
Abstract
Despite the efficacy of statins in reducing cardiovascular events in both primary and secondary prevention, the adherence to statin therapy is not optimal, mainly due to the occurrence of muscular adverse effects. Several risk factors may concur to the development of statin-induced myotoxicity, including patient-related factors (age, sex, and race), statin properties (dose, lipophilicity, and type of metabolism), and the concomitant administration of other drugs. Thus, the management of patients intolerant to statins, particularly those at high or very high cardiovascular risk, involves alternative therapies, including the switch to another statin or the use of intermittent dosage statin regimens, as well as nonstatin lipid lowering drugs (ezetimibe and fibrates) or new hypolipidemic drugs such as PCSK9 monoclonal antibodies, the antisense oligonucleotide against the coding region of human apolipoprotein B mRNA (mipomersen), and microsomal triglyceride transfer protein inhibitor lomitapide. Ongoing clinical trials will reveal whether the lipid-lowering effects of alternative therapies to statins can also translate into a cardiovascular benefit.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A large number of clinical trials have shown the efficacy of statins in reducing cardiovascular morbidity and mortality in both primary and secondary prevention [1–7]. Current guidelines recommend serum low density lipoprotein cholesterol (LDL-C) levels <100 mg/dL (2.59 mmol/L) and <70 mg/dL (1.8 mmol/L) for subjects at high and very high cardiovascular risk, respectively [8], leading to an increased use of statins at high doses. Despite the significant reduction of cardiovascular events in statin-treated subjects, the adhesion to therapy seems, however, not adequate, as many patients discontinue the statin therapy owing very often to the occurrence of drug-related side effects. In addition, an increasing number of subjects are treated with statins, often in combination with other drugs, thus increasing the probability of drug–drug interactions and the appearance of adverse side effects [9].
The most common adverse reactions among statin users are skeletal muscle-related disorders, generally referred to as myopathy, but the real incidence of this adverse effect is still uncertain. In fact, randomized clinical trials generally reported a 1.5–5 % incidence of statin-induced myopathy [10, 11], while in the clinical practice, the frequency appears to be higher [12]. This discrepancy can be attributed mainly to the fact that clinical trials tend to exclude older subjects, subjects with co-morbidities, with a history of muscle-related symptoms or at risk for myopathy or even have a run- in period that excludes subjects with side effects [12, 13]. Another explanation is the lack of a standard definition for statin-related myotoxicity and the common use of creatine kinase (CK) increase as a marker for statin-induced myopathy; however, the occurrence of myopathy is not always associated with CK elevations. Finally, less significant signs of myopathy (including fatigability or weakness) are often disregarded [13]. This results in an incidence of myopathy cases comparable to that observed in control subjects not taking statins and, as a consequence, in a possible underestimation of statin-induced risk of muscle-related side effects.
Statin-Induced Myopathy: Definition and Frequency
Statin-related myopathy occurs particularly with high-dosage statin regimens, and thus, its incidence is higher in high cardiovascular risk patients who need more aggressive lipid-lowering therapies, or in patients with co-morbidities due to metabolic interactions with other drugs [14, 15]. Clinically, statin-induced muscular adverse effects vary greatly, either as muscle symptoms and CK elevation degrees; recently, an algorithm to better define the type of statin-induced myotoxicity has been developed [16••]. Asymptomatic serum CK elevation <4× upper limit of normal (ULN) or muscle pain in the absence of CK elevation (referred to as myalgia) (Table 1) are the less severe manifestations and the most common muscle side effects associated with statin therapy [16••]; the last is characterized by muscle pain, weakness, and cramps, and if the subject can tolerate pain, statin therapy usually is not discontinued. On the contrary, in the presence of intolerable myalgia, characterized by muscle pain associated with CK elevation <4× ULN, statin discontinuation is usually applied leading to the complete resolution of the symptoms. Myopathy or myositis is defined by the presence of muscle pain associated with a significant increase in CK levels (>4× ULN and <10× ULN for less the severe form and >10 and <50 ULN for severe form) (Table 1), is less frequent, and generally, therapy discontinuation leads to symptom resolution [16••]. Rhabdomyolysis is a rare occurrence upon statin treatment (0.1–8.4/100,000 patient-year) defined as muscle pain associated with CK levels >10× ULN with evidence of renal function impairment or CK levels >50× ULN (Table 1) and is characterized by muscle necrosis and release of myoglobin in the circulation [16••]. Some studies have also reported the occurrence of autoimmune-mediated necrotizing myositis in statin-treated subjects characterized by progressive weakness, high levels of muscle enzymes, and autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A (HMGCoA) reductase, the pharmacological target of statins [17].
Evaluation of Statin Intolerance
Due to the exclusion of subjects with a history of muscular disorders or at risk for myalgia, the real frequency of statin-induced myotoxicity has been underestimated in the randomized clinical trials [10, 11]. In contrast, in the everyday clinical practice, a relevant number of subjects experiencing myalgia during statin therapy is observed; thus, the real clinical impact of muscular side effects induced by statins has been specifically evaluated in two studies.
