
Abstract
Purpose of Review
Apolipoprotein CIII (ApoCIII) is now recognized as a key regulator in severe hypertriglyceridemia, chylomicronemia, and conditions of triglyceride-rich lipoprotein (TRL) remnant excess due to its inhibition of lipoprotein lipase (LPL) and hepatic lipase, leading to decreased hepatic reuptake of TRLs, as well as enhanced synthesis and secretion of VLDL from the liver. ApoCIII gain-of-function mutations are associated with atherosclerosis and coronary heart disease (CHD), and contribute to the development of cardiometabolic syndrome, hypertriglyceridemia, and type 2 diabetes mellitus. Conversely, loss-of-function mutations in ApoCIII are associated with lower levels of plasma triglycerides (TG), attenuation of vascular inflammatory processes such as monocyte adhesion and endothelial dysfunction, and potentially, a reduction in the incidence and progression of atherosclerosis and cardioprotection.
Recent Findings
Evidence is now emerging that volanesorsen, a second-generation antisense oligonucleotide drug targeting ApoCIII messenger RNA resulting in decreases in TG in patients with familial chylomicronemia syndrome, severe hypertriglyceridemia, and metabolic dyslipidemia with type 2 diabetes giving support to the hypothesis that ApoCIII is a powerful inhibitor of LPL, and when reduced, endogenous clearance of TRLs can result in substantial reductions in TG levels.
Summary
Discovery of the ApoCIII inhibitor volanesorsen opens a new era of lipid-lowering drugs for reduction in TG and potentially for reduction in LDL-C. Herein, this review will provide an update on the pathophysiology of ApoCIII-linked atherosclerosis and the development of the first drug to target ApoCIII, volanesorsen, as a promising lipid-lowering agent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The critical impact of coronary heart disease (CHD) on morbidity and mortality worldwide has been the catalyst for remarkable scientific endeavors for over half a century [1,2,3]. Large population studies in the 1950s helped boost the lipid hypothesis in which total cholesterol, and, more specifically, LDL-C were associated with CHD risk and high-density lipoprotein cholesterol (HDL-C) had an inverse association with the incidence of CHD [4, 5]. This hypothesis remained debatable for years, but the controversy was settled in the 1990s when simvastatin was shown to definitively decrease cardiovascular mortality in the Scandinavian Simvastatin Survival Study [6]. Since then, a multitude of well-designed studies have established the benefit of different statins in reducing the risk of CHD events in a variety of populations, with a good safety profile [7,8,9]. Recently, evolocumab was shown to reduce LDL-C from 93 to 30 mg per deciliter (a 68% decrease) in patients with CHD on statins, but this conferred only a 20% reduction in nonfatal myocardial infarction, ischemic stroke, and cardiovascular death, suggesting a potential end to the limit in which LDL-C can be reduced to further lower CHD events [10•]. Despite these unique advances, it is well recognized that even when sizable reductions in LDL-C are achieved, the risk of CHD events is not abolished [10•, 11, 12]. While studies are ongoing, attempts to raise HDL-C have not been successful in reducing CHD events [13, 14]. Interestingly, it has been shown recently that HDL-C bound to apolipoprotein CIII (ApoCIII) did not reduce proatherogenic and proinflammatory properties in incident CHD and atherosclerosis, whereas HDL-C without ApoCIII did [15•], suggesting a possible impact in the atherogenicity-limiting potential of HDL-C depending on the presence or absence of ApoCIII. The revived interest in alternative targets brings triglycerides (TG) and apolipoproteins to the spotlight, along with a pressing need to understand how they affect CHD risk.
The association between TG and CHD has not been straightforward [16,17,18,19]. After years of discrepant study results, it is now recognized that reduction in both LDL-C and TG brings about a higher reduction in CHD risk when compared to LDL-C reduction alone and that lower levels of TG are associated with coronary plaque regression [19,20,21]. Triglyceride-rich lipoproteins (TRLs) include chylomicrons and very low-density lipoproteins (VLDL). Although these molecules are not small enough to penetrate the endothelium and lead to atherosclerosis, its remnants, which are a product of lipolysis by lipoprotein lipase (LPL) and hepatic lipase, can easily do so [22•]. After reaching the subendothelial space, they are taken up by macrophages, forming foam cells. Additionally, levels of remnant lipoprotein have been associated with nitric oxide-dependent coronary vasomotor function [23], and lipolysis of TRL generating remnants and free fatty acid (FFA) was shown to engender a proinflammatory environment leading to atherosclerosis [19, 22•].
