk of cirrhosis to a lesser extent (aHR 0.67), but was not associated with cirrhosis decompensation. Other lipid-lowering drugs had no effect on cirrhosis [48•].
Non-Alcoholic Fatty Liver Disease
In a cross-sectional study of 6358 healthy patients, the use of statins did not relate to the presence of NAFLD diagnosed by abdominal ultrasound, nor the presence or degree of fibrosis evaluated by the non-invasive markers FIB-4 and fatty liver index [49]. More recent data from a cross-sectional study evaluated the effect of statins in 1201 high-risk NAFLD patients (age 50, severe obese body mass index, half of them with impaired fasting glucose [IFG]/type 2 diabetes mellitus) without cirrhosis who underwent liver biopsy. At least 6 months of previous statins use were associated with less steatosis, showing an OR of 0.09, less inflammation according to NAS (OR 0.25), and decrease presence of fibrosis stage F2-F4 (OR 0.42) matched by age, gender, presence of IFG or type 2 diabetes, PNPLA3 I148M risk alleles, and TM6SF2 E167K variant with non-statin users. However, the statins’ beneficial effect in decreasing steatohepatitis histological score was lost in the presence of I148M PNPLA3 risk variant allele [50].
Cirrhosis and Portal Hypertension
Increasing amount of data have been accumulated focusing on the use of statins in cirrhotic patients regardless of etiology, additional to some of the abovementioned studies in HBV and HCV patients [47, 48]. A retrospective study by Kumar S. et al. evaluated statin use in 81 cirrhotic patients matched 1:2 to look for increased mortality or decompensation with a mean follow-up of 36 months, including most cirrhotic patients at an early stage (Child-Pugh A). They show that statins use was associated with lower mortality (HR 0.53) and fewer episodes of hepatic decompensation (HR 0.63) in multivariate analysis [51]. Of note, statin group patients had significantly higher prevalence of NASH, diabetes mellitus, and coronary artery disease.
In the same direction, the use of statins has been related to decreased risk of decompensation and mortality in HCV-related cirrhotic patients. A retrospective cohort using the Veteran Affairs Clinical Case Registry describes the effect of statins in 685 cirrhotic HCV patients matched 1:5 with statin non-users, finding fewer decompensation episodes (HR 0.55) and death (HR 0.56) in statin users. No differences in comorbidities, metabolic conditions, or hepatic function were reported between groups. The positive effect of statins in cirrhosis decompensation and mortality persisted at 10 years after adjustment for age, FIB-4 index score, serum level of albumin, model for end-stage liver disease (MELD), and Child-Turcotte-Pugh (CPT) scores [52•].
A recently published Taiwanese cohort study evaluates the effect of statin use in 1350 cirrhotic patients, where the dose-dependent reduction of statins in the risk of decompensation, mortality, and HCC was also described. According to etiology, the protective effects of statins on the risk of decompensation were present for HBV chronic infection (aHR 0.39) and HCV (aHR 0.51), but the statistical significance vanished in alcohol-related cirrhosis (aHR 0.69, CI 0.47–1.07). The effect of statins in mortality was only seen in HBV-related cirrhosis, but not in HCV and alcohol-related cirrhosis. Moreover, the risk of HCC was borderline significant in HCV-related cirrhosis, and non-significant in HBV and alcohol-related cirrhosis [53•].
A favorable effect of statins was also described for the incidence of infections requiring hospitalization in veteran compensated cirrhotic patients. Retrospective data showed an HR of 0.67 for serious infections in 154 patients using statins compared to other lipid-lowering drugs or non-users on 1194 days of mean follow-up. Patients on statins had lower comorbidity index, more alcohol, and less HCV-related cirrhosis [54].
As well as the findings in basic research models, the use of simvastatin has shown to enhance nitric oxide production and hepatosplenic output, while lowering the hepatic resistance in patients with cirrhosis. In a study including 30 compensated cirrhotic patients, acute infusion of 40 mg of simvastatin showed an increase in hepatic blood flow and decreased hepatic sinusoidal resistance, without affecting the hepatic venous pressure gradient (HVPG) accompanied by an increase in nitric oxide products in hepatic venous blood. A similar effect was found after the administration of simvastatin before food ingestion, where an attenuation in the hepatic venous pressure gradient was seen. No changes in systemic hemodynamics or peripheral nitric oxide products were observed [55].