The Prediction of Muscular Risk in Observational conditions (PRIMO) study was an observational survey of muscular symptoms in an unselected population of hyperlipidemic subjects taking high-dose statins [18]. This study revealed that mild to moderate muscular symptoms occurred more frequently (10.5 %) in patients treated with high-dose statins than established in randomized clinical trials and that the type of statin used was an independent predictor of muscular symptoms, being extended-release fluvastatin associated with the lowest occurrence (5.1 %) and simvastatin with the highest risk (18.2 %) [18]. The median time of onset for muscular symptoms was 1 month after statin therapy initiation or titration to a high dosage [18]; 57 % of the patients experiencing muscular symptoms were switched to another lipid-lowering therapy (61 % to another statin, 28 % to a fibrate), or to a lower dosage of the same statin (17 %), while 20 % of the patients completely discontinued statin therapy [18]. A relevant finding of this study was the observation that the occurrence of muscular symptoms in subjects taking high-dose statin impacts negatively the everyday life, as a high number of subjects (25 %) reported a continuous muscular pain, and 39 % needed an analgesic for pain relief [18]. These findings, confirmed by an observational survey conducted in a large unselected population of hypercholesterolemic subjects taking statins [19•], may explain why poor patient compliance represents a major issue for statin-treated subjects.
The Effects of Statins on Skeletal Muscle Function and Performance (STOMP) study evaluated the impact of high-dose statin therapy (atorvastatin, 80 mg) for 6 months in healthy, statin-naive subjects [20]. This study revealed that atorvastatin significantly increased the frequency of myalgia compared with placebo (9.4 % vs 4.6 %) [20]; CK levels increased also in asymptomatic atorvastatin-treated subjects (although none exceeded the 10× ULN), suggesting that high-dose statin may induce a low-grade muscle injury, although increased CK did not impact skeletal muscle function [20]. More atorvastatin-treated subjects at least doubled their baseline CK levels that, however, were not associated with deterioration of muscle function [21], thus confirming that increased CK is not necessarily suggestive of statin-induced muscle injury. Finally, subjects with myalgia tended to be older, and almost 60 % were women [20].
Statin-Induced Myopathy: Risk Factors and Mechanisms
A high number of risk factors may increase the probability of myotoxicity induced by statin therapy. Some of these risk factors are related to physicochemical and pharmacokinetic properties of statins or to concomitant therapies, but also patient characteristics may affect statin bioavailability and increase the vulnerability to high levels of statins.
Statin Properties
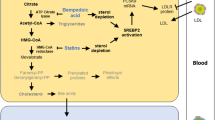
The main pharmacological target of statins is HMGCoA reductase, a key enzyme involved in the synthesis of cholesterol; its inhibition results in decreased cholesterol biosynthesis and depletion of other downstream metabolites, including ubiquinone (or coenzyme Q10), whose reduction in serum and muscle tissue has been hypothesized to affect the aerobic metabolism in muscle and potentially induce muscle injury [22], although final evidence is still lacking.
Statins exhibit different lipophilicity degrees, being pravastatin and rosuvastatin the most hydrophilic and the other statins (simvastatin, atorvastatin, lovastatin, fluvastatin, and pitavastatin) more lipophilic (Table 2). The risk of myopathy seems to be higher with lipophilic statins due to their ability to cross cell membranes and to impair mitochondrial functions in muscle tissues in a more relevant way compared with hydrophilic statins [23]; in fact, pravastatin seems to trigger myotoxicity to a lower extent compared with other statins [24]. Yet, hydrophilicity does not guarantee freedom from these effects, as pravastatin and rosuvastatin may be transported within cells by active uptake transporters; among them, organic anion-transporting polypeptide (OATP) 1B1 plays a key role in statin absorption in both liver and intestine (Table 2) [25, 26].
A dose-dependent effect of statins on skeletal muscles has been clearly established by in vitro studies, in animal models as well as in clinical studies [14]. Aggressive lipid-lowering therapies not only reduce significantly the risk of cardiovascular events but also increase the risk of adverse effects compared with moderate-dose statin therapy, suggesting that intensive therapy should be addressed to individuals at high cardiovascular risk, while other subjects may benefit also from moderate dose statin therapy [27].
Statin–Drug Interactions
Cerivastatin caused a high number of rhabdomyolysis cases before its withdrawal from the market; many of these cases were registered in subjects taking gemfibrozil and cerivastatin concurrently [28]. The possible mechanism was the inhibition of CYP2C8-mediated metabolism of cerivastatin by gemfibrozil [29, 30], suggesting that the pharmacokinetic interaction between these two drugs may have significantly increased the exposure to high levels of cerivastatin. The hepatic cytochrome P450 enzyme system plays a key role in the metabolism of lipophilic statins (with the exception of pitavastatin), being CYP3A4 isoenzyme responsible for the metabolism of simvastatin, lovastatin, and atorvastatin, while CYP2C9 is involved in the metabolism of fluvastatin and, to a lesser extent, rosuvastatin (Table 2) [14]. Pravastatin, rosuvastatin, and pitavastatin are excreted mainly as parent compounds [31, 32]. The bioavailability of different statins varies greatly, from <5 % of simvastatin to 60 % of pitavastatin [31, 32]. The administration of a statin along with other drugs that inhibit or are substrates of CYP isoenzymes, or interfere with drug transporters such as OATP1B1 or P-glycoprotein (P-gp, which on the contrary functions as an efflux pump), may lead to an interaction resulting in increased levels of statins in the circulation and a higher risk of statin-associated adverse effects [32]. For example, the plasma levels of simvastatin administered concurrently with cyclosporine, a substrate of CYP3A4 and OATP1B, increased significantly [33]. Thus, statin–drug interactions represent a risk factor for muscle-related adverse effects in statin users, as observed in a subanalysis of the Understanding Statin Use in America and Gaps in Education (USAGE) study, in which concomitant use of a CYP450 inhibitor or OATP1B1 or P-gp inhibitor significantly increased the risk of new or worse muscle symptoms and the probability to stop statin therapy due to muscle pain [34•]. Therefore, statin–drug interactions can be identified as a predictor of the occurrence of muscle pain and discontinuation of statin therapy [35, 36].