ApoCIII was first described in the late 1960s [24]. More recently, details regarding its exact genomic location and function have expanded greatly, generating hypotheses on therapeutic targets and possible secondary effects of its inhibition and intensification [24, 25•, 26]. It is recognized as a key factor in severe hypertriglyceridemia, chylomicronemia, and TRL remnant excess. Large population studies such as the Framingham Heart Study [27•] and the Verona Heart Study [27•] showed correlation between lower ApoCIII levels and favorable plasma lipid profile and cardioprotection [27•, 28•]. Gene analysis showed that loss-of-function (LOF) mutations in ApoCIII are associated with lower levels of plasma TG and a reduced risk of CHD [27•] and ischemic cardiovascular disease [28•]. Moreover, recent evidence has shown that ApoCIII promotes vascular inflammation and enhances atherogenicity of TRLs [29]. A null mutation of ApoCIII is associated with lower serum TG and cardioprotection [30, 31].
Thus, ApoCIII may become a much needed new target for severe hypertriglyceridemia where there is an unmet need given the modest effect of current lipid-lowering therapies on TG concentrations (fibrates 30–50% reduction, immediate-release niacin and omega-3 20–50%, extended-release niacin and statins 10–30%, and ezetimibe only 5–10%) [19].
Understanding ApoCIII and Triglyceride-Rich Lipoproteins
Several hepatic mechanisms have been proposed to explain ApoCIII’s role in maintaining TG, chylomicron, and TRL levels. Initially, studies had suggested that ApoCIII inhibits lipolysis of large molecules of triacylglycerol into smaller TRL by non-competitive inhibition of LPL. In recent human kinetic studies, modulation of hepatic uptake of remnants seemed to be the main effect, leading to clearance of TLR from the plasma, an event that was shown to possibly be independent of LPL. ApoCIII decreases hepatic uptake of TRL by interfering with the attachment of apolipoprotein B (ApoB) and apolipoprotein E to hepatic receptors. Lastly, it is associated with increased secretion of larger VLDL from the liver in patients with high body weight and reduced insulin sensitivity. Consequently, when ApoCIII is absent, lipolysis is more efficient and TRL levels are lower [21, 25•, 26, 29]. ApoCIII’s actions in the intestine are not yet fully described. It is possible that this apolipoprotein is a mediator of dietary lipid absorption, decreasing the transport of TG into lymph, and therefore, reducing chylomicrons in the blood stream [32].
As briefly mentioned, after undergoing lipolysis by LPL, TRL become small remnants that can permeate the arterial wall and be taken up by macrophages, generating foam cells [20]. Concomitantly, this by-product of lipolysis creates a concentration of oxidized FFA which triggers the production of proinflammatory molecules, such as TNFα, fibrinogen, and coagulation factors and contributes to endothelial dysfunction by increasing the expression of vascular cell adhesion molecule-1 (VCAM-1) and intracellular adhesion molecule 1 (ICAM1) [22•, 33, 34]. These molecules further aid in the attachment of monocytes, and thus, work to increase the numbers of proproliferative cells at the site of an atheroma. Lipolysis of TRL also increases endothelial permeability and induces the apoptotic cascade [35, 36] promoting greater cellular debris within the region of atherosclerosis.
