
Abstract
Statins are currently the primary treatment for hyperlipidemia, particularly for the treatment of high levels of low-density lipoprotein cholesterol (LDL-C), as many studies have proven benefit in a variety of populations. The benefits of statin treatment for high cholesterol have been proven in many trials. Forefront among different adverse events is statin-induced myopathy, which still eludes complete understanding, and may range anywhere from muscle soreness or fatigue to potentially extremely rare occurrence of rhabdomyolysis.
As most adverse events are rare and not life-threatening, in high-risk patients, a high-dose statin should be started initially as data suggests that clinicians rarely up titrate statin therapy after initial prescription leading to under-treatment of many patients requiring high-dose statin therapy. As we will discuss in this paper, musculoskeletal side effects are the main concern and reason for discontinuing statin therapy. The occurrence and true association of other adverse events in patients on statin such as new onset of diabetes, hepatic toxicity, or cognitive impairment are rare, controversial, and not proven. In placebo-controlled studies, abnormal liver function occurs to a similar degree in statin- and placebo-treated patients. This led to FDA removal of the requirement to monitor liver function tests in patients on statin therapy.
The combination of statins with other compounds such as ezetimibe or PCSK9 inhibitors has shown some additional benefits in the treatment of hypercholesterolemia. The goal of this manuscript is to conduct a comprehensive review about most commonly used statins and compare data on their history, structures, benefits, adverse effects, and clinical outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiovascular disease (CVD) is a major global health problem caused by a very complex process involving numerous risk factors. Inflammation along the walls of the vascular system plays a major role in the development of atherosclerosis [1]. Over time, atherosclerotic lesions composed of fats and cholesterol accumulate along vascular walls, eventually leading to myocardial infarction, stroke, and peripheral artery disease. In 2015, 39.5 million (70%) of 56.4 million deaths worldwide were due to noncommunicable diseases (NCDs) and CVD was the leading cause of death due to NCDs, with 17.7 million deaths attributed to it (45% of all NCD deaths). Not only is CVD the deadliest disease, it is also costly; the American Heart Association (AHA) estimates that CVD costs the American economy 329.7 billion dollars in direct health-related expenses and lost productivity between 2013 and 2014 [2]. This amount is projected to rise to 1.1 trillion dollars by 2035.
A key factor in determining one’s likelihood to develop CVD is high cholesterol levels. A risk calculator is available on the AHA website where one can get an estimate of their risk of CVD. The AHA has identified 4 groups at risk of atherosclerotic cardiovascular disease (ASCVD) that benefit from statin therapy in lowering LDL-C concentration. These include
Secondary prevention in patients previously diagnosed with ASCVD
Primary prevention for patients with [LDL-C] ≥ 190 mg/dL
Primary prevention for diabetic individuals (age 40 to 75) who have 189 mg/dL ≥ [LDL-C] ≥ 70 mg/dL
Primary prevention for non-diabetics (age 40 to 75) with an estimated 10-year ASCVD risk ≥ 7.5% who have 189 mg/dL ≥ [LDL-C] ≥ 70 mg/dL [3].
Recently, the guideline has suggested discussion about risk and benefit of treating hyperlipidemia in patient with ASCVD risk of more than 7.5 % if they are not at high risk for cardiac event before starting treatment.Total cholesterol encompasses the sum of both high- and low-density lipoprotein concentrations (HDL-C and LDL-C respectively), as well as 20% of one’s triglyceride levels. Of these, HDL-C is utilized to reverse cholesterol transfer from the arterial wall, which is then transported to the liver for clearance; this purpose leads to the widespread colloquialism, “good cholesterol.” [4] On the other hand, low-density lipoprotein-cholesterol (LDL-C), or “bad cholesterol,” shepherds cholesterol around the body and high levels of LDL-C are associated with increased risk of CVD.
Much attention has been placed on changing one’s cholesterol levels so that they fall more favorably, that is increasing one’s HDL-C levels or lowering LDL-C levels. Methods to effect this change can be as simple as altering one’s day-to-day habits. Positive habitual changes include adhering to a healthier diet, increasing physical activity, ceasing smoking, and regulating one’s weight. These lifestyle changes have been shown to decrease LDL-C levels by up to 30%. However, for some individuals, these changes might not be sufficient to bring their levels to healthy standards. However, many trials using HDL raising drugs have failed to show any benefit. Therefore, the focus on pharmacological treatment of hyperlipidemia has been the reduction of LDL-C and triglyceride levels.
The purpose of this paper is to examine statins, a pharmaceutical group used to combat high LDL-C levels and thus CVD. A background on statins including their history, biochemical mechanism, efficacy, and adverse effects will be given.
Statins
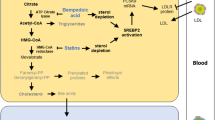
Statins, or 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, are a class of drugs that act by competitively inhibiting the HMG-CoA reductase enzyme (Fig. 1). HMG-CoA reductase (HMGR) catalyzes the synthesis of mevalonic acid from NADPH and HMG-CoA. This reaction is the committed step in the hepatic biosynthesis of cholesterol, a synthetic pathway that involves more than 27 reactions [5]. By inhibiting this step, production of cholesterol is diminished. This inhibition is achieved through an HMG-CoA—like moiety present in all statins. The intended mode of action is the inhibition of HMGR; however, statins have also been shown to retard the formation and accelerate removal of atherosclerotic plaque in the arteries [6].

3-Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA), the native substrate for HMGR
Type I Statins
Structure
The first statin, mevastatin, was identified in two species of fungi by independent groups of British and Japanese scientists in 1976. Mevastatin, pravastatin, simvastatin, and the first commercially prescribed statin, lovastatin (FDA approved in 1987), all share a bicyclic decalin-like motif (Fig. 2) [7]. Because of this shared structure, this subgroup of statins has been classified as type I statins.

Type I statins—mevastatin, pravastatin, simvastatin, and lovastatin
Type II Statins
Structure
While type I statins are all centered on a decalin motif, type II statins are not [8]. Instead, type II statins have their HMG-CoA motif bound to various heterocyclic aromatic ring systems. At the molecular level, the isopropyl group of type II statins takes part in hydrophobic interactions with HMGR similar to those of the decalin motif of type I statins. Furthermore, all type II statins share a para-fluorophenyl substituent relative to the aromatic center. Examples of type II statins include fluvastatin, cerivastatin, atorvastatin, and rosuvastatin (Fig. 3). Elucidation of various HMGR-statin structures has shown that rosuvastatin carries the largest number of enzyme-inhibitor bonds.

Several type II statins—fluvastatin, atorvastatin, cerivastatin, and rosuvastatin
Mode of Action
Kinetic studies and crystal structures of enzyme-statin complexes have shown that statins bind to the active site of HMGR, the enzyme which converts HMG-CoA to mevalonate, a precursor of cholesterol (Fig. 4) [6]. Initially, there was some confusion over how the larger substituents of statins, relative to the smaller pantothenic moiety of HMG-CoA, could fit in the pocket normally occupied by the substrate. However, the 28 amino acids of the carboxy-terminus that compose this pocket have been shown to be prone to disorder and mobility in human HMGR as well as bacterial homologs [9]. This mobility allows these 28 amino acids to adopt a conformation that permits inhabitation by the bulkier substituents of statins. By binding to the active site of HMGR, statins inhibit synthesis of mevalonic acid, a key step of de vivo synthesis of cholesterol. This action lowers the concentration of intracellular cholesterol. A lack of intracellular cholesterol stimulates the release of sterol regulating element-binding proteins (SREBP), which relocate to the hepatic nucleus. Once there, the SREBP serve to increase genetic expression of the LDL receptor, leading to increased receptor concentrations on the extracellular membrane. LDL receptors then remove LDL-C from the plasma, leading to decreased LDL-C concentrations in the circulatory system [5].