An effect in hepatic portal pressure after 1 month of continuous oral administration of escalating dose of simvastatin was demonstrated in a multicenter RCT in 59 cirrhotic patients, were almost half of the patients had ascites and esophageal varices as clinical signs of portal hypertension and with previous episode of decompensation, mainly CPT A and B patients. In the simvastatin-treated group, an 8.3% decrease in hepatic venous pressure gradient was observed, that was significant in patients receiving β-adrenergic blockers (11.0%) and in those who were not (5.9%). Nearly 30% of the patients in the experimental group reached the treatment target of a decrease in 20% from baseline or normalization of HVPG. Beneficial effects were also seen in previous decompensated patients. Compared to placebo, simvastatin administration improved clearance of indocyanine green, indicating improvements in liver perfusion and function. No effects on systemic hemodynamics or increased adverse events were observed in the experimental group [56].
Longer administration of simvastatin treatment compared to placebo for severe portal hypertension was assessed in a blinded randomized controlled trial. Three months of 40 mg simvastatin in 24 patients, most CPT A and B nearly, two thirds with use of non-selective β-blockers, and medium/large esophageal varices including 30% with previous variceal bleeding, showed a significant reduction in HVPG with greater effect in patients with previous variceal bleeding and medium/large esophageal varices. Again, no significant increase in adverse events from the use of simvastatin was reported [57].
This apparent beneficial effect of simvastatin in esophageal varices and rebleeding rates could not be confirmed in a recent multicenter, double-blinded, randomized clinical trial. The addition of 40 mg of simvastatin was compared to standard therapy for variceal rebleeding (β-blocker and esophageal band ligation) in 158 cirrhotic patients, stratified by CPT score, and followed by 24 months. The rebleeding rates were not different between the treatment and the control group (p = 0.58), but increased survival was seen in the simvastatin group (p = 0.03) for CPT A or B patients, mostly related to non-hepatic related death causes in the placebo group. The effect in mortality was not observed in CPT C-treated patients. No difference in the effect on secondary outcomes related to cirrhosis complications (e.g., ascites, spontaneous bacterial peritonitis, hepatorenal syndrome, or portal vein thrombosis) was observed in the simvastatin group compared to placebo [58••]. The occurrence of related adverse events in the simvastatin group was 8%, but not superior to the placebo group. Of note, two patients with advanced cirrhosis developed rhabdomyolysis, rising some concerns about the safety of statins in advanced chronic liver disease patients.
A recent systematic review and meta-analysis of existing data on statin use and the risk of cirrhosis development as well as the occurrence of cirrhosis-related complications in patients with CLD showed that there is a probable association between statin use and a lower risk of hepatic decompensation and mortality, and that statins might reduce portal hypertension in these patients [59••].
Hepatocellular Carcinoma
The use of statins has also been described as inversely associated with the incidence of hepatocellular carcinoma (HCC). Epidemiological retrospectives studies have shown a decreased rate of HCC with an OR of 0.32 in statin users compared to matched control in the US population, without a clear dose-related effect [60]. In a cohort of 27,883 HCV patients with HCC, the adjusted HR for statin users to develop HCC was 0.66 (95% CI, 0.59 to 0.74), 0.47 (95% CI, 0.40 to 0.56), and 0.33 (95% CI, 0.25 to 0.42) for patients with 28 to 89, 90 to 180, and 180 cumulative defined daily doses, respectively, showing a dose-related effect of statins in prevention of HCC [61]. The evidence about the role of statins in prevention of HCC is summarized in a systematic review and meta-analysis including 10 studies in quantitative synthesis with more than 1.4 million patients with 4298 cases of HCC, which indicated an adjusted OR of 0.63 (0.52–0.76) of the use of statins in the development of HCC, but with significant heterogeneity between studies included in the analysis. However, in East Asian population, the number needed to treat (NNT) could be estimated in 5209 patients needed to be treated with statins to prevent one of HCC per year. Moreover, in patients with HBV-associated cirrhosis, the NNT to prevent one case of HCC was estimated in 57 [62••].
The clinical findings from the use of statins in CLD patients are summarized in Table 1.
Statins and Drug-Induced Liver Injury
Initial trials and postmarketing reports of statins showed many patients having mild elevations in aminotransferases, which is a rising concern about potential hepatotoxicity [63]. However, a meta-analysis including nearly 50,000 patients showed no difference in alterations in liver function tests comparing statins with placebo [64].