ApoCIII Contributes to Atherosclerosis
It has long been suggested that ApoCIII in TLRs may directly contribute to the development of atherosclerosis by activating the proinflammatory signal transduction of vascular cells, emphasizing a novel role for ApoCIII that links dyslipidemia with atherosclerosis [37]. Basic science evidence has demonstrated that ApoCIII impacts several essential steps involved in atherosclerosis, such as monocyte adhesion to endothelial cells (EC) and smooth muscle cell (SMC) proliferation [29, 38]. Studies both under static conditions and under laminar flow have demonstrated that endothelial dysfunction and monocyte adhesion involve activation of nuclear factor κB (NF-κB) [39]. This nuclear factor leads to the expression of VCAM-1 and ICAM1, precipitating EC dysfunction and monocyte recruitment [40] resulting in greater degrees of cellular proliferation mentioned above. NF-κB also drives an enhancement of β1-integrin expression in monocytes, boosting their adhesion to EC [40]. A study in vivo involving statin use has been able to prove a similar interaction in the coronary artery endothelium [41]. An intriguing finding is that statins, despite their limited effects on ApoCIII levels, but by downregulation of VCAM-1, are able to hinder ApoCIII’s stimulation of vascular adhesiveness. Utilizing various transgenic and knockout animal models, Li et al. [38] showed that ApoCIII is working via Akt signaling pathway and reactive oxygen species (ROS) in cells that participate in atherosclerosis. ApoCIII stimulates SMC proliferation, precipitating worsening atherogenesis and coronary stenosis in mice with severe hypertriglyceridemia.
The interaction between ApoCIII and lipoproteins such as HDL-C, LDL-C, and VLDL may influence the relative degrees of atherogenicity of these particles. The presence or absence of ApoCIII attached to HDL-C might be the explanation for the failure of drugs that target this lipoprotein to show favorable outcomes. Evidence shows that obese patients and those with known CHD have a higher proportion of ApoCIII-containing HDL-C when compared to healthy subjects [42,43,44]. Studies with different methodologies have described the correlation between HDL-C enriched with ApoCIII and CHD, concluding that this form of HDL-C is likely not protective [44, 45, 46•]. Furthermore, the presence of ApoCIII on LDL-C confers a higher CHD risk [46•, 47]. Similarly, a prospective study with individuals initially free of CHD showed a linear association between LDL-C with ApoCIII and risk of CHD. The association persisted after adjustment for diverse risk factors [46•].
Therapies Targeting ApoCIII
Many of the currently available lipid-lowering drugs, such as fibrates, statins, omega-3 fatty acids, as well as the oral hypoglycemic pioglitazone, are able to decrease ApoCIII levels through different mechanisms [48, 49]. Fibrates prevent the transcription of ApoCIII mRNA [48], reducing its secretion. Atorvastatin and rosuvastatin decrease the generation of VLDL-ApoCIII and intensify its metabolism [50,51,52]. Pioglitazone reduces ApoCIII levels through its action as a peroxisome proliferator-activated receptors (PPARy) agonist [53]. However, none of the above mentioned therapies achieve a direct, robust reduction in ApoCIII plasma levels.
Volanesorsen
ApoCIII levels can be reduced significantly by a pharmacologic inhibitor of ApoCIII mRNA, volanesorsen (formally ISIS-APOCIIIRx and ISIS-304801). Volanesorsen is a new targeted form of therapy that has shown promising results on lowering ApoCIII levels as well as TG concentrations in phase 1, 2, and recently, in phase 3 studies [25•]. It is classified as a second-generation antisense oligonucleotide (ASO) which acts by selectively binding to ApoCIII mRNA and blocking protein synthesis as well as limiting mRNA availability due to enhanced ribonuclease H1-mediated degradation of the target mRNA [54•]. The result is a substantial reduction in ApoCIII production in the liver (Fig. 1).