Patient-Related Factors
Some patient characteristics represent a risk factor for statin-induced myopathy, including age (older subjects have higher probability to experience myalgia), female gender (women have a 2-fold risk compared with matched men), and Asian race [14]. For example, rosuvastatin exposure is 1.6- to 2.3-fold greater in Asians compared with matched white subjects [37], also when living in the same environment, suggesting the need of lower starting rosuvastatin dose in Asian patients [38]. In addition, the presence of comorbidities increases the incidence of myopathy [14]. Finally, some genetic factors may greatly affect the susceptibility to statin-induced myopathy [39]. Polymorphisms in the solute carrier organic anion transporter SLCO1B1, the gene encoding for OATP1B1, are associated with a reduced uptake of some statins, resulting in increased statin plasma levels [39]; the single nucleotide polymorphisms rs4149056 (c.521T>C) in SLCO1B1 has been associated with myopathy induced by simvastatin 80 mg/day, due to changes of plasma exposure to simvastatin, being the c.521CC homozygotes the most exposed [40–43]. In contrast, in patients taking atorvastatin, rs4149056 did not increase the risk of myopathy [43]; similar findings were observed in patients taking rosuvastatin [44]. These observations suggested a relevant role for SLCO1B1 in affecting simvastatin-induced myopathy, leading to the very recent dosing recommendations for simvastatin based on SLCO1B1 genotype [45] and to the hypothesis that subjects carrying this polymorphism, and thus at high risk for simvastatin-associated myopathy, could reduce the risk if treated with an alternative statin.
Furthermore, genetic variants of CYP isoenzymes might affect the individual response to statin therapy. Several statins are metabolized by CYP3A4; thus, alterations in this enzyme activity would translate into an altered statin metabolism and changes of circulating levels. A single nucleotide polymorphism (C>T) located in intron 6 of CYP3A4 (rs35599367, CYP3A4*22) has been associated with a reduced expression and activity of hepatic CYP3A4, which in turn was consistently associated with lower stable dose requirements of CYP3A4-metabolized statins (simvastatin, atorvastatin, and lovastatin) [46]; in a population-based cohort study, carriers of the T-variant allele had an increased lipid-lowering response to simvastatin [47]. However, this observation has not been confirmed in subjects with primary hypercholesterolemia [48], and no association of the CYP3A4*22 minor allele with the effectiveness of statins in reducing myocardial infarction risk was observed in two large population-based studies [49]. The reason for this lack of association of rs35599367 with the effectiveness of statins in reducing myocardial infarction seems to be due to the fact that the effect on LDL-C levels is too small to affect a clinical outcome [49]. Recently, the gene polymorphism CYP2D6*10 (C188T) has been related to the lipid-lowering efficacy of simvastatin, showing in the CC genotype an increased lipid-lowering response to simvastatin compared with CT/TT genotypes [50]. These observations suggest that subjects carrying particular genetic variants could respond better to low statin doses, thus decreasing the risk of adverse effects.
Some studies also observed that subjects with genetic muscle diseases have an increased risk of statin-induced myopathy [39, 51]. Thus, susceptible individuals, following the exposure to statin, may exacerbate a pre-existing muscle disorder, or may unmask an asymptomatic pre-existing disease.
Pharmacological Approaches to Statin Intolerance
In everyday clinical practice, discontinuation of statin therapy is a common event among patients experiencing muscle symptoms; in fact, a high number (59.2 %) of patients who had a documented statin-associated event (17.4 % of the total study patients) discontinued statins at least temporarily in a retrospective cohort study [52]. Among them, 59.1 % were rechallenged with a statin, and a high percentage (over 90 %) could tolerate it [52]. Discontinuation of statin therapy is of particular relevance in secondary prevention, or in high cardiovascular risk subjects; thus, it is of major importance to carefully evaluate a pharmacological approach that does not lead to an increased cardiovascular risk due to statin therapy discontinuation.
Statin Rechallenge
As established by some studies, a large number of subjects with statin intolerance can tolerate the rechallenge with a statin therapy [52, 53], and these findings suggested that reported adverse events might have other causes than statin therapy or that they could be related to a specific statin and not to the entire class. Statins can be reintroduced at a lower dose; as the incidence of muscle adverse effects is increased with high-dose statins, reduced dose may help in preventing the occurrence of myopathy. Alternatively, a different statin can be selected; one possibility is to change statin on the basis of its lipophilic degree, or on the basis of its metabolic route (for example, switching from a CYP450-dependent to a CYP450-independent statin) [54]. Another chance is to switch to a lower dose of a more potent statin; this possibility is supported by the observation that, in patients previously intolerant to a statin (primarily simvastatin or atorvastatin at any dose) and unable to meet LDL-C level goal when switched to a nonstatin lipid-lowering therapy, rosuvastatin 5 or 10 mg was well tolerated and safe, and, of the 61 subjects, only 1 discontinued the treatment due to unilateral muscular pain in the leg [55].