Compactin (shown in purple) HMGR crystal structure [7]
Clinical Effects of Statins
Statins as a primary or secondary method of prevention for cardiovascular disease are well established. With moderate statin therapy, LDL-C levels are expected to decrease between 30 and 45%. If more aggressive reductions are necessary, high-intensity therapy can generally decrease LDL-C levels by over 50% [10]. Meta-analysis for randomized clinical trials (RCTs) has shown that for every 1 mM reduction of LDL-C using standard statin treatments, the 5-year risk of CVD was decreased by 20% [11]. Because of this efficacy in reducing LDL-C levels, and thus the risk of CVD, statins have emerged as the primary method to treat high cholesterol. Statins have also been shown to have a positive effect on diminishing atherosclerotic plaque, as well as numerous other pleiotropic effects [12]. A study demonstrated that plaque reduction, visualized using fluorodeoxyglucose-positron emission tomography/imaging, was dose-dependent; patients on an 80-mg atorvastatin regimen exhibited more pronounced reductions than those on a 10-mg regimen [13]. Initial trials with the original statin, mevastatin, showed that doses between 15 and 60 mg/day reduced serum LDL-C by 20–40% [6]. In subsequent trials for type I statins, this reduction was further increased. In a 5-year study of over 700 patients, lovastatin decreased LDL-C levels by 44% [14]. In the PROSPER trial, 5804 patients were given either 40-mg doses of pravastatin or placebo. Those in the statin group saw a 34% reduction in LDL-C levels as well as reduced risk of cardiovascular events [15]. For some patients, this percentage of reduction might be satisfactory. For others, a more intensive statin regimen resulting in greater reduction in LDL-C levels might be needed. Rosuvastatin and atorvastatin, prime examples of type 2 statins, carry greater dose reductions and will be explored in the following section. In a recently updated meta-analysis of atorvastatin, 296 studies of almost 40,000 patients were analyzed to determine the dose-related efficacy of atorvastatin. Common doses of atorvastatin ranged from 10 to 80 mg/day, with a corresponding LDL-C reduction of 37.1 to 51.7% [16]. The CURVES study showed the efficacy of atorvastatin; 10, 20, or 40 mg doses of atorvastatin yielded greater reductions than a corresponding dosage of simvastatin, pravastatin, lovastatin, or fluvastatin [17]. Rosuvastatin, FDA approved in 2010, is another successful synthetic statin in improving patients’ LDL-C levels and has the lowest inhibition constant, Ki = 50 nM [7, 18]. Meta-analysis of 108 trials showed that rosuvastatin was able to achieve the same decrease in LDL-C as a dosage of atorvastatin three times greater [19]. Furthermore, 10–40 mg/day of rosuvastatin was shown to lower LDL-C by 48–55%, a significant increase relative to mevastatin. In the 6-week STELLAR trial, rosuvastatin was shown to have a greater impact on lowering LDL-C, compared to atorvastatin (8% more clearance), pravastatin (26%), and simvastatin (12–18%).
Pleiotropic effects are effects, both positive and negative, that occur outside a drug’s intended mode of action (Fig. 5). For statins, there are abundant suggested positive pleiotropic effects that serve to reinforce cardiovascular health, many of which are related to a reduction in protein isoprenylation that ultimately inhibits small GTP-binding proteins involved in transducing extracellular stimuli to various intracellular pathways [20]. These include increasing the bioavailability of nitric oxide and decreasing CRP concentrations. A report by Kavalipati shows statins’ efficacy in decreasing inflammatory cells in atherosclerotic plaques and increasing the stability of plaque through their combined reduction of lipids, macrophages, and MMPs (matrix metalloproteinase’s). Statins have also been reported to limit the expression of monocyte chemoattractant protein-1. This action helps reduce the interaction between monocytes and the vascular walls [21]. These trends are associated with better cardiovascular health. In the JUPITER trial, it was shown that statin therapy was also beneficial in those with “normal” LDL-C levels (< 130 mg/dL) who also had elevated high-sensitivity C-reactive protein (hsCRP) levels, a key bio-inflammatory marker. Risk of cardiovascular events in the JUPITER trial’s population was cut in half, attributed to lower hsCRP levels, which were lowered in an LDL-C-independent manner [22]. However, pleiotropic effects have not been clinically proven to be of benefit beyond LDL reduction.

Summary of pleiotropic effects due to statins
Adverse Events of Statins
Statins and Musculoskeletal Side Effects
Statin-Induced Myopathy/Musculoskeletal Pain
Due to statins being the primary method of combating hypercholesterolemia, adverse effects from statin treatment have been well reported. The mevalonate synthetic step which statins inhibit is utilized in the synthesis of other products, such as coenzyme Q10 and Heme A, both of which are important in physiology and cell biology [4]. This undesired inhibition, along with the fact that cholesterol itself has numerous beneficial uses in the body, highlights some of the dilemmas associated with inhibiting HMGR.
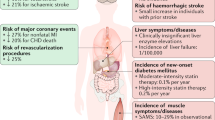
The main adverse effects associated with statins can be recalled with the mnemonic “5Ms” which include metabolism, muscle, medication interactions, major organ effects, and memory [23]. Of these, memory and major organ effects (specifically the liver) remain controversial. A meta-analysis of almost 50,000 patients in 13 trials showed no clinically significant increase in liver function test abnormalities when compared to placebo, suggesting that low to moderate dosages of statins do not present significant risk to the liver [24]. Cognitive impairment was evaluated in the RCT PROSPER trial, in which there was no significant difference in cognition for the 5804 elderly patients between the placebo and control groups [25]. The most common and well-documented adverse side effects are myopathic (muscle pain) in nature, including myalgia, myositis, and less frequently rhabdomyolysis. In the PRIMO study, 10.5% of the 7924 patients who underwent statin therapy reported muscle-related problems [26]. However, muscular adverse effects have been shown to be both statin and dose-dependent [19]. In the above-mentioned PRIMO study, those on fluvastatin had the lowest incidence (5.1%) of muscular adverse effects but are one of the weakest statins.
Vitamin D deficiency has been independently linked to muscle weakness and severe myopathy, and there is a hypothesis that inadequate vitamin D levels may worsen statin-induced myopathy. There is a school of thought that with vitamin D deficiency, CYP3A4 gets shunted into the vitamin D hydroxylation pathway, decreasing the amount of this metabolite available for statin metabolism. This is in turn suspected to increase statin-induced toxicity [27, 28]. A clinical study by Ahmed investigated whether there was an association between low vitamin D levels and myalgia in patients on statin therapy and whether the myalgia was reversible with vitamin D supplementation while keeping the patients on their statin therapy. They treated vitamin D deficient patients with 50,000 units of ergocalciferol per week for 12 weeks. Findings showed lower serum vitamin D levels in patients with statin-induced myalgia than in statin-treated patients without myalgia. Also, supplemental vitamin D therapy resolved myalgia in 92% of statin-taking patients who had myalgia and were deficient in vitamin D [27]. In a similar study to understand the effect of replenishing vitamin D levels on statin-induced myopathy, a group of pharmacists conducted a single-institution, retrospective cohort study of veteran patients in the Lipid Clinic. Their main aim was to determine whether patients who had previously been intolerable to statins due to myopathy would maintain their statin therapy without complaint following vitamin D supplementation. Though working with a sample size of only 27, 100% of the patients were able to continue their statin therapy following vitamin D supplementation, with the most frequently restated statins being atorvastatin (n = 15), pravastatin (n = 6), and rosuvastatin (n = 3). Despite the above findings, the vitamin D deficiency and statin-induced myopathy relationship remains a controversy requiring further investigation. While no potential association has been demonstrated by some retrospective reviews, prospective, cross-sectional, and case studies have shown a correlation between statin-induced myopathy and vitamin D deficiency. Studies that found a correlation suggest that each statin may have a different effect on vitamin D concentrations, with more lipophilic statins increasing vitamin D metabolites, while less lipophilic statins provide no improvement in vitamin D [24, 29, 30].