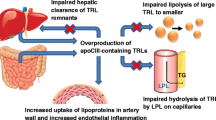
Metabolism of triglycerides. Chylomicrons are formed in the small intestine from dietary fat and contain triglycerides, Apo B48, ApoCIII, ApoCII, and ApoE. Chylomicrons enter the circulation, where they are hydrolyzed by lipoprotein lipase (LPL), which is activated by ApoCII and is inhibited by ApoCIII. Angiopoietin-like protein 3 (ANGPTL3) and angiopoietin-like protein 4 (ANGPTL4) (an antibody against ANGPTL4T is identified). Free fatty acids, which are produced by hydrolysis of triglycerides, are either used in muscle cells as source of energy or resynthesized into triglycerides and stored in adipose cells. Chylomicron remnant particles, also rich in triglycerides, are removed from the circulation by the liver. Triglycerides are also synthesized in liver cells and, together with ApoB100, ApoCIII, ApoCII, and ApoE, form very low-density lipoprotein (VLDL) particles, which are secreted in blood. VLDL particles are also hydrolyzed by LPL which is activated by ApoCII and is inhibited by ApoCIII and transformed into intermediate-density lipoprotein (IDL) particle. IDL particles are either catabolized in the liver or transformed into low-density lipoprotein (LDL) particle by LPL. Mechanism of action of volanesorsen (inset). ApoCIII inhibits LPL and hepatic lipase; thus, it inhibits hepatic uptake of triglyceride-rich lipoprotein (TRL) remnants (hepatocyte-VLDL remnants and enterocyte-chylomicron remnants). Volanesorsen is designed to specifically stimulate the catabolism of TRLs via LPL-independent pathway of triglyceride metabolism by targeting ApoCIII antisense oligonucleotide, thus lowering plasma ApoCIII levels. Reprinted and modified from Reiner [12] with permission
On July 6, 2015, the US Food and Drug Administration granted Orphan Drug Designation to volanesorsen for the treatment of patients with familial chylomicronemia syndrome (FCS). At present, there are eight studies of volanesorsen found in www.clinicaltrials.gov, including pre-clinical and clinical trials. Pre-clinical data with multiple rodent models and nonhuman primates showed ASO-mediated suppression of ApoCIII resulting in robust lowering of TG and VLDL levels [54•]. Importantly, in all of the preclinical studies, ApoCIII ASO was well tolerated and safe [54•]. Phase 1 clinical trials in healthy human volunteers showed concordant results with substantial (parenteral) dose and time-dependent decrement in plasma ApoCIII levels and TG levels (Table 1), without hepatic steatosis or transaminase elevation [54•]. Several phase 2 trials were conducted in patients with severe or uncontrolled hypertriglyceridemia [55••], in patients with FCS [56•, 57••], and in patients with type 2 diabetes [58•]. A phase 2 study of volanesorsen was a randomized, double-blind, placebo-controlled, dose-ranging trial with and without the addition of fibrate therapy. As a single agent, volanesorsen led to the reduction in plasma ApoCIII of 40 to 80%, and plasma TG levels dropped by 30 to 71% on a dose-dependent fashion. As an add-on to fibrates, volanesorsen resulted in reduction of ApoCIII levels by 60 to 71% and reduction of TG levels by 51 to 64% (Table 2) [55••]. Another phase 2 study of volanesorsen in patients with hypertriglyceridemia, including FCS, monotherapy with 100, 200, and 300 mg/dose resulted in reduction of ApoB-ApoCIII, ApoCIII-AI, and ApoCIII-LP(a) complex levels by 80% while the add-on to fibrate group resulted in reduction of ApoCIII-ApoB, ApoCIII-A1, and ApoCIII-Lp(a) complex levels by 75%. Similar reduction of the complex levels was found in the FCS group with 300 mg dose [56•]. Importantly, ApoCIII-ApoB levels correlated well with total ApoCIII, TG, VLDL-C, VLDL-ApoCIII, chylomicron-ApoCIII, chylomicron-TG, chylomicron-cholesterol, and LDL-C. A pilot phase 2 study was conducted in three FCS patients receiving 300 mg/week subcutaneous injection for 13 weeks [57••]. Plasma ApoCIII levels in the three patients were reduced by 70 to 90% and TG levels by 56 to 86% after 13 weeks of treatment (Table 3). In 2016, a randomized, double-blind, placebo-controlled trial with volanesorsen was published in 15 adult patients with type 2 diabetes (HbA1c > 7.5% [58 mmol/mol]) and hypertriglyceridemia (TG > 200 and < 500 mg/dL) [58•]. The results of the study showed that volanesorsen (300 mg/week, subcutaneously for 15 weeks) decreased ApoCIII (− 88%), TG (− 69%), VLDL-ApoCIII (− 90%), and increased HDL-C (+ 43%) compared with placebo [58•]. Additionally, these changes correlated well with improvement of insulin sensitivity [58•]. The Bruneck study was a prospective, population-based survey of the epidemiology and pathogenesis of atherosclerosis and cardiovascular disease [15•, 59•]. In a subsequent work by the Bruneck investigators, mass spectrometry was used in plasma samples gathered from two human intervention trials, one of which was randomized [55••, 57••]. They were able to demonstrate that ApoCIII mRNA inhibition by volanesorsen reduced plasma levels of ApoCIII by > 75%, ApoCII, triacylglycerol, diacylglycerols, and TG.