Another emerging option is the non-everyday statin administration. Several studies have established the efficacy and safety of this treatment option, mainly using rosuvastatin because of its long half-life (approximately 19 h) [56] and its lipid-lowering potency; in addition, rosuvastatin is minimally metabolized by the CYP450 system and thus is potentially less likely to be involved in the interaction with other drugs. Rosuvastatin (2.5–20 mg) once a week for 4 months in patients with statin intolerance was able to reduce significantly total cholesterol and LDL-C with a high tolerability, although this study could not prove that this regimen reduced cardiovascular risk [57]. Similarly, once a week high-dose rosuvastatin (80 mg) produced lipid changes comparable to those obtained with atorvastatin 10 mg daily [58], and rosuvastatin every other day increased the tolerability of the therapy among subjects previously intolerant to a statin [59]. Also atorvastatin, which has a half life of approximately 14 h and produces active metabolites with long half-lives, has been evaluated in nondaily dosing regimens. In patients intolerant to daily statin therapy and switched to ezetimibe without reaching LDL-C level goal, the addition of 10 mg atorvastatin twice a week significantly reduced LDL-C levels with a good tolerability [60]. Overall, nondaily statin use seems to be generally better tolerated than daily dosing regimens, probably due to the fact that not only intermittent statin administration may allow muscle toxicity recovery but also other components (including psychological factors) may increase the adherence to this type of therapy [61, 62]. However, further long-term studies are required to establish whether these alternative regimens may have cardiovascular effects similar to those obtained with daily therapies but with reduced adverse side effects.
Nonstatin Lipid-Lowering Drugs
The combination of nonstatin lipid-lowering drugs with the lowest statin dose tolerated or, as an alternative, a combination of nonstatin lipid-lowering drugs represent possible approaches for patients intolerant to statins. Among nonstatin lipid-lowering drugs, ezetimibe, fibrates, and bile acid sequestrants are largely used. Ezetimibe, a cholesterol absorption inhibitor, has been tested alone, showing the ability to reduce significantly LDL-C levels with a safety profile similar to that of placebo [63], or in combination with fenofibrate, which reduced LDL-C to a level similar to that observed with a 10-mg atorvastatin monotherapy [64]. The long-term consequences of this combination, either as effect on cardiovascular events or adverse effects, are still not known. When patients intolerant to standard statin therapy were treated with ezetimibe in combination with low dose of simvastatin (10 mg), the previously observed adverse effects did not occur during the 6-month period of treatment, while LDL-C levels were significantly decreased and a large number of subjects reached the LDL-C level goal [65]. The real efficacy of the combination therapy ezetimibe plus simvastatin in the reduction of cardiovascular events has been evaluated in the Improved Reduction of Outcomes: Vitoryn Efficay International Trial (IMPROVE-IT) [66], whose results were recently presented [67]. Patients who received combination therapy had a median LDL-C level lower than those who received statin monotherapy [54 mg/dL (1.4 mmol/L) and 69 mg/dL (1.78 mmol/L), respectively); this led to a statistically significant reduction of cardiovascular events in the combination therapy group compared with the statin group (32.7 % vs 34.7 %, p = 0.016) [67]. These results confirmed the safety profile of ezetimibe and demonstrated that a nonstatin drug may have a beneficial effect when added to a statin therapy [67].
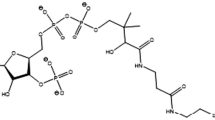
In addition mipomersen, a second-generation antisense oligonucleotide against the coding region of human apolipoprotein B mRNA, and lomitapide, a microsomal triglyceride transfer protein (MTP) inhibitor, are part of the therapeutic lipid-lowering approach in the treatment of familial hypercholesterolemia [68] but can also be considered in statin-intolerant patients. Mipomersen was shown to significantly reduce LDL-C levels in several studies [69–74]; mipomersen is not metabolized by CYP450 system, it does not inhibit major CYP isoenzymes, and it does not show clinically relevant interaction with simvastatin or ezetimibe [75]. Mipomersen is associated with frequent adverse reactions, being injection-site reaction following subcutaneous administration the most common, followed by increased hepatic enzyme levels (not associated with liver dysfunction) and flu-like symptoms; the most relevant side effect of mipomersen seems to be an increased content of triglycerides in the liver, but despite short-term studies revealed an increased steatosis (whose clinical relevance is considered low) in mipomersen-treated subjects, long-term clinical trials observed a return to normal of hepatic triglyceride level [76, 77]. When tested in statin-intolerant hypercholesterolemic patients, mipomersen (200 mg/week) significantly reduced apo-B100 and LDL-C levels as well as other lipids, without effect on HDL or apoA-I, and also affected LDL particle subclass distribution, decreasing significantly small dense proatherogenic LDL [78]; overall mipomersen was well tolerated [78].
Lomitapide inhibits the lipid transfer activity of MTP, thus inhibiting the assembly of intestinal chylomicrons and hepatic VLDL and results in reduced secretion of these lipoproteins into the circulation [79•]. Lomitapide was shown to be effective in reducing LDL-C levels in patients with moderate hypercholesterolemia in monotherapy as well as in combination with ezetimibe [80]; this drug is also effective in familial hypercholesterolemia patients when administered concurrently with background lipid-lowering therapies [81, 82]. Lomitapide is metabolized via CYP3A4; thus, it must be not administered with CYP3A4 inhibitors; in addition, lomitapide increases the exposure to simvastatin, atorvastatin, and rosuvastatin [76]. Major adverse effects are gastrointestinal disorders such as loose stools and increase in liver enzymes and fat content [76], although these effects result transient and reversed after drug discontinuation.
