
Abstract
Triglyceride-rich lipoproteins (TRLs) are causal contributors to the risk of developing coronary artery disease (CAD). Apolipoprotein C-III (apoC-III) is a component of TRLs that elevates plasma triglycerides (TGs) through delaying the lipolysis of TGs and the catabolism of TRL remnants. Recent human genetics approaches have shown that heterozygous loss-of-function mutations in APOC3, the gene encoding apoC-III, lower plasma TGs and protect from CAD. This observation has spawned new interest in therapeutic efforts to target apoC-III. Here, we briefly review both currently available as well as developing therapies for reducing apoC-III levels and function to lower TGs and cardiovascular risk. These therapies include existing options including statins, fibrates, thiazolidinediones, omega-3-fatty acids, and niacin, as well as an antisense oligonucleotide targeting APOC3 currently in clinical development. We review the mechanisms of action by which these drugs reduce apoC-III and the current understanding of how reduction in apoC-III may impact CAD risk.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The mainstay of reducing risk of developing coronary artery disease (CAD) has been to pharmacologically decrease circulating levels of low-density lipoprotein cholesterol (LDL-C), the predominant risk factor for atherosclerosis. Multiple effective therapies exist to lower blood LDL-C levels, including hydroxymethylglutaryl coenzyme A reductase inhibitors (statins), a cholesterol absorption inhibitor (ezetimibe), bile acid sequestrants, and most recently, inhibitors of proprotein convertase substilisin kexin 9 (PCSK9), a negative regulator of the LDL receptor pathway of LDL clearance [1]. While LDL-C lowering through these means has been remarkably effective, many patients are intolerant to the adverse effects of these drugs. Additionally, many high-risk patients continue to possess significant residual risk despite maximal LDL-C reduction [2–4]. This has prompted the search for additional targetable risk factors for CAD for novel therapeutic development.
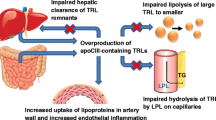
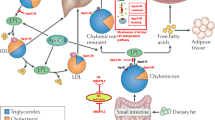
Plasma triglyceride (TG) levels may offer an orthogonal therapeutic target for lowering CAD risk. TGs, both fasting and randomly obtained, are directly and independently related to both the risk of CAD and adverse events in CAD patients [5•, 6•]. TGs are carried on TG-rich lipoproteins (TRLs) such as very low-density lipoproteins (VLDLs) and chylomicrons. TRLs are modified by lipolysis to yield remnants, which can be cleared from the circulation by hepatic receptors, or may deposit in the arterial wall similarly to LDL when in excess, contributing to lipid deposition and vascular inflammation [7–9]. Most recently, plasma TGs and TG-rich lipoproteins (TRLs) have been shown to contribute significantly to residual risk of CAD or adverse cardiac events in patients already achieving optimal LDL-C levels through other therapies [10–12]. These findings collectively suggest that TG and TRLs may be prime targets for novel treatments to reduce vascular risk on top of existing LDL-C lowering therapies.
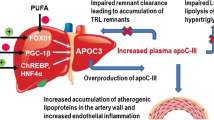
Recent findings from human genetic studies have strongly implicated the lipoprotein lipase (LPL) pathway of plasma TG clearance in modifying the risk of myocardial infarction [13–21]. LPL is an enzyme secreted by multiple peripheral tissues including adipose tissue, heart, and skeletal muscle. LPL is localized to the adjacent vascular endothelium, where it can hydrolyze TGs on circulating TRLs [22]. Released fatty acids from LPL-mediated hydrolysis are taken up by nearby tissues for storage or nutrition. Multiple circulating proteins may modulate LPL function, including apolipoproteins C-II and A-V, which promote LPL activity, and apolipoprotein C-III (apoC-III), and the angiopoietin-like proteins 3 and 4 (ANGPTL3 and 4), which inhibit LPL activity and TRL clearance. Both rare and common variants in the LPL gene and genes encoding the pathway regulators have been implicated in plasma TGs in humans.
Potential Promise of Targeting apoC-III
Recently, rare loss-of-function (LoF) variants in APOC3, the gene encoding apoC-III, have been identified and not only confer low TG levels but also protect against CAD. These studies include an initial identification in 2008 of the Arg19Ter (R19X) nonsense variant in APOC3 in the Lancaster County Amish that was associated with reduced TG and coronary artery calcification, a surrogate measure of atherosclerosis [23••]. More recently in 2014, two large sequencing studies showed a similar relationship between additional APOC3 LoF variants and cardiovascular diseases [24••, 25••]. One of these studies by the National Heart Lung and Blood Institute Exome Sequencing Project (NHLBI-ESP) identified four rare APOC3 LoF variants through exome sequencing in 3374 participants that were associated in aggregate with reduced TG of 38.5 %. Genotyping of these variants in an additional 110,970 participants (34,002 with CAD vs. 76,968 control subjects) demonstrated a concordant reduction in the incidence of CAD of 40 % in the carriers of any of the four APOC3 LoF variants (25). Further evaluation in the Framingham Heart Study demonstrated that these mutations were associated in aggregate with reduced apoC-III levels in plasma. Additionally, each 1 mg/dl decrease in apoC-III was associated with a 4 % decrease in risk of CAD incidence, a relationship that was dependent on other cardiovascular risk factors (25). The other effort from the Copenhagen Heart Study, published concurrently, showed through targeted APOC3 exon sequencing that three of the same LoF variants were associated with decreased TG and reduced incidence of ischemic vascular diseases [24••]. Importantly, neither study identified a relationship between the genetic variants with LDL-C levels, suggesting that reduction of APOC3 gene function may protect from vascular risk independently of LDL-C.