Phase 3 clinical trials include APPROACH [60••] study (A study of ISIS-APOCIIIRx in patients with FCS, NCT02211209), the COMPASS [61] study (A study of volanesorsen in patients with hypertriglyceridemia, NCT02300233), the BROADEN study (a study of volanesorsen in patients with partial lipodystrophy, NCT02527343), and the Approach Open Label Study (A study of volanesorsen in patients with FCS, NCT02658175). These studies are designed to investigate whether volanesorsen, when compared to placebo, is able to reduce plasma TG levels in patients with FCS, severe hypertriglyceridemia, and partial lipodystrophy. While the BROADEN trial is ongoing, with estimated primary completion in September 2017 and data release in 2019, both the APPROACH and the COMPASS trials have successfully met their primary endpoint as detailed below [60••, 61].
In December of 2016, the results of the COMPASS trial [61] were released, showing a statistically significant mean reduction in TG levels in patients treated with volanesorsen when compared to placebo (71.2 vs 0.9% reduction, p < 0.0001) which was sustained through 26 weeks of treatment. In this randomized controlled trial of 113 patients with severe hypertriglyceridemia, the average baseline TG level was 1261 mg/dl. Most patients were treated with volanesorsen (82%), including three of the patients with FCS achieved TG levels of < 500 mg/dl after 13 weeks of treatment (p < 0.0001, as compared to 14% of patients in the placebo group). Injection site reactions (ISR) were the most common adverse event in the treatment group. They were mostly mild but enough to lead to discontinuation of treatment in 13% of patients. No deaths and no serious platelet-related events were reported.
Consistent with the results of the COMPASS study, the APPROACH trial also met its primary endpoint [60••]. This study lasted 52 weeks, involved 66 patients with FCS and an average TG level of 2209 mg/dL. The mean reduction in TG levels was 77% in the volanesorsen group as compared to a mean increment in 18% (p < 0.0001) in the placebo-treated group. The authors also observed a reduction in the frequency of pancreatitis attacks and abdominal pain in the treatment arm. ISR was the most common adverse event along with thrombocytopenia, both of which combined led to a discontinuation rate of treatment of 30% (ten of 33 patients). Despite reaching their endpoint with the COMPASS and APPROACH trials, these results are preliminary and safety remains an issue. Large CHD outcome results will be needed to determine if these drugs will finally make it to the finish line and become the first TG-lowering medicine to reduce mortality.
Additional Targets to Lower Triglycerides
The understanding of the physiology behind LPL regulation coupled with human genetic evidence has identified other possible targets in the battle to lower TG levels. Analogous to ApoCIII, angiopoietin-like proteins (ANGPTL) are also established regulators of LPL. LOF mutation in the ANGPTL4 gene is significantly associated with lower TG levels as well as lower risk of coronary artery disease [62, 63]. Similarly, LOF variants in ANGPTL3 are associated with lower TG, LDL, and HDL-C in large population studies [64, 65]. In a recently published study involving ANGPTL3 exon sequencing in a large healthy adult population followed by a trial of the monoclonal antibody against ANGPTL3 evinacumab in healthy human volunteers with elevated LDL and TG levels, both genetic and therapeutic inhibition of ANGPTL3 in humans was associated not only with reduction in LDL, HDL-C, and TG but also with a decreased risk of CAD [66].
In contrast, ApoA5 (apolipoprotein A-V) seems to confer protection and loss-of-function mutation in its gene was shown to be related to higher TG levels and a higher myocardial infarction risk [63, 67]. Monoclonal antibodies against ANGPTL4 and ANGPTL4 are in development as a therapy to lower TRL and potentially influence the progression of atherosclerosis through yet another novel mechanism.