Myopathy has been shown to occur at a greater frequency with more intensive statins, but variations between each statins’ chemical structure have allowed some patients experiencing myopathy to switch statins without continued pain [31]. However, in extreme cases, myopathy can take the form of rhabdomyolysis, in which patients have creatine kinase (CK) levels > 10,000 IU/L [32]. About 1 in 10,000 statin-treated patients develop substantial elevations in CK levels and about 2–3 per 100,000 patients actually develop rhabdomyolysis with extremely high CK levels. Statins can cause either self-limited myotoxicity through their direct effects on the muscles, or autoimmune myopathy by triggering the body’s own antibodies to target HMGCR. Direct myotoxicity is rare with an incidence of about 10–20 cases in every 10,000 patients treated with statins each year. This condition is known to self-resolve when the patient is taken off statin treatment. Additionally, reports show that 2–3 per 100,000 statin-treated patients per year may develop autoimmune myopathy. Unlike direct myotoxicity, autoimmune toxicity does not reverse after statin therapy has been stopped; thus, immunosuppressive therapy is required to treat the condition.
Though the mechanism underlying statin myotoxicity is not well understood, some research suggests that statins cause muscle damage by inhibiting complex III and decreasing the production of ubiquinone, a protein that stabilizes the cell membrane and plays a role in muscle cell energy production through its effects on the mitochondrial respiratory chain. The latter triggers the release of pro-apoptotic proteins like cytochrome c and Smac/DIABLO which together with increased amounts of free radicals, cytosolic calcium, and inhibition of the prenylation of cell-signaling proteins like the Ras superfamily induce apoptosis (Fig. 6). Ubiquinone also increases muscle fiber sterol levels, which could increase the toxic effect of statins in the muscles or cause an overexpression of artrogen-1, a key gene involved in skeletal muscle atrophy [33, 34].

Statins inhibit complex III and the synthesis of ubiquinone. This interrupts the electron transport chain, preventing the oxidation of NADH to NAD+ (which is essential beta-oxidation of fatty acids) as well as reduce the potential of the mitochondrial membrane. This causes the release of pro-apoptotic proteins like cytochrome c and Smac/DIABLO which together with increased content of free radicals, cytosolic calcium and inhibition of prenylation of cell-signaling proteins like Ras superfamily induces apoptosis [34]
There is some data suggesting a low vitamin D level can contribute to statin-induced myalgia and correction of vitamin D levels can eliminate statin-induced myalgia in most patients. However, this still remains controversial.
In patients complaining of musculoskeletal symptoms on statin, permanent discontinuation of statin is a large mistake that occurs often. By modifying the dose or frequency or changing from one statin to another, most patients will not suffer from this adverse event. Checking CPK levels generally proves to be of little use, as many patients with myalgia have normal CPK; only in extreme cases, in which there is very high elevation of CPK levels be helpful for diagnosis. In patients with myalgia, a break for few weeks should be given, after which statin treatment should be resumed at lower dose or using a different statin. In rare cases where daily statin cannot be tolerated, it can be given every other day, twice, or even once a week. Rosuvastatin and atorvastatin have the longest half-lives and are therefore most suitable for biweekly statin therapy. Many musculoskeletal complaints are not related to statins and this should be discussed and evaluated individually. As is mentioned above, checking vitamin D levels and correcting it in patients with a deficiency may improve statin tolerance. If, despite vitamin D level correction, myalgia persists, statin therapy intensity should be reduced, or a different statin should be tried. If this remains unsuccessful, then statin frequency should be reduced to every other day or twice a week, which usually eliminates myopathy in majority of patients. This approach is preferable to stopping statin treatment completely.
Statin Use and Rhabdomyolysis
Rhabdomyolysis is the pathological syndrome in which muscles degenerate, and the circulatory system is inundated with various intracellular originating species such as myoglobin [35]. It is diagnosed by elevated creatine phosphokinase (CPK) levels, and despite the lack of an agreed-upon standard, clinicians often use five times the upper limit of normal CPK levels (~ 1000 U/I) for diagnosis. Excess myoglobin in the circulatory system leads to failure of the glomerulus, leading to kidney and possibly liver failure and correspondingly higher CK concentrations [30]. Although cases of statin-linked rhabdomyolysis have been observed, it is rather rare, occurring in less than 0.1% of statin users, and does not comprise the entirety of muscle-related complaints of patients on statins; less serious, non-rhabdomyolysis, muscle-related complaints have been shown to occur with greater frequency. Cerivastatin was discontinued due to an unacceptably high number of deaths directly attributed to rhabdomyolysis. However, this potentially fatal adverse event was quite rare and was reported to occur in less than 2 prescriptions per every 10 million prescriptions [36]. As mentioned above, ubiquinone, farnesyl, and other metabolites of mevalonate have been attributed to myopathy, but the exact cause of myopathic syndromes is currently unknown [37].
It is important to inform the patient to seek immediate medical attention in the case of severe myalgia together with the presence of dark urine as a sign of rhabdomyolysis. It is usually more occurring in patients on high-dose statin particularly in patients on high-dose simvastatin in the early phase of initiation of this medication. If the patient tolerates 80 mg of simvastatin for more than a year, this risk will be dramatically lower than in patients on this drug in less than 12 months. This is the main reason that the FDA does not recommend up titrating simvastatin above the 40-mg daily dose.
Statins and Myositis
Recent research suggests a link between statin use and the development of idiopathic inflammatory myositis (IIM), a group of rare, clinically heterogeneous autoimmune muscular disorders. Literature between the mid-1990s and early 2000s shows about 10 case reports of polymyositis or dermatomyositis in patients treated with statins. The incidence of IIM is estimated to be between 0.1 and 1.0 persons per 100,000 per year. Research shows that 1 in 18 patients with IIM between 2000 and 2004 were exposed to statins, and this number increased to 21 of 43 people between 2012 and 2014. Unlike other statin-induced musculoskeletal effects, myositis is irreversible, even if statin treatment is discontinued, and can result in permanent disability or death. However, aggressive treatment with steroids can help control myositis [38].
Lastly, statin therapy increases SREBP-2, a transcription factor, which in turn increases both LDL-R and PCSK9 levels [39]. Increasing concentrations of PCSK9 are correlated with increased levels of circulatory LDL-C. However, PCSK9 is affected in a greater fashion than LDL-R, and thus this unintended activity of statins promotes higher levels of LDL-C. In the JUPITER trial, participants on a 20-mg regimen of rosuvastatin had LDL-C levels 50% lower and PCSK9 levels 30% higher than those of the control [40]. This particular pleiotropic effect from statins, dubbed the “statin paradox,” shows one way in which a dual therapy of both statins and PCSK9 inhibitors might benefit the patient in a greater fashion than either therapy alone.
Statins and Neurocognition
The neurocognitive effect of statins is a potential issue that can be added onto the constant risk and reward analysis of these important drugs. However, unlike the more established effects of statins on measures such as LDL-C, onset of diabetes, and myalgia, their effects on cognitive functioning are still being disputed. In 2018, a systematic review highlighted the difficulty of understanding this topic [41]. In the past 15 years, many studies attempting to connect statins and cognition have been observational in nature and very few have even attempted to go through rigorous analysis using randomized controlled trials. For example, the authors cited only two large, double-blind, placebo-controlled RCTs of statins that examined cognition, namely the PROSPER and HEART studies [42]. These studies were far from conclusive on the issue. Therefore, firm conclusions remain elusive as to whether short-term or long-term statin regimens affect cognition. However, there is increasing concern about how statins may be a causative factor for cognitive problems [43]. It seems from available research that adverse effects appear to be a rare occurrence in certain vulnerable patient groups.