Some evidence of benefit from statins in liver disease comes from basic research. Chemotherapy agents are linked to idiosyncratic drug-induced liver injury (DILI), most commonly seen with kinase inhibitors, describing higher rates in CLD patients [65]. Anthracycline antibiotics, such as doxorubicin, has been associated to DILI. In a BALB/c mouse model, the use of lovastatin decreased oxidative stress and hepatotoxicity induced by doxorubicin [66].
Nevertheless, statins have been implied as idiosyncratic DILI agents. The Swedish DILI register states that statin-related DILI episode was reported in 1.2/100,000 users, where atorvastatin was implicated in 30/73 (41%) cases, simvastatin in 28/73 (38%), and fluvastatin 11/73 (15%), showing most commonly a cholestatic/mixed pattern, with 2 cases of death due to acute liver failure and 1 underwent liver transplantation [67]. In a report of 899 idiosyncratic DILI cases from the DILIN prospective study, atorvastatin was implicated in 1.0%, rosuvastatin in 0.8%, pravastatin in 0.4%, fluvastatin in 0.2%, and lovastatin in 0.2% of the definitive, highly likely, or probably total DILI cases. Atorvastatin and rosuvastatin showed a long latency period profile of DILI (>60 days postexposure) [68•].
Besides the low incidence of DILI in statin users, these agents have been implicated in triggering drug-induced autoimmune hepatitis, and are among the top of the list of drugs implicated in chronic outcome after an acute episode of DILI [69•]. This fact is, added to the greater mortality of DILI episodes in patients with pre-existing chronic liver diseases [68•], a matter of particular concern [70]. Accordingly, the use of statins in CLD patients is low, even when the indication for it use is established as preventive agents of cardiovascular disease, highlighted by the fact that 50% of the NAFLD patients with indication to statin treatment do not receive cholesterol lowering medication [71], regardless of the protective effect of statins described in the progression of liver damage in NAFLD [50].
In an attempt to settle the issue of the statin’s use in CLD patients the declaration from the National Lipid Association Statin Liver Safety Task Force, first published in 2006 [72] and updated in 2014 [8], indicates that follow-up liver enzyme testing was not uniformly required after statin initiation in CLD patients, unless clinically indicated for other reasons. Also, the 2014 update states that in CLD patients treated with statins, a liver enzyme increase should prompt diagnostic evaluation considering other causes of altered liver function tests, and mild elevations in liver enzymes should not be considered as a contraindication to the initiation or continued use of statins [8].
Current recommendations regarding the use of statins in patients with liver disease from the National Lipid Association Statin Liver Safety Task Force state that the presence of chronic liver disease or compensated cirrhosis is not a contraindication for the use of use of statins [8]. Moreover, statins can be used in patients with non-alcoholic fatty liver disease. However, decompensated cirrhosis or acute liver failure is still a contraindication, given that statins use are associated to a class effect elevation in aminotransferases [8]. More data on clinical studies are needed to support the use of statins in patients with decompensated cirrhosis. On the other hand, the occurrence of DILI associated with statins are rare and idiosyncratic and routine monitorization of liver enzymes is not effective in identifying patients who will develop liver injury from statin therapy. Before the start of statin therapy a baseline liver enzyme test is optional in patients without CLD. Drug interactions must be considered when prescribing statins in CLD patients due to their effects in cytochrome P450 metabolism, particularly with antiviral treatment for HCV and HBV patients [8].
To date, most pre-clinical and clinical data proceed from Simvastatin; however, no clear recommendation to use a specific statin in CLD patients could be make, and its use to reduce portal hypertension is limited to clinical trials. Atorvastatin has been used for a long time in transplant patients with few adverse effects.
Conclusions and Future Directions
The pattern of statin use in patients with CLD has been changing in recent years based on evidence from pre-clinical and clinical research. Statins were initially considered harmful agents for the liver, but current evidence now suggests that this drug class may have beneficial effects in some CLD as well as a potentially positive impact on the natural history and complications of cirrhosis. However, prospective interventional studies and randomized trial need to be carried out to confirm these observations. If the benefits of statins in CLD are confirmed, hepatologists will achieve better outcomes in their patients without significant additional costs.