Proprotein convertase subtilis/kexin type 9 (PCSK9) is a proteinase mainly synthesized and secreted by the liver and involved in the intracellular degradation of LDLR; its activity results in a decreased expression of hepatic LDLR and increase in circulating LDL-C levels. Thus, inhibiting PCSK9 expression/activity may result in the reduction of LDL-C levels. PCSK9 inhibition is actually achieved mainly by the use of monoclonal antibodies (mAbs) that have shown a significant efficacy to reduce LDL-C levels in experimental as well as in clinical studies [83]. The lipid-lowering efficacy of statins is partly limited by the induction of PCSK9 expression [84, 85], thus adding a PCSK9 mAb to a statin may increase the therapeutic effect. Several clinical studies have shown that anti-PCSK9 monoclonal antibody monotherapies significantly decreased total cholesterol and LDL-C, with an overall incidence of adverse effects similar between the treatment and placebo groups [86]. Evolocumab, alirocumab, and bococizumab are human monoclonal antibodies against PCSK9 that inhibit its interaction with the LDLR, leading to increased hepatic LDLR expression and LDL clearance; these antibodies have been all tested in hypercholesterolemic patients showing a great efficacy in reducing LDL-C levels [86]. Evolocumab has been evaluated also in a population of hypercholesterolemic patients intolerant to an effective dose of one or more statins due to muscle-related disorders in the Goal Achievement After Utilizing an Anti-PCSK9 Antibody in Statin-Intolerant Subjects (GAUSS) phase-2 study [87]. After 12-week treatment with evolocumab alone or in combination with ezetimibe, patients treated with evolocumab showed a dose-dependent reduction of LDL-C levels, and the association with ezetimibe resulted in a higher decrease compared with ezetimibe alone (−63 vs −15 %); the maximal reduction occurred within 2 weeks after evolocumab treatment [87]. The reduction obtained with every dose of evolocumab was greater than with ezetimibe, and a high proportion of patients reached the recommended LDL-C target [87]. The administration of evolocumab was well tolerated, and myalgia was the most common side effect occurring in 7.4 % taking evolocumab alone, in 20 % taking evolocumab+ezetimibe, and in 3.1 % taking ezetimibe [87]; the overall incidence of all adverse effects was similar among the groups, and the occurrence of four serious adverse events was not considered treatment-related. The GAUSS-2 phase 3 study compared the effect of evolocumab or ezetimibe 12-week treatment in a population at higher cardiovascular risk with a high proportion of patients intolerant to at least two statins [88•]; evolocumab reduced significantly LDL-C levels compared with ezetimibe (53–56 % vs 37–39 %), with muscle-related side effects occurring in 12 % of evolocumab group and 23 % of ezetimibe group [88•]. A higher proportion of evolocumab-treated patients reached the LDL-C level target compared with ezetimibe-treated patients [88•]. Myalgia was reported in 8 % of evolocumab group and 18 % of ezetimibe group; patients using low-dose statin developed myalgia more likely in the ezetimibe group than in evolocumab group [88•]. However, the benefit of evolocumab on cardiovascular outcomes is still under investigation (ClinicalTrials.gov Identifier NCT01764633) and the tolerability of long-term treatment remains to be addressed. The ongoing GAUSS-3 study will include a placebo-controlled re-exposure to statin (atorvastatin) before a 24-week double-blind comparison of evolocumab and ezetimibe, followed by 2-year open-label evolocumab extension (ClinicalTrials.gov Identifier: NCT01984424). Several ongoing phase III trials are evaluating the effects of alirocumab (ODYSSEY studies) [86] and bococizumab (SPIRE-1, ClinicalTrials.gov Identifier: NCT01975376; SPIRE-2, ClinicalTrials.gov Identifier: NCT01975389) in several populations and will provide further insights into the long-term efficacy and safety of these monoclonal antibody based-therapies.
Diet Supplements
Red yeast rice (RYR) is a diet supplement containing several components, including monacolin K (lovastatin), which exhibit LDL-C-lowering properties [89] and thus may represent an alternative approach for the treatment of hypercholesterolemic patients. No significant differences were observed in hepatic enzymes as well as serum creatinine levels between groups [89]. RYR seems to be effective in statin-intolerant patients and well tolerated by a high number of subjects that exhibited myalgia following statin or ezetimibe therapy [90–92]. The presence of statin-like components at low dose in RYR may, however, raise doubts about the reduced incidence of myalgia; the additional components having LDL-C-lowering properties may further contribute to the effect on LDL-C levels. The administration of a nutraceutical combination containing berberine, RYR, and policosanol to hypercholesterolemic patients previously intolerant to statins resulted in the reduction of cholesterolemia [93]. Berberine, which acts by increasing LDLR expression and by reducing PCSK9 expression in the liver [94, 95], significantly improved blood lipid profile with a good tolerability [96] and was more effective in reducing LDL-C compared with ezetimibe in patients with primary hypercholesterolemia and a history of statin intolerance [97].
Conclusions
As an increasing number of patients are treated with high-dose statins, the incidence of muscle-related side effects is expected to raise. Thus, given the cardiovascular benefits of statin therapy, it appears crucial to find efficient pharmacological alternatives for the management of statin-intolerant patients, particularly for those at high or very high cardiovascular risk. The use of intermittent nondaily statin regimens may be a valid approach to treat patients with previous intolerance to daily therapy. In addition, the newer lipid-lowering therapies currently under clinical investigation might be helpful in the management of these patients; the ongoing trials will help to define whether their lipid-lowering efficacy will also translate into a cardiovascular benefit.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Naci H, Brugts JJ, Fleurence R, et al. Comparative benefits of statins in the primary and secondary prevention of major coronary events and all-cause mortality: a network meta-analysis of placebo-controlled and active-comparator trials. Eur J Prev Cardiol. 2013;20:641–57.
Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81.
Mills EJ, Wu P, Chong G, et al. Efficacy and safety of statin treatment for cardiovascular disease: a network meta-analysis of 170,255 patients from 76 randomized trials. QJM. 2011;104:109–24.
Tonelli M, Lloyd A, Clement F, et al. Efficacy of statins for primary prevention in people at low cardiovascular risk: a meta-analysis. CMAJ. 2011;183:E1189–202.