Biochemical and animal model studies of apoC-III function suggest that, in addition to its LPL inhibitory role, it delays TRL remnant clearance [26, 27] and may facilitate hepatic VLDL-TG secretion [28]. Notably, of the four LoF variants described above, one was the R19X variant and two were splice-site variants, all of which would be expected to reduce the biosynthesis of full-length apoC-III. The fourth variant was a missense A43T variant, and the mechanistic basis by which this single amino acid substitution results in impaired apoC-III concentration and/or function is very unclear. Importantly, homozygotes for APOC3 LoF have not yet been reported. In any case, because of the multifaceted roles apoC-III may play, a critical question arising from the human genetics efforts is which nodes of TG metabolism regulated by apoC-III are critical for its proatherogenicity.
Motivated in part by these human genetics findings, interest in therapeutically targeting apoC-III to reduce CAD risk has surged in recent years. This has spawned novel apoC-III-focused therapies currently under development as well as reevaluation of existing lipid-lowering treatments that in part act through modulating circulating apoC-III levels. Here, we review the existing therapies that impact apoC-III and new therapies in clinical development. We divide the therapies into those that accelerate apoC-III catabolism and those that reduce apoC-III production.
Therapies That Reduce apoC-III Primarily by Enhancing its Catabolism
Statins
Statins inhibit the rate-limiting step of cholesterol biosynthesis catalyzed by HMG-CoA reductase, thereby altering intracellular membrane cholesterol content and upregulating expression of the LDL receptor [29]. This causes increased hepatic LDL-C uptake, markedly reduced plasma LDL-C and decreased coronary disease risk and is the fundamental reason that statins are the mainstay of treatment for preventing cardiovascular events in at-risk patients. Statins also lower plasma TGs, and this may be due to their ability to promote apoC-III reduction through increasing catabolism of apoC-III containing lipoproteins. In a randomized study of atorvastatin (10 or 80 mg) vs. placebo in 217 patients with type 2 diabetes (T2DM), statin treatment resulted in a dose-dependent reduction in total apoC-III, apoB-associated apoC-III, and HDL-associated apoC-III, and concomitant reduction in plasma TGs [30]. Another study examined the kinetic properties of apoC-III and TRLs in the setting of rosuvastatin treatment [31]. Twelve men with the metabolic syndrome were randomized to placebo or rosuvastatin (10 or 40 mg) for 5 weeks in a double-blinded crossover study and kinetics were monitored using stable isotopes during each phase. Rosuvastatin caused dose-dependent increases in the apoC-III fractional catabolic rate compared to placebo. Another study by these investigators compared the impact of atorvastatin (40 mg) and fenofibrate (200 mg) on apoC-III kinetics in eleven men with the metabolic syndrome and found comparable increases in apoC-III clearance in both treatment arms [32]. While this study suggested that statins might also lower VLDL-associated apoC-III production but not the total apoC-III production rate, the rapid exchangeability of apoC-III between VLDL and HDL may complicate interpretation of this measurement [33]. A subsequent study of atorvastatin by the same investigators in 39 obese men supported the previous findings that statins increase apoC-III catabolic rates without altering apoC-III production [34]. Thus, statins augment apoC-III clearance and TRL turnover, an effect that may contribute to their atheroprotective effect.
Omega-3 Polyunsaturated Fatty Acids
Omega-3 polyunsaturated acids (n-3 PUFAs), including docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), are essential fatty acids derived from fish that can be obtained through diet, prescription supplementation or over-the-counter [35]. N-3 PUFAs may confer cardioprotective effects though this remains a debatable issue [35, 36]. The JELIS study, a randomized trial of EPA in 18,645 Japanese hypercholesterolemic subjects, showed that EPA and statin combination therapy reduced incidence of cardiovascular events by 18 % in patients with a history of CAD compared to patients treated with statins alone [37]. A post hoc analysis of this trial showed that the CAD-protective effect of EPA and statins was as high as 53 % in the group of patients with high TGs, low HDL-C and increased CAD hazard ratio [38]. However, some larger studies and meta-analyses of n-3 PUFAs and cardiovascular events have not supported a clear reduction in CV risk [39, 40]. None of the completed CV event trials were performed in individuals with elevated TG levels at baseline, though two such studies are currently ongoing. These include the STRENGTH trial (NCT02104817) involving Epanova, a prescription formulation of EPA and DHA, and the REDUCE-IT trial (NCT01492361) of AMR101, a purified ethyl ester of EPA.
At high pharmacological doses, n-3 PUFAs robustly decrease plasma TGs by 25–45 %. The TG-lowering properties of n-3 PUFAs are multi-factorial, but may involve apoC-III reduction. The EVOLVE trial evaluated the impact of Epanova, a prescription formulation of EPA and DHA, on plasma lipids over 12 weeks in 399 subjects with severe hypertriglyceridemia (500–2000 mg/dL) who maintained a low-fat diet [41]. This study found a dose-dependent reduction of 25 to 31 % in plasma TGs in n-3 PUFA treated subjects relative to those receiving placebo, as well as an 11 to 14 % reduction in apoC-III levels. The mechanisms by which n-3 PUFAs reduce apoC-III levels remain unclear, though the balance of data supports an increase in apoC-III clearance from the circulation [34, 42].