References
Papers of particular interest, published recently, have been highlighted as: •Of importance ••Of major importance
GBD 2015 DALYs and HALE Collaborators. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1603–58.
Berzigotti A, Erice E, Gilabert R, Reverter E, Abraldes JG, García-Pagan JC, et al. Cardiovascular risk factors and systemic endothelial function in patients with cirrhosis. Am J Gastroenterol. 2013;108(1):75–82.
Francque SM, van der Graaff D, Kwanten WJ. Non-alcoholic fatty liver disease and cardiovascular risk: pathophysiological mechanisms and implications. J Hepatol. 2016;65(2):425–43.
Chou R, Dana T, Blazina I, Daeges M, Jeanne TL. Statins for prevention of cardiovascular disease in adults: evidence report and systematic review for the US preventive services task force. JAMA. 2016;316(19):2008–24.
Shalev V, Chodick G, Silber H, Kokia E, Jan J, Heymann AD. Continuation of statin treatment and all-cause mortality: a population-based cohort study. Arch Intern Med. 2009;169(3):260–8.
Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet. 2016;388(10059):2532–61.
Blaha MJ, Martin SS. How do statins work?: changing paradigms with implications for statin allocation. J Am Coll Cardiol. 2013;62(25):2392–4.
•• Bays H, Cohen DE, Chalasani N, Harrison SA. The National Lipid Association’s Statin Safety Task Force. An assessment by the Statin Liver Safety Task Force: 2014 update. J Clin Lipidol. 2014;8(3 Suppl):S47–57. Provides recommendations for statins use and monitoring in healthy patients and CLD patients.
Schierwagen R, Uschner FE, Magdaleno F, Klein S, Trebicka J. Rationale for the use of statins in liver disease. Am J Physiol Gastrointest Liver Physiol. 2017;312(5):G407–12.
Sirtori CR. The pharmacology of statins. Pharmacol Res. 2014;88:3–11.
Germershausen JI, Hunt VM, Bostedor RG, Bailey PJ, Karkas JD, Alberts AW. Tissue selectivity of the cholesterol-lowering agents lovastatin, simvastatin and pravastatin in rats in vivo. Biochem Biophys Res Commun. 1989;158(3):667–75.
Arnaboldi L, Corsini A. Do structural differences in statins correlate with clinical efficacy? Curr Opin Lipidol. 2010;21(4):298–304.
Desai CS, Martin SS, Blumenthal RS. Non-cardiovascular effects associated with statins. BMJ. 2014;349:g3743.
Trebicka J, Schierwagen R. Statins, Rho GTPases and KLF2: new mechanistic insight into liver fibrosis and portal hypertension. Gut. 2015;64(9):1349–50.
Izadpanah R, Schächtele DJ, Pfnür AB, Lin D, Slakey DP, Kadowitz PJ, et al. The impact of statins on biological characteristics of stem cells provides a novel explanation for their pleiotropic beneficial and adverse clinical effects. Am J Physiol, Cell Physiol. 2015;309(8):C522–31.
Gracia-Sancho J, Russo L, García-Calderó H, García-Pagán JC, García-Cardeña G, Bosch J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut. 2011;60(4):517–24.
• Marrone G, Maeso-Díaz R, García-Cardena G, Abraldes JG, García-Pagán JC, Bosch J, et al. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut. 2015;64(9):1434–43. Describes the benefitial effects of KFL2 induction by simvastatin in an animal model of liver fibrosis and portal hypertension.
Marrone G, Russo L, Rosado E, Hide D, García-Cardeña G, García-Pagán JC, et al. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J Hepatol. 2013;58(1):98–103.
Russo L, Gracia-Sancho J, García-Calderó H, Marrone G, García-Pagán JC, García-Cardeña G, et al. Addition of simvastatin to cold storage solution prevents endothelial dysfunction in explanted rat livers. Hepatology. 2012;55(3):921–30.
Chong L-W, Hsu Y-C, Lee T-F, Lin Y, Chiu Y-T, Yang K-C, et al. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015;15:22.
Klein S, Klösel J, Schierwagen R, Körner C, Granzow M, Huss S, et al. Atorvastatin inhibits proliferation and apoptosis, but induces senescence in hepatic myofibroblasts and thereby attenuates hepatic fibrosis in rats. Lab Investig. 2012;92(10):1440–50.