Mills EJ, O’Regan C, Eyawo O, et al. Intensive statin therapy compared with moderate dosing for prevention of cardiovascular events: a meta-analysis of >40 000 patients. Eur Heart J. 2011;32:1409–15.
Chan DK, O’Rourke F, Shen Q, et al. Meta-analysis of the cardiovascular benefits of intensive lipid lowering with statins. Acta Neurol Scand. 2011;124:188–95.
Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013;1:CD004816.
Catapano AL, Reiner Z, de Backer G, et al. ESC/EAS guidelines for the management of dyslipidaemias the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis. 2011;217:3–46.
Corrao G, Conti V, Merlino L, et al. Results of a retrospective database analysis of adherence to statin therapy and risk of nonfatal ischemic heart disease in daily clinical practice in Italy. Clin Ther. 2010;32:300–10.
Joy TR, Hegele RA. Narrative review: statin-related myopathy. Ann Intern Med. 2009;150:858–68.
Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97:52C–60.
Fernandez G, Spatz ES, Jablecki C, et al. Statin myopathy: a common dilemma not reflected in clinical trials. Cleve Clin J Med. 2011;78:393–403.
Maningat P, Breslow JL. Needed: pragmatic clinical trials for statin-intolerant patients. N Engl J Med. 2011;365:2250–1.
Taha DA, de Moor CH, Barrett DA, et al. Translational insight into statin-induced muscle toxicity: from cell culture to clinical studies. Transl Res. 2014;164:85–109.
Chatzizisis YS, Koskinas KC, Misirli G, et al. Risk factors and drug interactions predisposing to statin-induced myopathy: implications for risk assessment, prevention and treatment. Drug Saf. 2010;33:171–87.
Alfirevic A, Neely D, Armitage J, et al. Phenotype standardization for statin-induced myotoxicity. Clin Pharmacol Ther. 2014;96:470–6. In this paper, the authors defined phenotypic criteria to standardize statin-induced myotoxicity phenotypes, based on a previously described consensus approach.
Mohassel P, Mammen AL. Statin-associated autoimmune myopathy and anti-HMGCR autoantibodies. Muscle Nerve. 2013;48:477–83.
Bruckert E, Hayem G, Dejager S, et al. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients—the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–14.
Rosenbaum D, Dallongeville J, Sabouret P, et al. Discontinuation of statin therapy due to muscular side effects: a survey in real life. Nutr Metab Cardiovasc Dis. 2013;23:871–5. This survey revealed the higher frequency of muscular symptoms associated with statin therapy in real life than in clinical trials.
Parker BA, Capizzi JA, Grimaldi AS, et al. Effect of statins on skeletal muscle function. Circulation. 2013;127:96–103.
Ballard KD, Parker BA, Capizzi JA, et al. Increases in creatine kinase with atorvastatin treatment are not associated with decreases in muscular performance. Atherosclerosis. 2013;230:121–4.
Marcoff L, Thompson PD. The role of coenzyme Q10 in statin-associated myopathy: a systematic review. J Am Coll Cardiol. 2007;49:2231–7.
Kaufmann P, Torok M, Zahno A, et al. Toxicity of statins on rat skeletal muscle mitochondria. Cell Mol Life Sci. 2006;63:2415–25.
Masters BA, Palmoski MJ, Flint OP, et al. In vitro myotoxicity of the 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors, pravastatin, lovastatin, and simvastatin, using neonatal rat skeletal myocytes. Toxicol Appl Pharmacol. 1995;131:163–74.
Ho RH, Tirona RG, Leake BF, et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology. 2006;130:1793–806.
Nakai D, Nakagomi R, Furuta Y, et al. Human liver-specific organic anion transporter, LST-1, mediates uptake of pravastatin by human hepatocytes. J Pharmacol Exp Ther. 2001;297:861–7.
Silva M, Matthews ML, Jarvis C, et al. Meta-analysis of drug-induced adverse events associated with intensive-dose statin therapy. Clin Ther. 2007;29:253–60.
Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med. 2002;346:539–40.
Wang JS, Neuvonen M, Wen X, et al. Gemfibrozil inhibits CYP2C8-mediated cerivastatin metabolism in human liver microsomes. Drug Metab Dispos. 2002;30:1352–6.
Ogilvie BW, Zhang D, Li W, et al. Glucuronidation converts gemfibrozil to a potent, metabolism-dependent inhibitor of CYP2C8: implications for drug–drug interactions. Drug Metab Dispos. 2006;34:191–7.
Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19:117–25.
Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid-lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80:565–81.
Ichimaru N, Takahara S, Kokado Y, et al. Changes in lipid metabolism and effect of simvastatin in renal transplant recipients induced by cyclosporine or tacrolimus. Atherosclerosis. 2001;158:417–23.
Ito MK, Maki KC, Brinton EA, et al. Muscle symptoms in statin users, associations with cytochrome P450, and membrane transporter inhibitor use: a subanalysis of the USAGE study. J Clin Lipidol. 2014;8:69–76. This study showed the relevance of drug-drug interaction as a risk factor for muscle-related side effects in patients taking statins and concomitant therapies that interfere with statin metabolism.
Tragni E, Casula M, Pieri V, et al. Prevalence of the prescription of potentially interacting drugs. PLoS One. 2013;8:e78827.
Kellick KA, Bottorff M, Toth PP. A clinician’s guide to statin drug–drug interactions. J Clin Lipidol. 2014;8:S30–46.
Lee E, Ryan S, Birmingham B, et al. Rosuvastatin pharmacokinetics and pharmacogenetics in white and Asian subjects residing in the same environment. Clin Pharmacol Ther. 2005;78:330–41.