To add to the debate, there are studies that find a neuroprotective effect of statins [44, 45]. For example, they examined how dysregulation of cholesterol homeostasis is a major contributor to Alzheimer’s disease. They argue that statins help prevent accumulation of beta-amyloid (Aβ) peptide and tau hyperphosphorylation by preventing the propagation of excess free cholesterol that is converted to cholesteryl esters by the enzyme ACAT1. Essentially, they claim that, similar to reducing LDL-C levels, statins reduce the hallmark biomarkers of Alzheimer’s through the same HMG-CoA pathway. A systematic meta-analysis corroborated these findings by looking at 25 clinical studies and assessed how statins might contribute to cognitive decline in adults [46]. Using “random-effects meta-analyses calculating relative risks,” they determined that statins had an association with reducing Alzheimer’s disease and mild cognitive impairment. However, it must be noted that they did not find a protective effect of statins on vascular dementia. This type of dementia is caused by atherosclerosis of arteries leading to the brain. This is a contradictory finding considering the robust effects of statins in lowering LDL-C found throughout the literature. As we know, cholesterol is important for many functions involved in memory such as myelin sheath formation, neurotransmitter propagation, receptor and synapse creation, and steroid hormones. Therefore, more research on the disconnect between statins and vascular dementia in particular is needed. In support of the neuroprotective effect of statins, they cite a positive effect of statins that ultimately weakens beta-amyloid formation. Additionally, they cite a decrease in inflammatory cytokines in the brain such as IL-1 beta, IL-6, and tumor necrosis factors in the hippocampus.
In contrast to the claim of the neuroprotective effects of statins, there is research suggesting the potential short-term detrimental effect statins have on cognitive performance [47]. This research originates from the FDA’s changes to statin drugs’ safety labels in 2012, which warns of “non-serious and reversible cognitive side effects” from statin use. The essential argument of these studies centers on the lipophilicity of statins, which allows them easier passage through the blood-brain barrier, and how this creates a concentration gradient that lowers cholesterol below that required for normal cognitive functioning in the central nervous system [48]. As stated earlier, cholesterol is important for building the physical structures of learning and memory (i.e., myelin sheaths), and lower cholesterol in the central nervous system is not the same as lower LDL-C levels in the peripheral tissues. Statins such as atorvastatin and simvastatin are said to have the highest cognitive impairment reports, which may be due to this mechanism. Additionally, in patients with mitochondrial conditions like metabolic syndrome and thyroid disease, statins are argued to be risk factors for exacerbating these conditions [49]. Mitochondria are primarily important in aerobic respiration and muscle function, but also have a role in the functioning of the brain. Nevertheless, more research is needed to delineate the detrimental and beneficial effects of statins on short-term and long-term cognitive functioning.
It is also worth mentioning that in some of the studies that associate statin use with various health-related problems including cognitive impairment, participants were also on non-statin lipid-lowering drugs (LLDs). A study by Strom et al. compared the effect of statin versus non-statin LLDs on memory and reported no significant difference between these drugs. After their observation that all LLDs, regardless of the class, were associated with memory loss, they concluded that singling out statins as being responsible for this defect was most likely due to selection bias. They also reported an increased risk of detection bias in patients on statins who visited their physicians more frequently [38].
Statins and Diabetes
Statin effect on inducing diabetes is controversial and is not consistent. Recent research on the effect of statins on incident diabetes has provided more insight into this potential adverse effect [50]. Across 18 studies from 1994 to 2010, there was an average reduction of 0.89 mmol/L in LDL-C associated with statin use. However, an average hazard ratio of 1.102 was found for the development of incident diabetes in these studies, meaning that at any given time, patients taking statins have approximately a 10% greater chance of developing diabetes than their control counterparts. Simvastatin, pravastatin, lovastatin, fluvastatin, and atorvastatin were the main statins observed in these studies. Based on various meta-analyses and clinical trials, the authors determined that the risk of developing new-onset diabetes from statins was 0.1% annually. In comparison, the reduction of CVD events was approximately 0.42% annually [51]. They concluded that although the risk of developing diabetes increased with statin use, the benefit of reduced major coronary events outweighed this potential adverse effect.
This is corroborated by a retrospective cohort study done in the UK examining 12,725 insulin initiators with type 2 diabetes using a primary care database called “The Health Improvement Network” (THIN) [52]. At 6-month intervals for 3 years, they compared the HbA1c levels of previous statin users with those of non-users who were beginning insulin treatment. In the first 6 months, previous statin users had a 0.26% reduction in HbA1c versus non-users with a 0.34% HbA1c reduction. A similar and significant difference between the cohort groups’ HbA1c levels was also seen at 12 months with statin users displaying a 0.29% reduction versus a 0.37% reduction in non-users. Overall, previous statin users maintained a higher HbA1c throughout the 3 years. Essentially, this study claimed that the previous use of statins for those receiving insulin treatment for type 2 diabetes decreased their insulin sensitivity, rendering such treatment less effective in lowering HbA1c levels.
The mechanism explaining how this occurs involves statins blocking the synthesis of cholesterol in the HMG-CoA pathway, with a downstream impairment effect on the pancreas through calcium channels and ultimately decreasing insulin sensitivity. Isolated single-islet cultures have been attained in vitro from the pancreas using a microfluidic technique that mimics the “glucose-stimulated insulin secretion pathway” in β cells [53]. In these single-islet cells, they found that 1 μl of statin was all that was needed to impair insulin response even at a relatively high glucose concentration of 11 mM. It was found that statins such as simvastatin decreased the secretion of insulin in response to glucose stimuli at the cellular level. Additionally, a previous study using mouse pancreatic MIN6 β cells found that simvastatin, but not pravastatin, reduced insulin secretion by 59 to 79% at 5.5 mmol/L and 16.7 mmol/L [54]. Another possible mechanism suggests that statin-induced NLRP3 inflammasome activation contributes to insulin resistance (Fig. 7). In the first step known as priming, transcriptional events induced by NF-κB following PRR stimulation increase levels of inflammasomes like NLRP3 and inflammasome effectors like pro-IL-1beta. This leads to immune activation where HMGCR inhibition with statins decreases protein prenylation, causing pleiotropic effects. Decreased protein prenylation is suspected to trigger signals that promote NLRP3 inflammasome activity. At this point, statins come in with a variety of effects including promotion of intracellular ATP release. While out of the cell, ATP promotes potassium efflux, a key trigger for increased NLRP3 inflammasome activity. This activity causes cleavage of pro-IL-1beta into active IL-1beta by caspase-1, promoting metabolic modulation that inhibits downstream signaling through a suspected number of pathways [55].