Rombouts K, Kisanga E, Hellemans K, Wielant A, Schuppan D, Geerts A. Effect of HMG-CoA reductase inhibitors on proliferation and protein synthesis by rat hepatic stellate cells. J Hepatol. 2003;38(5):564–72.
Trebicka J, Hennenberg M, Odenthal M, Shir K, Klein S, Granzow M, et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J Hepatol. 2010;53(4):702–12.
Shirin H, Sharvit E, Aeed H, Gavish D, Bruck R. Atorvastatin and rosuvastatin do not prevent thioacetamide induced liver cirrhosis in rats. World J Gastroenterol. 2013;19(2):241–8.
Uschner FE, Ranabhat G, Choi SS, Granzow M, Klein S, Schierwagen R, et al. Statins activate the canonical hedgehog-signaling and aggravate non-cirrhotic portal hypertension, but inhibit the non-canonical hedgehog signaling and cirrhotic portal hypertension. Sci Rep. 2015;5:14573.
Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology. 2007;46(1):242–53.
Arab JP, Shah VH. Statins and portal hypertension: a tale of two models. Hepatology. 2016;63(6):2044–7.
Hsu S-J, Wang S-S, Hsin I-F, Huang H-C, Lee F-Y, Lee J-Y, et al. Effects of simvastatin on the portal-systemic collateral vascular response to endothelin-1 and shunting degree in portal hypertensive rats. Scand J Gastroenterol. 2013;48(7):831–8.
Huang H-C, Wang S-S, Lee J-Y, Chen Y-C, Lee F-Y, Lin H-C, et al. Simvastatin effects on portal-systemic collaterals of portal hypertensive rats. J Gastroenterol Hepatol. 2010;25(8):1401–9.
Chang C-C, Wang S-S, Huang H-C, Lee J-Y, Lee F-Y, Lin H-C, et al. Pravastatin administration does not induce detrimental effects on hemodynamics and collaterals of portal hypertensive rats. J Gastroenterol Hepatol. 2010;25(8):1394–400.
Rodríguez S, Raurell I, Torres-Arauz M, García-Lezana T, Genescà J, Martell M. A nitric oxide-donating statin decreases portal pressure with a better toxicity profile than conventional statins in cirrhotic rats. Sci Rep. 2017;7:40461.
Nie L, Imamura M, Itoh H, Ueno H. Pitavastatin enhances the anti-fibrogenesis effects of candesartan, an angiotensin II receptor blocker, on CCl4-induced liver fibrosis in rats. J UOEH. 2004;26(2):165–77.
El-Ashmawy NE, El-Bahrawy HA, Shamloula MM, Ibrahim AO. Antifibrotic effect of AT-1 blocker and statin in rats with hepatic fibrosis. Clin Exp Pharmacol Physiol. 2015;
Nseir W, Mahamid M. Statins in nonalcoholic fatty liver disease and steatohepatitis: updated review. Curr Atheroscler Rep. 2013;15(3):305.
Schierwagen R, Maybüchen L, Hittatiya K, Klein S, Uschner FE, Braga TT, et al. Statins improve NASH via inhibition of RhoA and Ras. Am J Physiol Gastrointest Liver Physiol. 2016;311(4):G724–33.
Park HS, Jang JE, Ko MS, Woo SH, Kim BJ, Kim HS, et al. Statins increase mitochondrial and peroxisomal fatty acid oxidation in the liver and prevent non-alcoholic steatohepatitis in mice. Diabetes Metab J. 2016;40(5):376–85.
Okada Y, Yamaguchi K, Nakajima T, Nishikawa T, Jo M, Mitsumoto Y, et al. Rosuvastatin ameliorates high-fat and high-cholesterol diet-induced nonalcoholic steatohepatitis in rats. Liver Int. 2013;33(2):301–11.
Kamada Y, Kiso S, Yoshida Y, Chatani N, Kizu T, Hamano M, et al. Estrogen deficiency worsens steatohepatitis in mice fed high-fat and high-cholesterol diet. Am J Physiol Gastrointest Liver Physiol. 2011;301(6):G1031–43.
Kamada Y, Kiso S, Yoshida Y, Chatani N, Kizu T, Hamano M, et al. Pitavastatin ameliorated the progression of steatohepatitis in ovariectomized mice fed a high fat and high cholesterol diet. Hepatol Res. 2013;43(4):401–12.