Therapies That Reduce apoC-III Primarily by Reducing its Production
Fibrates
Fibrates are amphipathic carboxylic acids that serve as ligands for the peroxisome-proliferator activated receptor alpha (PPARα) [35, 36, 43]. They reduce circulating TGs by 20–50 % through reducing both VLDL production and increasing TRL catabolism. In 1995, several groups showed that fibrates suppress the expression of APOC3 in hepatocytes [44–46]. One study in rats demonstrated that fenofibrate conferred a dose-dependent and PPARα-mediated reduction in in hepatic apoC-III expression [45]. Another study also in rats showed that this PPARα-mediated effect involved disruption of hepatocyte nuclear factor 4α binding to the APOC3 promoter in the liver [46]. Consistent with this effect, radioisotope tracer studies in hypertriglyceridemic human subjects showed that fenofibrate reduced apoC-III production rate and VLDL-associated apoC-III in humans [47].
Fibrate monotherapy has been demonstrated in some trials to reduce cardiovascular events [48–50], though the effects have in some cases been modest. Studies of the addition of a fibrate to a statin compared with statin alone have not indicated a clear reduction in cardiovascular events [51]. However, none of the fibrate trials were specifically performed in individuals with elevated TG levels at baseline. Subgroup analyses have suggested that individuals with high TG levels may be the most likely to benefit from fibrate therapy [48, 52].
Thiazoledinediones
Like fibrates, thiazoledinediones (TZDs) are nuclear receptor ligands [43]. TZDs are selective for PPARγ and are indicated for the treatment of T2DM due to their insulin-sensitizing effects. Though TZDs have largely fallen out of favor due to their adverse effects, their risks must be considered along with their potent ability to sustain insulin sensitivity and potentially maintain pancreatic cell function, which could merit utility in select populations of patients prone to diabetes and the metabolic syndrome [53]. A recent elegant study comparing two mouse strains and human adipose demonstrated an important role for natural genetic variation in affecting PPARγ binding to target sizes and also the importance of this variation in governing the ability of TZDs to exert their effects through PPARγ [54]. This work supports the potential of pharmacogenomics to help identify subsets of patients most likely to benefit from these drugs. Among the TZDs, pioglitazone is favored because of its comparatively fewer side effects and demonstrated efficacy in preventing T2DM and reducing nonalcoholic steatohepatitis [36, 53].
The impact of TZDs on plasma TGs are drug-dependent [55]. Studies in non-diabetic patients have suggested that both pioglitazone and rosiglitazone have minimal effects on plasma TGs directly in these populations [56, 57]. However, in a direct comparison of these TZDs in diabetic subjects, pioglitazone caused a 52 mg/dl reduction in plasma TGs while rosiglitazone increased TGs by 13 mg/dl. In another study using stable isotope tracers to measure lipoprotein kinetics in 8 subjects with T2DM, pioglitazone effectively lowered circulating TGs due to increased VLDL-TG clearance [58]. This study also showed that pioglitazone reduced apoC-III levels, apparently due to decreased apoC-III production [58]. However, he mechanism of PPARγ-mediated regulation of apoC-III expression, and the suggested selective regulation by pioglitazone among the TZDs remains unknown. Because apoC-III expression increases with insulin resistance, it is possible that increased insulin sensitivity due to pioglitazone may help suppress aberrantly high apoC-III expression and secretion in T2DM. Interestingly, dual PPARα/γ agonists, such as tesaglitazar and aleglitazar, reduce proatherogenic lipids and in some cases apoC-III levels [59], but they have been discontinued from clinical development due to increased adverse events [36].
Niacin
Niacin, also known as nicotinic acid or vitamin B3, is a water-soluble vitamin. At pharmacological doses, niacin reduces TGs by 25–40 % (as well as reduces LDL-C and increases HDL-C) [35]. The exact mechanism of niacin’s lipoprotein-altering effects is unresolved. Niacin activates the G-protein-coupled receptor 109A (GPR109A) in adipocytes and inhibits TG lipolysis and free fatty acid release [60–62]; historically it was believed that the reduced flux of FFA to the liver resulted in reduced VLDL-TG secretion [63–71]. Animal studies suggested that niacin’s anti-lipolytic actions in the adipose suppress hepatic APOC3 expression by reducing PPARγ coactivator-1β (PGC-1β), a circadian transcriptional coactivator induced by fatty acids that coordinates VLDL secretion and hepatic lipogenesis [72]. However, studies with niacin receptor agonists in animals and humans strongly suggest that activation of this receptor is unlikely to be the mechanism of TG-lowering [73]. An early study of niacin showed that it reduced plasma apoC-III associated with VLDL in humans through promoting VLDL clearance [74]. Thus, the exact mechanisms of action of niacin on TRL metabolism and specifically on apoC-III production vs. clearance remain unknown and will require additional studies, such as incorporation of stable isotopes to study apoC-III production vs. clearance in humans treated with niacin in order to resolve its roles.
Interest in using niacin clinically to reduce cardiovascular risk has dwindled. The AIM-HIGH study, a study of 3414 patients with cardiovascular disease and managed for LDL-C reduction, evaluated the impact of niacin vs. placebo on cardiovascular events and was stopped early after 3 years due to futility to demonstrate any benefit from niacin [75]. Another larger study, HPS2-THRIVE, evaluated extended-release niacin vs. placebo in 25,673 cardiovascular disease patients on simvastatin for LDL-C control and similarly found no reduction in the incidence of cardiovascular events compared to placebo-treated control subjects [76]. Both studies reported increased incidence of certain adverse events in the niacin-treated groups, adding additional concern [76, 77]. However, neither study focused on patients with elevated baseline TG levels. There remains interest in the development of niacin-based approaches that may be more efficacious with less adverse effects. For example, CAT-2003 is a conjugate of niacin and EPA that through linker-technology is targeted specifically to the liver; it reduces TG and apoC-III levels substantially and is currently in clinical development for the treatment of severe hypertriglyceridemia (NCT02098278). The basic mechanisms by which niacin modulates plasma TG and apoC-III still warrant further study as they may inform future therapeutic developments.