Liao JK. Safety and efficacy of statins in Asians. Am J Cardiol. 2007;99:410–4.
Needham M, Mastaglia FL. Statin myotoxicity: a review of genetic susceptibility factors. Neuromuscul Disord. 2014;24:4–15.
Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy—a genomewide study. N Engl J Med. 2008;359:789–99.
Pasanen MK, Neuvonen M, Neuvonen PJ, et al. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006;16:873–9.
Stewart A. SLCO1B1 Polymorphisms and statin-induced myopathy. PLoS Curr 2013;5. doi:10.1371/currents.eogt.d21e7f0c58463571bb0d9d3a19b82203
Brunham LR, Lansberg PJ, Zhang L, et al. Differential effect of the rs4149056 variant in SLCO1B1 on myopathy associated with simvastatin and atorvastatin. Pharmacogenomics J. 2012;12:233–7.
Danik JS, Chasman DI, MacFadyen JG, et al. Lack of association between SLCO1B1 polymorphisms and clinical myalgia following rosuvastatin therapy. Am Heart J. 2013;165:1008–14.
Ramsey LB, Johnson SG, Caudle KE, et al. The clinical pharmacogenetics implementation consortium guideline for SLCO1B1 and simvastatin-induced myopathy: 2014 update. Clin Pharmacol Ther. 2014;96:423–8.
Wang D, Guo Y, Wrighton SA, et al. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 2011;11:274–86.
Elens L, Becker ML, Haufroid V, et al. Novel CYP3A4 intron 6 single nucleotide polymorphism is associated with simvastatin-mediated cholesterol reduction in the Rotterdam Study. Pharmacogenet Genomics. 2011;21:861–6.
Ragia G, Kolovou V, Tavridou A, et al. No effect of CYP3A4 intron 6 C>T polymorphism (CYP3A4*22) on lipid-lowering response to statins in Greek patients with primary hypercholesterolemia. Drug Metabol Drug Interact 2015; in press. doi:10.1515/dmdi-2014-0021
Leusink M, de Keyser CE, Onland-Moret NC, et al. No association between CYP3A4*22 and statin effectiveness in reducing the risk for myocardial infarction. Pharmacogenomics. 2014;15:1471–7.
Li J, Wang X, Zhang Z, et al. Statin therapy correlated CYP2D6 gene polymorphism and hyperlipidemia. Curr Med Res Opin. 2014;30:223–8.
Vladutiu GD, Simmons Z, Isackson PJ, et al. Genetic risk factors associated with lipid-lowering drug-induced myopathies. Muscle Nerve. 2006;34:153–62.
Zhang H, Plutzky J, Skentzos S, et al. Discontinuation of statins in routine care settings: a cohort study. Ann Intern Med. 2013;158:526–34.
Mampuya WM, Frid D, Rocco M, et al. Treatment strategies in patients with statin intolerance: the Cleveland Clinic experience. Am Heart J. 2013;166:597–603.
Arca M, Pigna G. Treating statin-intolerant patients. Diabetes Metab Syndr Obes. 2011;4:155–66.
Glueck CJ, Aregawi D, Agloria M, et al. Rosuvastatin 5 and 10 mg/d: a pilot study of the effects in hypercholesterolemic adults unable to tolerate other statins and reach LDL cholesterol goals with nonstatin lipid-lowering therapies. Clin Ther. 2006;28:933–42.
Martin PD, Warwick MJ, Dane AL, et al. Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin Ther. 2003;25:2822–35.
Ruisinger JF, Backes JM, Gibson CA, et al. Once-a-week rosuvastatin (2.5 to 20 mg) in patients with a previous statin intolerance. Am J Cardiol. 2009;103:393–4.
Backes JM, Gibson CA, Ruisinger JF, et al. The high-dose rosuvastatin once weekly study (the HD-ROWS). J Clin Lipidol. 2011;6:362–7.
Backes JM, Venero CV, Gibson CA, et al. Effectiveness and tolerability of every-other-day rosuvastatin dosing in patients with prior statin intolerance. Ann Pharmacother. 2008;42:341–6.
Athyros VG, Tziomalos K, Kakafika AI, et al. Effectiveness of ezetimibe alone or in combination with twice a week Atorvastatin (10 mg) for statin intolerant high-risk patients. Am J Cardiol. 2008;101:483–5.
Elis A, Lishner M. Non-every day statin administration—a literature review. Eur J Intern Med. 2012;23:474–8.
Marcus FI, Baumgarten AJ, Fritz WL, et al. Alternate-day dosing with statins. Am J Med. 2013;126:99–104.
Pandor A, Ara RM, Tumur I, et al. Ezetimibe monotherapy for cholesterol lowering in 2,722 people: systematic review and meta-analysis of randomized controlled trials. J Intern Med. 2009;265:568–80.
Kumar SS, Lahey KA, Day A, et al. Comparison of the efficacy of administering a combination of ezetimibe plus fenofibrate versus atorvastatin monotherapy in the treatment of dyslipidemia. Lipids Health Dis. 2009;8:56.
Derosa G, D’Angelo A, Franzetti IG, et al. Efficacy and safety of ezetimibe/simvastatin association on non-diabetic and diabetic patients with polygenic hypercholesterolemia or combined hyperlipidemia and previously intolerant to standard statin treatment. J Clin Pharm Ther. 2009;34:267–76.
Blazing MA, Giugliano RP, Cannon CP, et al. Evaluating cardiovascular event reduction with ezetimibe as an adjunct to simvastatin in 18,144 patients after acute coronary syndromes: final baseline characteristics of the IMPROVE-IT study population. Am Heart J. 2014;168:205–12.