a) Priming: PRR stimulation causes NF-kB to stimulate transcriptional events that increase inflammasome and inflammasome effectors like NLRP3 and pro-IL-1beta respectively. b) Immune activation: Statins inhibit HMGCR, causing pleiotropic effects through decreased protein prenylation (suspected cause for signals that promote increased NLRP3 inflammasome activity, but mechanism is unknown). This inflammasome activation by statins is known to lead to mitochondrial membrane dysfunction, increase intracellular reactive oxygen species (ROS) and promote release of cellular ATP. Extracellular ATP can bind P2X7 receptor and promote efflux of K+, increasing NLRP3 inflammasome activity. This activation causes caspace-1 to cleave pro-IL-1beta into active IL-1beta. c) Metabolic modulation: IL-1beta-mediated inflammation and activation of MAPKs inhibit insulin signaling either at receptor substrate-1 level (IRSI), or through an unknown target of caspace-1 which may alter insulin signaling at the level of PTEN (phosphatase and tensin homolog) or another site such as AKT phosphorylation [55]
Generally, these studies and reviews have come to the consensus that statins’ role in inducing or exacerbating type 2 diabetes remains controversial and unproven. Various mechanisms have been proposed including a direct or indirect effect on calcium channels of pancreatic β cells, translocation of GLUT4 transporter, and decreased downstream production of coenzyme Q10 that impairs intracellular signaling [46, 56]. However, more random-controlled trials and basic research are needed to elucidate the mechanisms of this relatively understudied effect of statin use. Although current evidence points to a moderate statin-induced diabetogenic effect, the efficacy of statins in reducing LDL-C and preventing adverse cardiovascular events, at this time, outweighs the risk of new-onset diabetes. Pravastatin has never showed to have any diabetic effects, but it is one of the weak statins and is not recommended in patients with established cardiovascular disease who require high-intensity statin therapy.
Based on the above studies and reviews, statins’ role in inducing or exacerbating type 2 diabetes remains controversial. If it is true, various mechanisms have been proposed including a direct or indirect effect on calcium channels of pancreatic β cells, translocation of GLUT4 transporter, and decreased downstream production of coenzyme Q10 that impairs intracellular signaling [46, 57]. However, more random-controlled trials and basic research are needed to elucidate the mechanisms of this relatively understudied effect of statin use. Although current evidence points to a moderate statin-induced diabetogenic effect, the efficacy of statins in reducing LDL-C and preventing adverse cardiovascular events, at this time, outweighs the risk of new-onset diabetes.
Statins and Hepatoxicity
In vitro models have shown a dose- and time-dependent impairment of mitochondrial function by statins. Mitochondrial toxicity assay after statin treatment has revealed significant increase in mitochondrial superoxide, one of the important cytotoxic and signaling mediators in mitochondrial/liver damage. Other findings report statin use could lead to apoptosis. Statins could also inhibit the respiratory chain complexes I and II, trigger the release of excess calcium ions, or cause mitochondrial membrane depolarization in the liver. The incidence of statin-induced liver disease if any, increases when statins are used at maximum doses together with other lipid-lowering drugs like fibrates, other hepatotoxic drugs or drugs with similar enzymatic pathways, or used by elderly subjects and subjects with considerable liver or kidney dysfunction. Experiments using higher doses of lovastatin in rabbits and simvastatin in guinea pigs resulted in liver cell necrosis. A study to investigate statin associated liver injury revealed that for patients on statins the latency period to the onset of liver injury ranged from 34 days to 10 years [58, 59].
In addition, factors such as concomitant diseases, concomitant drug use, individual immunity, lifestyle and genetics have a role in liver health. Drug-induced liver injury in itself is relatively rare and requires the presence of liver damage biomarkers such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma glutamyl transferase (GGT), serum total bilirubin, and alkaline phosphatase (ALP) [60]. In this context, active liver monitoring in patients asymptomatic of hepatoxicity is not recommended. Looking at liver enzyme elevations in isolation could be misleading to the actual extent of damage. Despite elevated liver enzyme levels, adverse liver effects have a low risk of occurring. However, action should be taken if symptoms of hepatotoxicity manifest, such as fatigue, weakness, loss of appetite, yellowing of skin or sclera of the eyes, and dark-colored urine [61].
In a meta-analysis of 135 RCT looking at 246,000 patients taking atorvastatin, lovastatin, and simvastatin, 50% of these patients showed a higher risk of transaminase elevation compared to placebo [50]. However, the caveat is that these elevations were minor did not necessarily lead to serious adverse liver effects and usually stabilized over time. It must be noted that there is a dose-response relationship to transaminase levels and higher doses lead to a higher risk of transaminase elevation. Additionally, a study in 2016 examining the UK General Practice Research Database, with patient data from 1997 to 2006, had similar findings in regard to liver effects [62]. Their criteria for drug-induced liver damage was defined as transaminase elevation > 5× ULN and/or ALP > 2× ULN, serum bilirubin > 60 μmol/L, AST or ALT > 200 U/L, and alkaline phosphatase > 1200 U/L. This study found that 71/164,407 patients taking simvastatin and 101/76,411 patients taking atorvastatin developed moderate or severe hepatotoxicity.
In conclusion, statin-induced hepatic abnormalities have not been proven and have been the same between placebo and statin-treated patients. This is the main reason behind the removal of liver enzyme monitoring by the FDA in statin-treated patients. When liver dysfunction exists, or a statin is combined with other hepatotoxic drugs, then liver enzyme monitoring is warranted. It is important not to reduce intensity or discontinue statin therapy if liver function test elevation remains below three times upper normal values. Hepatotoxicity initially was thought to be a problem with statins, but many large randomized trials did not show any differences between statins and placebo in the occurrence of liver enzyme abnormalities. This is the reason why the FDA removed routine checking of liver enzymes after statin therapy as a requirement.
Statins and Grapefruit Juice
Since the accidental discovery in 1989 of grapefruit juice’s interaction with medications, it has been reported to interact with about 85 drugs, including statins. Bergamottin and its derivative, 6′,7′-dihydroxy bergamottin (DHB), which are furanocoumarins, are the main agents in grapefruit juice that mediate the statin-grapefruit juice interaction. Bergamottin and DHB inactivate cytochrome P450 3A4 (CYP3A4), the major enzyme involved in the metabolic degradation of some statins. Studies show that the intestinal concentration of this enzyme reduces by 50% within 4 h of drinking juice from a whole grapefruit. Inactivation of intestinal CYP3A4 affects the pre-systemic degradation of statins, thereby increasing their systemic availability [63]. This grapefruit juice effect is solely felt by those statins metabolized by CYP3A4 (lovastatin, simvastatin, and atorvastatin). Fluvastatin, rosuvastatin, and pravastatin are therefore not affected, as they are metabolized by a different enzyme. A pharmaceutical study showed that increasing the amount of grapefruit juice consumption, for the same dose of a statin, increases the blood level of that statin an observation that was represented by the area under a curve (AUC). This grapefruit juice effect with statins has been shown to decrease to 10% of its maximum 24 h after intake of the juice. This suggests a half-life of about 7–8 h for the grapefruit juice effect. Thus, for statins like simvastatin and lovastatin, with a relatively short half-life, taking grapefruit juice just in the evening produces about half the effect of juice taken in the morning. This action by grapefruit juice enhances the efficacy of related statins at reducing LDL-C by about 6 percentage points [64].
Though enhancing, some studies report dangerous effects of grapefruit juice on statins. According to Harvard Medical School, however, these studies used large amounts of furanocoumarins. Using about a quart or more of the juice poses potential danger as it increases the amount of drug entering the bloodstream. Thus, grapefruit juice may pose a threat when more than this recommended amount is consumed. Patients who absolutely cannot stand the effects of this combination can be put on statins like fluvastatin, rosuvastatin, and pravastatin that are not affected by grapefruit juice [63].
Timing of Statin Intake
Due to the varying half-life of statins, the benefits from this class of medicines, especially those with a short half-life, greatly depends on what time of the day they are administered. Cholesterol biosynthesis varies diurnally with peak hours between midnight and 5 a.m. and having a statin in the system during those peak hours can greatly impact patients’ lipid profile. Most of the current summarized evidence supports that short half-life statins like simvastatin, pravastatin, lovastatin, and fluvastatin be taken in the evening for maximum effect. The few studies that contradict this used small sample sizes, which after being pulled together in a meta-analysis supported the evening administration of these short-acting statins. Long half-life statins like atorvastatin and rosuvastatin, on the contrary, are not affected by time of administration. A systematic review and meta-analysis by Awad showed that morning and evening statin administration produced the highest therapeutic efficacy on lipid profile. The flexibility in choosing the time of the dose according to patient’s preference is also likely to improve compliance and reduce drug discontinuation [63, 65].