Antisense Oligonucleotides Against APOC3
None of the above currently available therapies directly target apoC-III but rather reduce apoC-III through mechanisms that are indirect. Approaches to directly targeting apoC-III could theoretically include nucleic-acid based approaches (antisense oligonucleotides or siRNA) or monoclonal antibody based approaches. Antisense oligonucleotides (ASOs) are small, single-stranded DNA- and RNA-based oligonucleotides that pair with complementary mRNA targets and prevent their expression through triggering ribonuclease H-mediated RNA cleavage or inhibiting translation [78–80]. Because of the addition of chemical modifications to the nucleotide backbones, ASOs escape rapid degradation by endogenous nucleases [80, 81]. ASOs are administered subcutaneously or intravenously and are primarily taken up by kidneys, liver, and spleen but have a widespread distribution of tissue uptake [80, 82]. Mipomersen, targeting the APOB gene, was the first systemic ASO approved for clinical use and is on the market in the US for the treatment of homozygous familial hypercholesterolemia [83].
In 2013, Ionis Pharmaceuticals (then Isis Pharmaceuticals) reported the evaluation of ASOs targeting APOC3 in rodents and humans [84•]. Generating 26 candidate ASOs against the human APOC3 mRNA, the investigators screened these in vivo in human APOC3 transgenic mice and selected the candidate ISIS308401 because of its ability to reduce APOC3 mRNA, apoC-III protein and triglycerides in vivo with high tolerability. They further explored the physiological implications of ASO-mediated APOC3 silencing through knockdown studies in rodent and nonhuman primate models of metabolic dysfunction and dyslipidemia. In all models, ASO-mediated knockdown reduced circulating apoC-III levels and plasma TG over 6 weeks of ASO treatment. Importantly, despite reports of insulin resistance in mice with APOC3 deficiency [85], no differences in fasting glucose were seen with ASO treatment in all models. Likewise, while prior work in hepatocyte cell models suggested that apoC-III might regulate intracellular TG metabolism and facilitate VLDL-TG secretion [86, 87], no effect of the ASO was observed on VLDL-TG secretion or hepatic TG content in WT or Apoc3 KO mice. The authors further demonstrated the safety, tolerability and pharmacokinetics of the lead human APOC3 ASO ISIS308401 in a randomized phase I clinical study in healthy human volunteers. Participants received either a single dose or three doses of 50, 100, 200, or 400 mg of the ASO or placebo by subcutaneous injection and were studied for 50 days following the initiation of dosing. No serious adverse events were noted, and participants receiving the ASO demonstrated a dose-dependent reduction in both circulating apoC-III levels and TG, which were sustained for more than 4 weeks after administration of the last dose, though for some measurements statistical significance was not achieved likely due to insufficient power. Thus, this proof-of-principle study served as the first demonstration of an apoC-III-specific therapeutic and showed efficacy in preclinical and early clinical investigations.
Following the success of the preclinical and phase I study, ISIS308401 was tested as a potential treatment for patients with familial chylomicronemia syndrome, an autosomal recessive disorder caused by inactivating mutations in the LPL gene leading to severe hypertriglyceridemia, recurrent pancreatitis and additional potentially fatal complications [88•]. Three patients with this disorder were treated with 300 mg of the ASO once weekly for 13 weeks and monitored for this duration and an additional 13 weeks after the last dose. The 3 participants all demonstrated a robust >70 % reduction in plasma apoC-III levels and >55 % reduction in plasma TGs within the first 2 weeks of treatment, with a high correlation of apoC-III levels and TG levels at each study time point. These patients also demonstrated large reductions in apoB48 levels (surrogate measure of chylomicron particle number), VLDL particle number, apoE, and non-HDL-C levels. These findings, albeit in a limited number of patients, offer support to the notion that apoC-III modulates TRL metabolism in a manner that is partially independent of its LPL inhibitory role. This work also provided further support for hepatic APOC3 mRNA targeting as a means to reduce both fasting and postprandial TRL levels in humans.
In a subsequent randomized phase II monotherapy trial in 57 hypertriglyceridemic patients treated with 100, 200, or 300 mg of ISIS308401 once weekly vs. a placebo control group, the investigators further demonstrated the dose-dependent apoC-III and TG-lowering effect of the ASO [89••]. In addition, the authors reported substantial decreases in VLDL particle number, VLDL-C, and increased HDL-C and LDL-C in ASO-treated subjects, which were also dose-dependent. The cause of the increased LDL-C was unclear but may have resulted from increased conversion of VLDL to LDL due to improved LPL function, or additional particle remodeling. Interestingly, in a parallel study of 28 hypertriglyceridemic subjects treated with either fenofibrate alone or in combination with the APOC3 ASO at either 200 mg or 300 mg weekly doses, the ASO lowered apoC-III, TG, and VLDL-C but without elevations in total non-HDL-C or LDL-C. This finding was thought to be due to the LDL-lowering ability of fibrates. In a recent follow-up analysis of lipoprotein-associated apoC-III from this phase II trial, the highest dose of APOC3 ASO resulted in >80 % reductions in apoC-III associated with all lipoprotein classes measured (ApoB, ApoA-I, and lipoprotein (a)) [90]. Additionally, a preliminary evaluation of this ASO in 15 hypertriglycerdemic T2DM patients demonstrated a 57 % improvement in whole-body insulin sensitivity that was related to the degree of apoC-III and TG reduction over 15 weeks [91]. Currently, three phase III studies are ongoing to study the efficacy of ISIS308401 (now termed Volanesorsen) for the treatment of hypertriglyceridemic states. The Approach Extension study (NCT02658175) is a trial evaluating the ASO for the treatment of familial chylomicronemia syndrome, LPL deficiency, or familial lipoproteinemia type I. Participants will be randomized to placebo or 300 mg weekly doses of the APOC3 ASO for 52 weeks and evaluated for TG reduction. The COMPASS study (NCT02300233) is another study evaluating the ASO for 26 weeks in hypertriglyceridemic patients of multiple etiologies that demonstrate fasting TG of ≥500 mg/dL. The third ongoing trial is the BROADEN study (NCT02527343), an evaluation of the ASO in patients with partial lipodystrophy and hypertriglyceridemia (fasting TG ≥ 500 mg/dL). Results from these studies have the potential to offer a new treatment for severe hypertriglyceridemia due to myriad causes. The potential of this ASO targeting apoC-III to reduce atherosclerotic risk and cardiovascular diseases associated with TRLs has yet to be determined in preclinical models and humans.