Wu W, Zhao L, Yang P, Zhou W, Li B, Moorhead JF, et al. Inflammatory stress sensitizes the liver to atorvastatin-induced injury in ApoE−/− mice. PLoS One. 2016;11(7):e0159512.
Kamisako T, Adachi Y. Marked improvement in cholestasis and hypercholesterolemia with simvastatin in a patient with primary biliary cirrhosis. Am J Gastroenterol. 1995;90(7):1187–8.
Stojakovic T, Claudel T, Putz-Bankuti C, Fauler G, Scharnagl H, Wagner M, et al. Low-dose atorvastatin improves dyslipidemia and vascular function in patients with primary biliary cirrhosis after one year of treatment. Atherosclerosis. 2010;209(1):178–83.
Cash WJ, O’Neill S, O’Donnell ME, McCance DR, Young IS, McEneny J, et al. Randomized controlled trial assessing the effect of simvastatin in primary biliary cirrhosis. Liver Int. 2013;33(8):1166–74.
Henderson LM, Patel S, Giordano TP, Green L, El-Serag HB. Statin therapy and serum transaminases among a cohort of HCV-infected veterans. Dig Dis Sci. 2010;55(1):190–5.
Simon TG, King LY, Zheng H, Chung RT. Statin use is associated with a reduced risk of fibrosis progression in chronic hepatitis C. J Hepatol. 2015;62(1):18–23.
Butt AA, Yan P, Bonilla H, Abou-Samra A-B, Shaikh OS, Simon TG, et al. Effect of addition of statins to antiviral therapy in hepatitis C virus-infected persons: results from ERCHIVES. Hepatology. 2015;62(2):365–74.
Yang Y-H, Chen W-C, Tsan Y-T, Chen M-J, Shih W-T, Tsai Y-H, et al. Statin use and the risk of cirrhosis development in patients with hepatitis C virus infection. J Hepatol. 2015;63(5):1111–7.
• Huang Y-W, Lee C-L, Yang S-S, Fu S-C, Chen Y-Y, Wang T-C, et al. Statins reduce the risk of cirrhosis and its decompensation in chronic hepatitis B patients: a nationwide cohort study. Am J Gastroenterol. 2016;111(7):976–85. Study from a large cohort of patients with chronic hepatitis B showing a dose-dependent reduction in the risk of cirrhosis and its decompensation.
Oni ET, Sinha P, Karim A, Martin SS, Blaha MJ, Agatston AS, et al. Statin use is not associated with presence of and severity of nonalcoholic fatty liver disease. Arch Med Res. 2014;45(1):52–7.
Dongiovanni P, Petta S, Mannisto V, Mancina RM, Pipitone R, Karja V, et al. Statin use and non-alcoholic steatohepatitis in at risk individuals. J Hepatol. 2015;63(3):705–12.
Kumar S, Grace ND, Qamar AA. Statin use in patients with cirrhosis: a retrospective cohort study. Dig Dis Sci. 2014;59(8):1958–65.
• Mohanty A, Tate JP, Garcia-Tsao G. Statins are associated with a decreased risk of decompensation and death in veterans with hepatitis C-related compensated cirrhosis. Gastroenterology. 2016;150(2):430–40.e1. Retrospective database analysis including a large number of patients showing an effect of statins in cirrhosis decompensation and death.
• ChangF-M, WangY-P, LangH-C, TsaiC-F, HouM-C, LeeF-Y, et al. Statins decrease the risk of decompensation in HBV- and HCV-related cirrhosis: a population-based study. Hepatology. 2017. Epidemiological study suggesting that the effect of statins in cirrhosis progression and complications is related to etiology.
Motzkus-Feagans C, Pakyz AL, Ratliff SM, Bajaj JS, Lapane KL. Statin use and infections in veterans with cirrhosis. Aliment Pharmacol Ther. 2013;38(6):611–8.
Zafra C, Abraldes JG, Turnes J, Berzigotti A, Fernández M, Garca-Pagán JC, et al. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology. 2004;126(3):749–55.
Abraldes JG, Albillos A, Bañares R, Turnes J, González R, García-Pagán JC, et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology. 2009;136(5):1651–8.
Pollo-Flores P, Soldan M, Santos UC, Kunz DG, Mattos DE, da Silva AC, et al. Three months of simvastatin therapy vs. placebo for severe portal hypertension in cirrhosis: a randomized controlled trial. Dig Liver Dis. 2015;47(11):957–63.