Individual Statins
Lovastatin
Exposure to this medication increases when it is taken with a strong CYP3A4 inhibitor. For instance, a literature review showed that lovastatin exposure is increased by up to 20-fold by itraconazole. This drug interaction that apparently results in rhabdomyolysis and subsequent kidney problems has been extrapolated to other strong CYP3A4 inhibitors like nefazodone, ketoconazole, posaconazole, erythromycin, clarithromycin, telithromycin, HIV protease inhibitors, boceprevir, and telaprevir. This resulted in the FDA recommending that these drug-drug interactions, contraindications, and dose limitations of lovastatin be added to its label.
Due to its high rate of drug-drug interaction, it is also recommended that patients do not take any other medications, including prescription and nonprescription medicine and herbal or vitamin supplements while on lovastatin without discussing it with their doctor. Lovastatin is also not recommended for pregnant women as it can cause harm to the unborn baby. Women are therefore recommended to use an effective form of birth control while on this medication in order to avoid pregnancy.
In addition to its drug-drug interactions, lovastatin, like atorvastatin, simvastatin, fluvastatin, and cerivastatin, is lipophilic. This characteristic allows these statins to easily cross the blood-brain barrier (BBB) compared to hydrophilic statins. It is suspected that this lipophilicity increases the risk of intracranial hemorrhage, especially when these statins are used as secondary prevention of ischemic stroke. Though studies by Gaist to investigate this suspicion reported no influence of statin lipophilicity on the risk of intracranial hemorrhage, this controversy still needs to be addressed. Lovastatin’s lipophilicity also gives it easier access into muscles, increasing its likelihood of causing myopathy [66, 67].
Pravastatin
Pravastatin is likely to cause fewer musculoskeletal issues due to its low lipophilicity, which makes it more difficult for pravastatin to access muscle but this is not definitely proven. Additionally, due to its short half-life, its effect would be best felt if taken at night. In a drug-level meta-analysis directly comparing pravastatin to atorvastatin and fluvastatin, participants randomized to pravastatin had significantly lower odds of transaminase elevations. The odds of a statin elevating transaminase levels increased with increased dose of the medication [68]. This is a relatively low strength statin which does not produce adverse reactions commonly, and thus can be tried in statin-intolerant patients. However, in clinical trials the most common adverse reactions, regardless of whether patients were in the placebo or statin group, were myalgia, nausea, vomiting, upper respiratory infection, diarrhea, and headache.
Simvastatin
As mentioned above, the effect of simvastatin is enhanced by grapefruit juice. In a pharmaceutical study where 40 mg of simvastatin was taken with juice from about 6 grapefruits a day, the blood levels of this drug increased by about 13.5-fold [55].
Also, in a study directly comparing individual statins, simvastatin was found to be significantly more tolerable than atorvastatin (OR, 0.61; 95% CI, 0.42–0.89; I2, 71.9%) and rosuvastatin (OR, 0.49; 95% CI, 0.27–0.88; I2, 0.0%); thus, it is less likely to be discontinued because of adverse events [61]. It also showed significantly low odds of transaminase elevation compared to atorvastatin and fluvastatin. Simvastatin also produces the highest amounts of reactive oxygen species, which are important mediators in the pathophysiology of inflammatory liver disease [54].
Simvastatin’s equipotent dose compared to rosuvastatin is 7–8 mg simvastatin to 1 mg rosuvastatin [69]. At doses at or above 80 mg, increases in reported myopathy have been seen in comparison to a lower 20-mg dose. However, it was found in a double-blind randomized trial of 12,064 men and women with a history of myocardial infarction that the 80-mg dose produced a 0.35 mmol/L greater reduction in LDL-C than the 20-mg dose [70]. Due to increased myopathy with a higher dose, moderate and low dosing regimens are recommended by the ACC and AHA statin dosing guidelines [71]. In 2011, the FDA issued a statement prohibiting de novo prescription of simvastatin at 80 mg. It stated that simvastatin 80 mg be continued only in patients who have tolerated that dose for at least a year. This decision came after reported cases like that of a 64-year old woman who experienced severe rhabdomyolysis within 24 h of her first dose of simvastatin 80 mg. This happened after an increase in her dose of simvastatin from 40 to 80 mg, following an elevation of her fasting lipid profile. It is therefore recommended that an alternative statin therapy be used in patients whose lipid level is not well controlled by simvastatin 40 mg, rather than increasing the dose [72, 73]. Other common adverse effects include upper respiratory tract infection, headache, abdominal pain, constipation, and nausea in clinical trials. It also has drug interactions with strong CYP3A4 inhibitors, verapamil, diltiazem, dronedarone, amiodarone, amlodipine, ranolazine, and lomitapide.
Atorvastatin
Its effect is enhanced by the presence of grapefruit juice. A study observed a 1.8-fold increase in the blood levels of 10 mg atorvastatin when taken with grapefruit juice [55]. Atorvastatin’s equipotent dose ratio, compared to rosuvastatin, is 3–3.5 mg atorvastatin to 1 mg rosuvastatin in order to achieve the same levels of LDL-C reduction [64]. In general, atorvastatin has a linear dose-response reduction of approximately 5.3% in LDL-C with every twofold increase in dosage. Additionally, it has shown to produce the highest LDL-C reduction in females as opposed to males, while having less reduction in familial or genetic hypercholesteremia versus non-familial-associated hypercholesteremia []. According to ACC and AHA statin dosing guidelines, atorvastatin has a higher dose-potency ratio in comparison to simvastatin, pravastatin, and lovastatin [74, 75]. Like rosuvastatin, the high intensity and effectiveness of atorvastatin must be implemented strategically depending on patient needs. For example, discontinuation of high-intensity atorvastatin doses due to muscular adverse effects was significantly higher than simvastatin doses [76]. The most common adverse effects of atorvastatin are myalgia and myopathy. With increasing dose and age, adverse effects include hypothyroidism, renal impairment, and rhabdomyolysis. It has interactions with cyclosporine, fibrates, and strong CYP3A4 inhibitors.
In a head to head randomized “PROVE it” trial, high-dose atorvastatin was superior to high-dose pravastatin in patients with cardiovascular disease, confirming the hypothesis that lower LDL is better for clinical outcome. It also has the best effect on triglyceride reduction. Therefore, in patients with severe hypertriglyceridemia, atorvastatin is the best choice in combining it with other anti-lipid drugs.
Rosuvastatin
Rosuvastatin differs from other statins in that it contains a sulfone (SOOCH3) substituent and takes part in a polar interaction with HMGR [77]. It is less lipophilic than other statins, limiting its capability to cross the BBB into the central nervous system and therefore diminishing its effect on cognition. Additionally, rosuvastatin has a long half-life, allowing it to be taken at any time of the day or even less frequently if necessary, to reduce the chance of adverse effects. Rosuvastatin is the highest intensity statin in use today; it is more effective in binding to HMG-CoA reductase, and thus reducing cholesterol levels, than other statins, but, in contrast, rosuvastatin also carries a greater risk of potentially fatal statin-induced myopathy. In 2018, a Korean research group examined the overall effects of rosuvastatin on both healthy and hyperlipidemic patients. The group found that total LDL and cholesterol significantly decreased in both groups, even when they were at acceptable levels before rosuvastatin treatment, as they were in the healthy group, suggesting that rosuvastatin effectively reduces cholesterol and LDL levels regardless of their initial levels. The research group also noted that the overall levels of fatty acids increased, and beta-oxidation decreased in both groups of patients. Additionally, the production of polyunsaturated fatty acids increased in the hyperlipidemic group. There was no observed effect on HDL levels due to rosuvastatin, and only a slight reduction in triglycerides in both groups was observed. Effects related to myopathy were only noted in the hyperlipidemic group, and they usually subsided within a week of their appearance.