Another approach to targeting APOC3 could be using siRNA. Alnylam Therapeutics reported that a GalNAc-conjugated siRNA targeting apoC-III in mice resulted in knockdown of apoC-III levels of up to 95 % and a reduction in triglyceride levels of up to 68 % with durability out to over 20 days [92]. In addition, because apoC-III is a secreted protein, it could in theory be targeted with a monoclonal antibody as in the example of PCSK9. In practice this may be challenging because of the relatively high abundance of apoC-III and its association with lipoproteins, but efforts toward this approach are likely.
Conclusions
Since its discovery more than 40 years ago, apoC-III has been an important and intensely studied regulator of TG and TRL metabolism. Recent human genetics advances have solidified the connection between apoC-III and coronary disease risk in humans, reigniting interest in therapeutically targeting this protein. As discussed, many existing therapies already in clinical use effectively reduce apoC-III levels, either by promoting catabolism of apoC-III and associated TRLs or by suppressing APOC3 gene expression. Perhaps most exciting is the potential of apoC-III directed therapies, such as the ASO currently in clinical development by Ionis Therapeutics. Such approaches will help address the specific contribution of apoC-III to cardiometabolic disease in humans. As interest in targeting apoC-III grows, critical questions arise. For example, can apoC-III specific therapies work well with existing lipid-lowering medications such as statins to further reduce cardiovascular risk or residual vascular risk in patients already controlled for LDL-C? Likewise, are there any adverse consequences of sustained, near-zero levels of circulating apoC-III and does the source of apoC-III (liver vs. small intestine) dictate its function and contribution to vascular risk? Also, can human genetics be leveraged to identify additional strategies to target circulating apoC-III (e.g., through disruption of lipoprotein binding or partitioning between TRLs and HDL)? And finally, do any emerging therapies for cardiovascular risk reduction that reduce circulating apoB-containing lipoproteins, such as glucagon like peptide analogues and receptor agonists as well as PCSK9 inhibitory antibodies, act in part through perturbing apoC-III levels or function? Further investigation through preclinical studies focused on the complex physiology of apoC-III coupled with innovative drug design, perhaps informed by disease-protective APOC3 genetic variants, will help answer these and many other questions surrounding the potential promise of apoC-III as a new target for CAD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kastelein JJ. Decade in review—dyslipidaemia: resurgence of targets and compounds to treat dyslipidaemia. Nat Rev Cardiol. 2014;11:629–31.
Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78.
Cholesterol Treatment Trialists C, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81.
Boekholdt SM, Arsenault BJ, Mora S, Pedersen TR, LaRosa JC, Nestel PJ, et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: a meta-analysis. JAMA. 2012;307:1302–9.
Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. This large prospective study in almost 14,000 subjects demonstrated the significant association of nonfasted TGs and remnant cholesterol with risk of cardiovascular morbidity and mortality.
Sarwar N, Danesh J, Eiriksdottir G, Sigurdsson G, Wareham N, Bingham S, et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450–8. This meta-analysis of data from more than 10,000 participants demonstrated the significant association of triglycerides with coronary heart disease risk.
Thorin E. Vascular disease risk in patients with hypertriglyceridemia: endothelial progenitor cells, oxidative stress, accelerated senescence, and impaired vascular repair. Can J Cardiol. 2011;27:538–40.
Yu KC, Cooper AD. Postprandial lipoproteins and atherosclerosis. Front Biosci. 2001;6:D332–354.
Schwartz EA, Reaven PD. Lipolysis of triglyceride-rich lipoproteins, vascular inflammation, and atherosclerosis. Biochim Biophys Acta. 2012;1821:858–66.
Carey VJ, Bishop L, Laranjo N, Harshfield BJ, Kwiat C, Sacks FM. Contribution of high plasma triglycerides and low high-density lipoprotein cholesterol to residual risk of coronary heart disease after establishment of low-density lipoprotein cholesterol control. Am J Cardiol. 2010;106:757–63.
Faergeman O, Holme I, Fayyad R, Bhatia S, Grundy SM, Kastelein JJ, et al. Plasma triglycerides and cardiovascular events in the Treating to New Targets and Incremental Decrease in End-Points through Aggressive Lipid Lowering trials of statins in patients with coronary artery disease. Am J Cardiol. 2009;104:459–63.
Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E, et al. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. 2008;51:724–30.
Global Lipids Genetics Consortium, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83.
Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52.
Rip J, Nierman MC, Ross CJ, Jukema JW, Hayden MR, Kastelein JJ, et al. Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol. 2006;26:1236–45.
Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36.