•• Abraldes JG, Villanueva C, Aracil C, Turnes J, Hernandez-Guerra M, Genesca J, et al. Addition of simvastatin to standard therapy for the prevention of variceal rebleeding does not reduce rebleeding but increases survival in patients with cirrhosis. Gastroenterology. 2016;150(5):1160–70.e3. Randomized controlled trial evaluating the benefit of addition of simvastatin to standard therapy in prevention of recurrent variceal bledding.
•• KimRG, LoombaR, ProkopLJ, SinghS. Statin use and risk of cirrhosis and related complications in patients with chronic liver diseases: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2017. Quantitative summary of the risk and benefits of the use of statins in CLD patients.
McGlynn KA, Divine GW, Sahasrabuddhe VV, Engel LS, VanSlooten A, Wells K, et al. Statin use and risk of hepatocellular carcinoma in a U.S. population. Cancer Epidemiol. 2014;38(5):523–7.
Tsan Y-T, Lee C-H, Ho W-C, Lin M-H, Wang J-D, Chen P-C. Statins and the risk of hepatocellular carcinoma in patients with hepatitis C virus infection. J Clin Oncol. 2013;31(12):1514–21.
•• Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144(2):323–32. Quantitative summary of the effect of statins in HCC in CLD patients.
Demyen M, Alkhalloufi K, Pyrsopoulos NT. Lipid-lowering agents and hepatotoxicity. Clin Liver Dis. 2013;17(4):699–714. x
Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97(8A):52C–60C.
Thatishetty AV, Agresti N, O’Brien CB. Chemotherapy-induced hepatotoxicity. Clin Liver Dis. 2013;17(4):671–86. ix
Henninger C, Huelsenbeck J, Huelsenbeck S, Grösch S, Schad A, Lackner KJ, et al. The lipid lowering drug lovastatin protects against doxorubicin-induced hepatotoxicity. Toxicol Appl Pharmacol. 2012;261(1):66–73.
Björnsson E, Jacobsen EI, Kalaitzakis E. Hepatotoxicity associated with statins: reports of idiosyncratic liver injury post-marketing. J Hepatol. 2012;56(2):374–80.
• Chalasani N, Bonkovsky HL, Fontana R, Lee W, Stolz A, Talwalkar J, et al. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015;148(7):1340–52.e7. Data from the US registry of drug induced liver injury.
• Medina-Caliz I, Robles-Diaz M, Garcia-Muñoz B, Stephens C, Ortega-Alonso A, Garcia-Cortes M, et al. Definition and risk factors for chronicity following acute idiosyncratic drug-induced liver injury. J Hepatol. 2016;65(3):532–42. Data from the Spanish registry of drug induced liver injury.
Slim M, Ruiz-Cabello E, Robles-Díaz M, Lucena MI, Andrade RJ. A new hepatoprotective effect of statins: are they always safe for the liver? Am J Gastroenterol. 2017;112(2):384–5.
Del Ben M, Baratta F, Polimeni L, Pastori D, Loffredo L, Averna M, et al. Under-prescription of statins in patients with non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2017;27(2):161–7.
Bays H. Statin safety: an overview and assessment of the data—2005. Am J Cardiol. 2006;97(8A):6C–26C.
Acknowledgments
This work was supported by grant(s) NIH DK59615 and AA021171 (VHS), the Clinical Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567). Arab JP was founded by an award from AASLD Foundation (AASLD/LIFER Clinical and Translational Research Fellowship in Liver Diseases 2016). Support from the government of Chile through the Fondo Nacional de Desarrollo Cientıfico y Tecnologico (FONDECYT 1150327 to M.A.) and the Comision Nacional de Investigacion Cientıfica y Tecnologica (grant CONICYTPIA/Basal PFB12, Basal Centre for Excellence in Science and Technology to M.A.) is also acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Liver
Rights and permissions
About this article

Cite this article
Vargas, J.I., Arrese, M., Shah, V.H. et al. Use of Statins in Patients with Chronic Liver Disease and Cirrhosis: Current Views and Prospects. Curr Gastroenterol Rep 19, 43 (2017). https://doi.org/10.1007/s11894-017-0584-7
Published:
DOI: https://doi.org/10.1007/s11894-017-0584-7