Statin Use in Elderly
Since statins have not been adequately studied in elderly with no cardiovascular disease. There is a notion that elderly will not benefit from statin therapy [78]. This has led to indiscriminate statin discontinuation by many providers despite the fact that cardiovascular disease remains the number one cause of mortality and morbidity in elderly. There is a recent study published by Giral showing that discontinuation of statin in elderly over age of 75 without a history of cardiovascular disease who have been on statin for at least 2 years has led to 33% increased risk of hospital admission related to cardiovascular events. Therefore, it is recommended not to discontinue statin therapy in elderly unless the life expectancy is very short or patient has significant adverse event to statin therapy [78] (Table 1).
Conclusion
The LDL-C reducing effect with reduction in clinical events of statins is well documented in the literature. However, positive and negative pleiotropic effects outside of this LDL-C reducing function are still being investigated. Positive effects include increased bioavailability of nitric oxide, lowered inflammatory biomarkers in patients with high levels, and normal levels of LDL-C leading to event reduction. Despite clear benefits of statins in many patients with or without cardiovascular diseases, statin use is underutilized. Recently published American Heart Association/American College of Cardiology guidelines have emphasized to increase the use of statins and the fact that an LDL-C level < 70 is desirable in high-risk ACVD patients and additional ezetimibe and/or PCSK9 inhibitor should be utilized if needed to reach this goal.
References
Dimmler S. Cardiovascular disease review series. Embo Mol Med. 2011;3:697.
Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation. 2018; 137(12).
Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults. J Am Coll Cardiol. 2014;63:2889–934.
Patel B. Pharmacology of statins: a brief overview. Nurse Prescribing. 2014;12(9):451–6.
Chakravartri R, Sahai V. Compactin—a review. Appl Microbiol Biotechnol. 2004;64:618–24.
Stancu C, Sima A. Statins: mechanism of action and effects. J Cell Mol Med. 2001;5(4):378–87.
Endo A. The origin of the statins. Atherosclerosis (Supplements) (Component). 2004;5:125–30.
Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Am Heart J. 2002;144(6 suppl):S27–32.
Lawrence CM, Rodwell VW, Stauffacher CV. Crystal structure of Pseudomonas mevalonii HMG-CoA reductase at 3.0 angstrom resolution. Science. 1995;268(1758).
Benjamin EJ, Virani SS, Callaway CW, et al. Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation. 2018; 137(12).
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366(9493):1267–78.
Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118.
Tawakol A, Fayad ZA, Mogg R, Alon A, Klimas MT, Dansky H, et al. Intensification of statin therapy results in a rapid reduction in atherosclerotic inflammation: results of a multicenter fluorodeoxyglucose-positron emission tomography/computed tomography feasibility study. J Am Coll Cardiol. 2013;62(10):909–17.
Lovastatin Study Groups I Through IV. Lovastatin 5-year safety and efficacy study. Arch Intern Med. 1993;153:1079–87.
Shepherd J, Blauw GJ, Murphy MB, Bollen ELEM, Buckley BM, Cobbe SM, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–30.
Adams SP, Tsang M, Wright JM. Lipid-lowering efficacy of atorvastatin. Cochrane Database Syst Rev. 2015;3:CD008226.
Jones P, Kafonek S, Laurora I, Hunninghake D. Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study). Am J Cardiol. 1998;81(5):582–7.
Adams SP, Sekhon SS, Wright JM. Lipid-lowering efficacy of rosuvastatin. Cochrane Database Syst Rev. 2014:11.
Jones P, Davidson M, Stein E, Bays HE, McKenney J, Miller E, et al. Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR trial). Am J Cardiol. 2003;92:152–60.
Wang C-Y, Liu P-Y, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14(1):37–44.
Kavalipati N, Shah J, Ramakrishan A, Vasnawala H. Pleiotropic effects of statins. Advances in pediatrics. Indian J Endocrinol Metab. 2015;19(5):554–62.
Ridker PM. The JUPITER trial: results, controversies, and implications for prevention. Circulation Cardiovasc Qual Outcomes. 2009;2:279–85.
Katz DH, Intwala SS, Stone NJ. Addressing statin adverse effects in the clinic: the 5 Ms. J Cardiovasc Pharmacol Ther. 2014;19(6):533–42.
Denus S, Spinler SA, Miller K, Peterson AM. Statins and liver toxicity: a meta-analysis. Pharmacotherapy. 2004;24:584–91.
Trompet S, van Vliet P, de Craen AJM, et al. Pravastatin and cognitive function in the elderly. Results of the PROSPER study. J Neurol. 2010;257:85–90.
Bruckert E, Hayem G, Dejager S, Yau C, Bégaud B. Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients the PRIMO study. Cardiovasc Drugs Ther. 2005;19:403–14.
Ahmed W, Khan N, Glueck CJ, Pandey S, Wang P, Goldenberg N, et al. Low serum 25 (OH) vitamin D levels (<32 ng/mL) are associated with reversible myositis-myalgia in statin-treated patients. Transl Res. 2009;153(1):11–6.
Ovesjö M-L, Skilving I, Bergman P, Rane A, Ekström L, Björkhem-Bergman L. Low vitamin D levels and genetic polymorphism in the vitamin D receptor are associated with increased risk of statin-induced myopathy. Basic ClinPharmacol Toxicol. 2015;118(3):214–8.
Riche KD, Arnall J, Rieser K, East HE, Riche DM. Impact of vitamin D status on statin-induced myopathy. J Clin Transl Endocrinol. 2016;6:56–9.
Kang J, Nguyen Q, Mutka J, Le Q. Rechallenging statin therapy in veterans with statin-induced myopathy post vitamin D replenishment. J Pharm Pract. 2016;30(5):521–7.
Golomb BA, Evans MA. Statin adverse effects: a review of the literature and evidence for a mitochondrial mechanism. Am J Cardiovasc Drugs. 2008;8(6):373–418.
Bitzur R, Cohen H, Kamari Y, Harats D. Intolerance to statins: mechanisms and management. Diabetes Care. 2013;36(Suppl 2):S325–30.
Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Statins: pros and cons. Med Clin (Barc). 2018;150(10):398–402.
Ahmadi Y, Ghorbanihaghjo A, Naghi-Zadeh M, Yagin N. Oxidative stress as a possible mechanism of statin-induced myopathy. Inflammopharmacology. 2018;26.3:667–74 Web.
Bagley WH, Yang H, Shah KH. Rhabdomyolysis. Intern Emerg Med. 2007;2:210–8.
Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med. 2002;346(7):539–40.
Thompson PD, Clarkson P, Karas RH. Statin-associated myopathy. JAMA. 2003;289(13):1681–90.
Mohassel P, Mammen AL. Anti-HMGCR Myopathy. J Neuromuscular Dis. 2018;5(1):11–20.
Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–9.
Awan Z, Seidah NG, MacFadyen JG, et al. Rosuvastatin, proprotein convertase subtilisin/kexin type 9 concentrations, and LDL cholesterol response: the JUPITER trial. Clin Chem. 2012;58:183–9.
Lyu P-Y, Li R, Wang T-J, et al. Effects of plasma lipids and statins on cognitive function. Chin Med J. 2018;131(4):471.