Wittrup HH, Tybjaerg-Hansen A, Nordestgaard BG. Lipoprotein lipase mutations, plasma lipids and lipoproteins, and risk of ischemic heart disease. A meta-analysis Circulation. 1999;99:2901–7.
Gagne SE, Larson MG, Pimstone SN, Schaefer EJ, Kastelein JJ, Wilson PW, et al. A common truncation variant of lipoprotein lipase (Ser447X) confers protection against coronary heart disease: the Framingham Offspring Study. Clin Genet. 1999;55:450–4.
Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–6.
Myocardial Infarction, G., and Investigators, C.A.E.C. 2016. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N Engl J Med
Dewey, F.E., Gusarova, V., O’Dushlaine, C., Gottesman, O., Trejos, J., Hunt, C., Van Hout, C.V., Habegger, L., Buckler, D., Lai, K.V., et al. 2016. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N Engl J Med.
Kersten S. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta. 2014;1841:919–33.
Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5. This is the initial study of genetic loss-of-function in APOC3 showing that heterozygous APOC3 deficiency in humans may correlate not only with reduced TGs but also reduced atherosclerosis.
Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41. This is one of two large sequencing studies from 2014 showing that coding variants in APOC3 that confer loss-of-function cause lower TGs, higher HDL-C, minimal impact on LDL-C but protection from ischemic vascular diseases.
Tg, Hdl Working Group of the Exome Sequencing Project, N.H.L., Blood, I., Crosby, J., Peloso, G.M., Auer, P.L., Crosslin, D.R., Stitziel, N.O., Lange, L.A., Lu, Y., et al. 2014. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 371:22–31. This study is the second of two large sequencing efforts in 2014 that showed that genetic inactivation of APOC3 through loss-of-function coding variants protects from coronary disease risk despite a minimal impact on LDL-C but a lifelong reduction in TGs.
Aalto-Setala K, Fisher EA, Chen X, Chajek-Shaul T, Hayek T, Zechner R, et al. Mechanism of hypertriglyceridemia in human apolipoprotein (apo) CIII transgenic mice. Diminished very low density lipoprotein fractional catabolic rate associated with increased apo CIII and reduced apo E on the particles. J Clin Invest. 1992;90:1889–900.
Aalto-Setala K, Weinstock PH, Bisgaier CL, Wu L, Smith JD, Breslow JL. Further characterization of the metabolic properties of triglyceride-rich lipoproteins from human and mouse apoC-III transgenic mice. J Lipid Res. 1996;37:1802–11.
Qin W, Sundaram M, Wang Y, Zhou H, Zhong S, Chang CC, et al. Missense mutation in APOC3 within the C-terminal lipid binding domain of human ApoC-III results in impaired assembly and secretion of triacylglycerol-rich very low density lipoproteins: evidence that ApoC-III plays a major role in the formation of lipid precursors within the microsomal lumen. J Biol Chem. 2011;286:27769–80.
Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. 2015;161:161–72.
Dallinga-Thie, G.M., Berk, P., II, Bootsma, A.H., Jansen, H., and Diabetes Atorvastatin Lipid intervention Study, G. Atorvastatin decreases apolipoprotein C-III in apolipoprotein B-containing lipoprotein and HDL in type 2 diabetes: a potential mechanism to lower plasma triglycerides. Diabetes Care. 2004;27:1358–64.
Ooi EM, Watts GF, Chan DC, Chen MM, Nestel PJ, Sviridov D, et al. Dose-dependent effect of rosuvastatin on VLDL-apolipoprotein C-III kinetics in the metabolic syndrome. Diabetes Care. 2008;31:1656–61.
Chan DC, Watts GF, Ooi EM, Ji J, Johnson AG, Barrett PH. Atorvastatin and fenofibrate have comparable effects on VLDL-apolipoprotein C-III kinetics in men with the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1831–7.
Ginsberg HN, Ramakrishnan R. Kinetic studies of the metabolism of rapidly exchangeable apolipoproteins may leave investigators and readers with exchangeable results. Arterioscler Thromb Vasc Biol. 2008;28:1685–6.
Chan DC, Nguyen MN, Watts GF, Ooi EM, Barrett PH. Effects of atorvastatin and n-3 fatty acid supplementation on VLDL apolipoprotein C-III kinetics in men with abdominal obesity. Am J Clin Nutr. 2010;91:900–6.
Sando KR, Knight M. Nonstatin therapies for management of dyslipidemia: a review. Clin Ther. 2015;37:2153–79.
Sahebkar A, Chew GT, Watts GF. Recent advances in pharmacotherapy for hypertriglyceridemia. Prog Lipid Res. 2014;56:47–66.
Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369:1090–8.
Saito Y, Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Ishikawa Y, et al. Effects of EPA on coronary artery disease in hypercholesterolemic patients with multiple risk factors: sub-analysis of primary prevention cases from the Japan EPA Lipid Intervention Study (JELIS). Atherosclerosis. 2008;200:135–40.
Kwak, S.M., Myung, S.K., Lee, Y.J., Seo, H.G., and Korean Meta-analysis Study, G. Efficacy of omega-3 fatty acid supplements (eicosapentaenoic acid and docosahexaenoic acid) in the secondary prevention of cardiovascular disease: a meta-analysis of randomized, double-blind, placebo-controlled trials. Arch Intern Med. 2012;172:686–94.
Rizos EC, Ntzani EE, Bika E, Kostapanos MS, Elisaf MS. Association between omega-3 fatty acid supplementation and risk of major cardiovascular disease events: a systematic review and meta-analysis. JAMA. 2012;308:1024–33.