Cannon CP on behalf of the IMPROVE-IT Investigators. IMPROVE-IT Trial: a comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes after acute coronary syndromes. Presented at the American Heart Association Scientific Session. Chicago, United States of America; November 15–19, 2014.
Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–57.
Akdim F, Stroes ES, Sijbrands EJ, et al. Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy. J Am Coll Cardiol. 2010;55:1611–8.
Akdim F, Visser ME, Tribble DL, et al. Effect of mipomersen, an apolipoprotein B synthesis inhibitor, on low-density lipoprotein cholesterol in patients with familial hypercholesterolemia. Am J Cardiol. 2010;105:1413–9.
Kastelein JJ, Wedel MK, Baker BF, et al. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114:1729–35.
McGowan MP, Tardif JC, Ceska R, et al. Randomized, placebo-controlled trial of mipomersen in patients with severe hypercholesterolemia receiving maximally tolerated lipid-lowering therapy. PLoS One. 2012;7:e49006.
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006.
Stein EA, Dufour R, Gagne C, et al. Apolipoprotein B synthesis inhibition with mipomersen in heterozygous familial hypercholesterolemia: results of a randomized, double-blind, placebo-controlled trial to assess efficacy and safety as add-on therapy in patients with coronary artery disease. Circulation. 2012;126:2283–92.
Yu RZ, Geary RS, Flaim JD, et al. Lack of pharmacokinetic interaction of mipomersen sodium (ISIS 301012), a 2′-O-methoxyethyl modified antisense oligonucleotide targeting apolipoprotein B-100 messenger RNA, with simvastatin and ezetimibe. Clin Pharmacokinet. 2009;48:39–50.
Sahebkar A, Watts GF. New LDL-cholesterol lowering therapies: pharmacology, clinical trials, and relevance to acute coronary syndromes. Clin Ther. 2013;35:1082–98.
Toth PP. Emerging LDL, therapies: mipomersen-antisense oligonucleotide therapy in the management of hypercholesterolemia. J Clin Lipidol. 2013;7:S6–10.
Visser ME, Wagener G, Baker BF, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2012;33:1142–9.
Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129:1022–32. This review describes 2 drugs that reduce LDL-C independently of LDLR, and thus effective in familial hypercholesterolemic patients who are at higher cardiovascular risk.
Samaha FF, McKenney J, Bloedon LT, et al. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2008;5:497–505.
Cuchel M, Bloedon LT, Szapary PO, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–56.
Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6.
Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis. 2013;228:18–28.
Careskey HE, Davis RA, Alborn WE, et al. Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res. 2008;49:394–8.
Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–9.
Abifadel M, Elbitar S, el Khoury P, et al. Living the PCSK9 adventure: from the identification of a new gene in familial hypercholesterolemia towards a potential new class of anticholesterol drugs. Curr Atheroscler Rep. 2014;16:439.
Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA. 2012;308:2497–506.
Stroes E, Colquhoun D, Sullivan D, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–8. This clinical trial identified evolocumab as a promising therapy for statin-intolerant patients at high cardiovascular risk.
Li Y, Jiang L, Jia Z, et al. A meta-analysis of red yeast rice: an effective and relatively safe alternative approach for dyslipidemia. PLoS One. 2014;9:e98611.
Venero CV, Venero JV, Wortham DC, et al. Lipid-lowering efficacy of red yeast rice in a population intolerant to statins. Am J Cardiol. 2010;105:664–6.
Becker DJ, Gordon RY, Halbert SC, et al. Red yeast rice for dyslipidemia in statin-intolerant patients: a randomized trial. Ann Intern Med. 2009;150:830–9.
Halbert SC, French B, Gordon RY, et al. Tolerability of red yeast rice (2,400 mg twice daily) versus pravastatin (20 mg twice daily) in patients with previous statin intolerance. Am J Cardiol. 2010;105:198–204.
Marazzi G, Cacciotti L, Pelliccia F, et al. Long-term effects of nutraceuticals (berberine, red yeast rice, policosanol) in elderly hypercholesterolemic patients. Adv Ther. 2011;28:1105–13.
Kong W, Wei J, Abidi P, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10:1344–51.
Cameron J, Ranheim T, Kulseth MA, et al. Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis. 2008;201:266–73.
Dong H, Zhao Y, Zhao L, et al. The effects of berberine on blood lipids: a systemic review and meta-analysis of randomized controlled trials. Planta Med. 2013;79:437–46.
Pisciotta L, Bellocchio A, Bertolini S. Nutraceutical pill containing berberine versus ezetimibe on plasma lipid pattern in hypercholesterolemic subjects and its additive effect in patients with familial hypercholesterolemia on stable cholesterol-lowering treatment. Lipids Health Dis. 2012;11:123.
Compliance with Ethics Guidelines
Conflict of Interest
Angela Pirillo has no conflicts of interest.
Alberico Luigi Catapano is on the advisory board or a member of the speaker bureau for AstraZeneca, Amgen, Aegerion, Eli-Lilly, Genzyme, Mediolanum, Merck-MSD, Pfizer, Recordati, Rottapharm, Sanofi, and Sigma-Tau.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is part of the Topical Collection on Lipid Abnormalities and Cardiovascular Prevention
Rights and permissions
About this article

Cite this article
Pirillo, A., Catapano, A.L. Statin Intolerance: Diagnosis and Remedies. Curr Cardiol Rep 17, 27 (2015). https://doi.org/10.1007/s11886-015-0582-z
Published:
DOI: https://doi.org/10.1007/s11886-015-0582-z