MRC/BHF heart protection study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomized placebo-controlled trial. Lancet. 2002; 360(9326):7–22.
Strom BL, Schinnar R, Karlawish J, Hennessy S, Teal V, Bilker WB. Statin therapy and risk of acute memory impairment. JAMA Intern Med. 2015;175(8):1399–405.
Huang C-N, Li H-H, Lin C-L. Neuroprotective effects of statins against amyloid β-induced neurotoxicity. Neural Regen Res. 2018;13(2):198–206.
Chan D, Binks S, Nicholas JM, Frost C, Cardoso MJ, Ourselin S, et al. Effect of high-dose simvastatin on cognitive, neuropsychiatric, and health-related quality-of-life measures in secondary progressive multiple sclerosis: secondary analyses from the MS-STAT randomised, placebo-controlled trial. Lancet Neurol. 2017;16(8):591–600.
Xiaoxue L, Min Y, Shuya L, Zheng L. The association between apathy and mild cognitive impairment: a systematic review and meta-analysis. Alzheimers Dement. 2017;13(7).
Schultz BG, Patten DK, Berlau DJ. The role of statins in both cognitive impairment and protection against dementia: a tale of two mechanisms. Transl Neurodegener. 2018;7(1).
Orth M, Bellosta S. Cholesterol: its regulation and role in central nervous system disorders. Cholesterol. 2012;2012:1–19.
Golomb BA, Evans MA. Statin adverse effects. Am J Cardiovasc Drugs. 2008;8(6):373–418.
Agarwala A, Kulkarni S, Maddox T. The association of statin therapy with incident diabetes: evidence, mechanisms, and recommendations. Curr Cardiol Rep. 2018;20:50.
Anyanwagu U, Mamza J, Donnelly R, Idris I. Effects of background statin therapy on glycemic response and cardiovascular events following initiation of insulin therapy in type 2 diabetes: a large UK cohort study. Cardiovasc Diabetol. 2017;16:107.
Scattolini V, Luni C, Zambon A, et al. Simvastatin rapidly and reversibly inhibits insulin secretion in intact single-islet cultures. Diabetes Ther. 2016;7(4):679–93.
Betteridge DJ, Carmera R. The diabetogenic action of statins - mechanisms and clinical implications. Nat Rev Endocrinol. 2016;12(2):99–110.
Yaluri N, Modi S, Rodríguez ML, et al. Simvastatin impairs insulin secretion by multiple mechanisms in MIN6 cells. PLoS One. 2015;10(11).
Henriksbo B, Schertzer J. Is immunity a mechanism contributing to statin-induced diabetes? Adipocyte. 2015;4(4):1–7.
Brault M, Ray J, Gomez Y-H, Mantzoros CS, Daskalopoulou SS. Statin treatment and new-onset diabetes: a review of proposed mechanisms. Metabolism. 2014;63(6):735–45.
Brault M, Ray J, Gomez Y-H, Mantzoros CS, Daskalopoulou SS. Statin treatment and new-onset diabetes: a review of proposed mechanisms. Metabolism. 2014;63(6):735–45.
Karahalil B, Hare E, Koç G, Uslu I, Şentürk K, Özkan Y. Hepatotoxicity associated with statins. Arh Hig Rada Toksikol. 2017;68(4):254–60.
Farrag S, Hamzawy M, El-Yamany M, Saad M, Nassar N. Atorvastatin in nano-particulate formulation abates muscle and liver affliction when coalesced with coenzyme Q10 and/or vitamin E in hyperlipidemic rats. Life Sci. 2018;203:129–40.
Mach F, Ray KK, Wiklund O, Corsini A, Catapano AL, Bruckert E, et al. Adverse effects of statin therapy: perception vs. the evidence – focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur Heart J. 2018;39(27):2526–39.
Naci H, Brugts J, Ades T. Comparative tolerability and harms of individual statins: a study-level network meta-analysis of 246 955 participants from 135 randomized, controlled trials. Circ Cardiovacs Qual Outcomes. 2013;6(4):390–9.
Clarke AT, Johnson PCD, Hall GC, Ford I, Mills PR. High dose atorvastatin associated with increased risk of significant hepatotoxicity in comparison to simvastatin in UK GPRD cohort. PLoS One. 2016;11(3).
Awad K, Serban M-C, Penson P. Effects of morning vs evening statin administration on lipid profile: a systematic review and meta-analysis. J Clin Lipidol. 2017;11(4):972–985.e9.
Lee J, Morris J, Wald N. Grapefruit juice and statins. Am J Med. 2016;129(1):26–9.
Awad K, Banach M. The optimal time of day for statin administration: a review of current evidence. Curr Opin Lipidol. 2018;29(4):340–5.
Quinn KL, Macdonald EM, Mamdani MM, Diong C, Juurlink DN. Lipophilic statins and the risk of intracranial hemorrhage following ischemic stroke: a population-based study. Drug Saf. 2017;40(10):887–93.
Gaist D, Goldstein LB, Soriano LC, Rodríguez LAG. Statins and the risk of intracerebral hemorrhage in patients with previous ischemic stroke or transient ischemic attack. Stroke. 2017;48(12):3245–51.
Naci H, Brugts J, Ades T. Comparative tolerability and harms of individual statins: a study-level network meta-analysis of 246955 participants from 135 randomized controlled trials. Circ Cardiovasc Qual Outcomes. 2013;6(4):390–9.
Karlson BW, Palmer MK, Nicholls SJ, Lundman P, Barter PJ. Doses of rosuvastatin, atorvastatin and simvastatin that induce equal reductions in LDL-C and non-HDL-C: results from the VOYAGER meta-analysis. Eur J Prev Cardiol. 2016;23(7):744–7.
Welty FK, Lewis SJ, Friday KE, Cain VA, Anzalone DA. A comparison of statin therapies in hypercholesterolemia in women: a subgroup analysis of the STELLAR study. J Women's Health. 2016;25(1):50–6.
Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12 064 survivors of myocardial infarction: a double-blind randomized trial. The Lancet. 2010; 376(9753): 1658–1669.
Tayal U, Carroll R. Should anyone still be taking simvastatin 80 mg? BMJ Case Rep. 2013;bcr2013200415.
Tayal U, Carroll R. Should anyone still be taking simvastatin 80 mg? BMJ Case Rep. 2013;bcr2013200415.
Wong N. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014;63(25 Pt B):2889–934.
Adams SP, Tsang M, Wright JM. Lipid lowering efficacy of atorvastatin. Cochrane Database Syst Rev. 2015;3:CD008226.
Karlson BW, Palmer MK, Nicholls SJ, Lundman P, Barter PJ. To what extent do high-intensity statins reduce low-density lipoprotein cholesterol in each of the four statin benefit groups identified by the 2013 American College of Cardiology/American Heart Association guidelines? A VOYAGER meta-analysis Atherosclerosis. 2015;241(2):450–4.
Lee H, Choi J, Cho J, Kim T, Lee H, Jung B. Regulation of endogenic metabolites by rosuvastatin in hyperlipidemia patients: an integration of metabolomics and lipidomics. Chem Phys Lipids. 2018;214:69–83.
Giral P, Neumann A, Weill A, Coste J. Cardiovascular effect of discontinuing statins for primary prevention at the age of 75 years: a nationwide population-based cohort study in France. Eur Heart J. 2019;Epub ahead of print.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Azemawah, V., Movahed, M.R., Centuori, P. et al. State of the Art Comprehensive Review of Individual Statins, Their Differences, Pharmacology, and Clinical Implications. Cardiovasc Drugs Ther 33, 625–639 (2019). https://doi.org/10.1007/s10557-019-06904-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-019-06904-x