Kastelein JJ, Maki KC, Susekov A, Ezhov M, Nordestgaard BG, Machielse BN, et al. Omega-3 free fatty acids for the treatment of severe hypertriglyceridemia: the EpanoVa fOr Lowering Very high triglyceridEs (EVOLVE) trial. J Clin Lipidol. 2014;8:94–106.
Maki KC, Bays HE, Dicklin MR, Johnson SL, Shabbout M. Effects of prescription omega-3-acid ethyl esters, coadministered with atorvastatin, on circulating levels of lipoprotein particles, apolipoprotein CIII, and lipoprotein-associated phospholipase A2 mass in men and women with mixed dyslipidemia. J Clin Lipidol. 2011;5:483–92.
Kersten S. Peroxisome proliferator activated receptors and lipoprotein metabolism. PPAR Res. 2008;2008:132960.
Haubenwallner S, Essenburg AD, Barnett BC, Pape ME, DeMattos RB, Krause BR, et al. Hypolipidemic activity of select fibrates correlates to changes in hepatic apolipoprotein C-III expression: a potential physiologic basis for their mode of action. J Lipid Res. 1995;36:2541–51.
Staels B, Vu-Dac N, Kosykh VA, Saladin R, Fruchart JC, Dallongeville J, et al. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest. 1995;95:705–12.
Hertz R, Bishara-Shieban J, Bar-Tana J. Mode of action of peroxisome proliferators as hypolipidemic drugs. Suppression of apolipoprotein C-III. J Biol Chem. 1995;270:13470–5.
Malmendier CL, Lontie JF, Delcroix C, Dubois DY, Magot T, De Roy L. Apolipoproteins C-II and C-III metabolism in hypertriglyceridemic patients. Effect of a drastic triglyceride reduction by combined diet restriction and fenofibrate administration. Atherosclerosis. 1989;77:139–49.
Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317:1237–45.
Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–8.
Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–61.
Group, A.S., Ginsberg, H.N., Elam, M.B., Lovato, L.C., Crouse, J.R., 3rd, Leiter, L.A., Linz, P., Friedewald, W.T., Buse, J.B., Gerstein, H.C., et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74.
Reyes-Soffer G, Ngai CI, Lovato L, Karmally W, Ramakrishnan R, Holleran S, et al. Effect of combination therapy with fenofibrate and simvastatin on postprandial lipemia in the ACCORD lipid trial. Diabetes Care. 2013;36:422–8.
Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014;20:573–91.
Soccio RE, Chen ER, Rajapurkar SR, Safabakhsh P, Marinis JM, Dispirito JR, et al. Genetic variation determines PPARγ function and anti-diabetic drug response in vivo. Cell. 2015;162:33–44.
Yki-Jarvinen H. Thiazolidinediones and the liver in humans. Curr Opin Lipidol. 2009;20:477–83.
Samaha FF, Szapary PO, Iqbal N, Williams MM, Bloedon LT, Kochar A, et al. Effects of rosiglitazone on lipids, adipokines, and inflammatory markers in nondiabetic patients with low high-density lipoprotein cholesterol and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2006;26:624–30.
Szapary PO, Bloedon LT, Samaha FF, Duffy D, Wolfe ML, Soffer D, et al. Effects of pioglitazone on lipoproteins, inflammatory markers, and adipokines in nondiabetic patients with metabolic syndrome. Arterioscler Thromb Vasc Biol. 2006;26:182–8.
Nagashima K, Lopez C, Donovan D, Ngai C, Fontanez N, Bensadoun A, et al. Effects of the PPARγ agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J Clin Invest. 2005;115:1323–32.
Schuster H, Fagerberg B, Edwards S, Halmos T, Lopatynski J, Stender S, et al. Tesaglitazar, a dual peroxisome proliferator-activated receptor α/γ agonist, improves apolipoprotein levels in non-diabetic subjects with insulin resistance. Atherosclerosis. 2008;197:355–62.
Wise A, Foord SM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M, et al. Molecular identification of high and low affinity receptors for nicotinic acid. J Biol Chem. 2003;278:9869–74.
Soga T, Kamohara M, Takasaki J, Matsumoto S, Saito T, Ohishi T, et al. Molecular identification of nicotinic acid receptor. Biochem Biophys Res Commun. 2003;303:364–9.
Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–5.
Carlson LA. Nicotinic acid: the broad-spectrum lipid drug. A 50th anniversary review. J Intern Med. 2005;258:94–114.
Carlson LA, Havel RJ, Ekelund LG, Holmgren A. Effect of nicotinic acid on the turnover rate and oxidation of the free fatty acids of plasma in man during exercise. Metabolism. 1963;12:837–45.
Carlson LA, Oro L. The effect of nicotinic acid on the plasma free fatty acid; demonstration of a metabolic type of sympathicolysis. Acta Med Scand. 1962;172:641–5.
Carlson LA, Ostman J. Inhibition of the mobilization of free fatty acids from adipose tissue in diabetes. II. Effect of nicotinic acid and acetylsalicylate on blood glucose in human diabetics. Acta Med Scand. 1965;178:71–9.
Carlson LA, Ostman J. Inhibition of the mobilization of free fatty acids from adipose tissue in diabetes. I. Effect of nicotinic acid on the alloxan-diabetic state in rats. Acta Med Scand. 1965;177:631–7.
Duncan CH, Best MM, Robertson GL. A comparison of the effects of ethyl chlorophenoxyisobutyrate and nicotinic acid on plasma free fatty acids. Lancet. 1965;1:191–3.
Froeberg S, Oroe L. The effects of nicotinic acid, phentolamine and nethalide on the plasma free fatty acids and the blood pressure in the dog. A comparative study. Acta Med Scand. 1963;174:635–41.
Havel RJ, Carlson LA, Ekelund LG, Holmgren A. Studies on the relation between mobilization of free fatty acids and energy metabolism in man: effects of norepinephrine and nicotinic acid. Metabolism. 1964;13:1402–12.
Nestel PJ. Plasma triglyceride concentration and plasma free fatty acid changes in response to norepinephrine in man. J Clin Invest. 1964;43:77–82.
Hernandez C, Molusky M, Li Y, Li S, Lin JD. Regulation of hepatic APOC3 expression by PGC-1beta mediates hypolipidemic effect of nicotinic acid. Cell Metab. 2010;12:411–9.
Lauring B, Taggart AK, Tata JR, Dunbar R, Caro L, Cheng K, et al. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med. 2012;4:148ra115.
Yovos JG, Patel ST, Falko JM, Newman HA, Hill DS. Effects of nicotinic acid therapy on plasma lipoproteins and very low density lipoprotein apoprotein C subspecies in hyperlipoproteinemia. J Clin Endocrinol Metab. 1982;54:1210–5.
Investigators A-H, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
Group, H.T.C, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12.
Anderson TJ, Boden WE, Desvigne-Nickens P, Fleg JL, Kashyap ML, McBride R, et al. Safety profile of extended-release niacin in the AIM-HIGH trial. N Engl J Med. 2014;371:288–90.
Kurreck J. Antisense technologies. Improvement through novel chemical modifications. Eur J Biochem. 2003;270:1628–44.
Crooke ST. Progress in antisense technology: the end of the beginning. Methods Enzymol. 2000;313:3–45.
Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51.
Brown DA, Kang SH, Gryaznov SM, DeDionisio L, Heidenreich O, Sullivan S, et al. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J Biol Chem. 1994;269:26801–5.
Yu RZ, Kim TW, Hong A, Watanabe TA, Gaus HJ, Geary RS. Cross-species pharmacokinetic comparison from mouse to man of a second-generation antisense oligonucleotide, ISIS 301012, targeting human apolipoprotein B-100. Drug Metab Dispos. 2007;35:460–8.
Agarwala A, Jones P, Nambi V. The role of antisense oligonucleotide therapy in patients with familial hypercholesterolemia: risks, benefits, and management recommendations. Curr Atheroscler Rep. 2015;17:467.
Graham MJ, Lee RG, Bell III TA, Fu W, Mullick AE, Alexander VJ, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–90. This study provided comprehensive preclinical evaluation and phase I study of the anti-APOC3 antisense oligonucleotide, the newest apoC-III focused therapy in development. It showed in mice, rats, nonhuman primates and humans that ASO treatment was safe and efficacious in reducing circulating apoC-III levels.
Duivenvoorden I, Teusink B, Rensen PC, Romijn JA, Havekes LM, Voshol PJ. Apolipoprotein C3 deficiency results in diet-induced obesity and aggravated insulin resistance in mice. Diabetes. 2005;54:664–71.
Sundaram M, Zhong S, Bou Khalil M, Zhou H, Jiang ZG, Zhao Y, et al. Functional analysis of the missense APOC3 mutation Ala23Thr associated with human hypotriglyceridemia. J Lipid Res. 2010;51:1524–34.
Sundaram M, Zhong S, Bou Khalil M, Links PH, Zhao Y, Iqbal J, et al. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J Lipid Res. 2010;51:150–61.
Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–6. This small clinical study was the first to assess the APOC3 ASO in hypertriglyceridemic patients. It showed that apoC-III inhibition in patients with familial chylomicronemia syndrome genetically lacking LPL potently lowered TG levels, suggesting that apoC-III modulates TGs at least in part by LPL-independent mechanisms.
Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. 2015;373:438–47. This phase II study of the APOC3 ASO in 57 hypertriglyceridemic patients showed that the ASO was efficacious in reducing apoC-III levels and TG while raising HDL-C.
Yang, X., Lee, S.R., Choi, Y.S., Alexander, V.J., Digenio, A., Yang, Q., Miller, Y.I., Witztum, J.L., and Tsimikas, S. 2016. Reduction in lipoprotein-associated apoC-III levels following volanesorsen therapy: phase 2 randomized trial results. J Lipid Res
Digenio, A., Dunbar, R.L., Alexander, V.J., Hompesch, M., Morrow, L., Lee, R.G., Graham, M.J., Hughes, S.G., Yu, R., Singleton, W., et al. 2016. Antisense-mediated lowering of plasma apolipoprotein C-III by volanesorsen improves dyslipidemia and insulin sensitivity in type 2 diabetes. Diabetes Care.
Kevin Fitzgerald, A.B., William Querbes, Jessica Sutherland, Renta Hutabarat, Stuart Milstein, Satya Kuchimanchi, Rajeev Kallanthottathil, Klaus Charisse, Kristina Yucias, Abigail Liebow, Andrew Sprague, Martin Maier, David Kallend2 Jay Horton, Amy Simon. 2014. A subcutaneous, potent and durable RNAi platform targeting metabolic diseases, genes PCSK9, APOC3 and ANGPLT3. In Arterioscler Thromb Vasc Biol.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Sumeet A. Khetarpal, Arman Qamar, John S. Millar, and Daniel J. Rader declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Nonstatin Drugs
Rights and permissions
About this article

Cite this article
Khetarpal, S.A., Qamar, A., Millar, J.S. et al. Targeting ApoC-III to Reduce Coronary Disease Risk. Curr Atheroscler Rep 18, 54 (2016). https://doi.org/10.1007/s11883-016-0609-y
Published:
DOI: https://doi.org/10.1007/s11883-016-0609-y