Conclusions
Genome-wide association studies have been instrumental in the identification of therapeutic hypotheses to further decrease the progression of atherosclerosis. Evidence is accumulating that targeting TRL may be a treatment target not addressed by current lipid-lowering therapies. Activation or enhancement of lipoprotein lipase may be a key mechanism to accomplish not only reductions in TRL. There is now clear evidence that ApoCIII is not only strongly associated with TRL but is also linked to inflammation, proliferation, and the progression of atherosclerosis. Volanesorsen is an antisense inhibitor of ApoCIII production and effectively reduces plasma ApoCII and TG concentrations in the rare syndrome of FCS. Discovery of the ApoCIII inhibitor volanesorsen opens a new era of lipid-lowering drugs targeting TRL and potentially serving as non-statin agents to reduce the progression and consequences of atherosclerosis.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302(18):1993–2000. https://doi.org/10.1001/jama.2009.1619.
Reiner Z. Statins in the primary prevention of cardiovascular disease. Nat Rev Cardiol. 2013;10(8):453–64. https://doi.org/10.1038/nrcardio.2013.80.
Tobert JA. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2(7):517–26. https://doi.org/10.1038/nrd1112.
Steinberg D, Gotto AM Jr. Preventing coronary artery disease by lowering cholesterol levels: fifty years from bench to bedside. JAMA. 1999;282(21):2043–50.
Keys A, Menotti A, Aravanis C, Blackburn H, Djordevic BS, Buzina R, et al. The seven countries study: 2,289 deaths in 15 years. Prev Med. 1984;13(2):141–54.
Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet (London, England). 1994;344(8934):1383–9
Pedersen TR, Berg K, Cook TJ, Faergeman O, Haghfelt T, Kjekshus J, et al. Safety and tolerability of cholesterol lowering with simvastatin during 5 years in the Scandinavian Simvastatin Survival Study. Arch Intern Med. 1996;156(18):2085–92.
Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med. 1995;333(20):1301–7. https://doi.org/10.1056/nejm199511163332001.
Sever PS, Dahlof B, Poulter NR, Wedel H, Beevers G, Caulfield M, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial—Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet (London, England). 2003;361(9364):1149–58. https://doi.org/10.1016/s0140-6736(03)12948-0.
• Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376(18):1713–22. https://doi.org/10.1056/NEJMoa1615664. First cardiovascular outcomes trial with one of the new cholesterol-lowering PCSK9 inhibitors.
Superko HR, King S 3rd. Lipid management to reduce cardiovascular risk: a new strategy is required. Circulation. 2008;117(4):560–568; discussion 8. https://doi.org/10.1161/circulationaha.106.667428.
Reiner Z. Managing the residual cardiovascular disease risk associated with HDL-cholesterol and triglycerides in statin-treated patients: a clinical update. Nutr Metabolism Cardiovasc Dis: NMCD. 2013;23(9):799–807. https://doi.org/10.1016/j.numecd.2013.05.002.
Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365(24):2255–67. https://doi.org/10.1056/NEJMoa1107579.
Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet (London, England). 2012;380(9841):572–80. https://doi.org/10.1016/s0140-6736(12)60312-2.
• Pechlaner R, Tsimikas S, Yin X, Willeit P, Baig F, Santer P, et al. Very-low-density lipoprotein-associated apolipoproteins predict cardiovascular events and are lowered by inhibition of APOC-III. J Am Coll Cardiol. 2017;69(7):789–800. https://doi.org/10.1016/j.jacc.2016.11.065. Prospective, population-based study showing strong associations of VLDL-associated apolipoproteins with incident CVD in the general community.
Hulley SB, Rosenman RH, Bawol RD, Brand RJ. Epidemiology as a guide to clinical decisions. The association between triglyceride and coronary heart disease. N Engl J Med. 1980;302(25):1383–9. https://doi.org/10.1056/nejm198006193022503.
NIH Consensus conference. Triglyceride, high-density lipoprotein, and coronary heart disease. NIH consensus development panel on triglyceride, high-density lipoprotein, and coronary heart disease. JAMA. 1993;269(4):505–10.
Carroll MD, Lacher DA, Sorlie PD, Cleeman JI, Gordon DJ, Wolz M, et al. Trends in serum lipids and lipoproteins of adults, 1960–2002. JAMA. 2005;294(14):1773–81. https://doi.org/10.1001/jama.294.14.1773.
Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011;123(20):2292–333. https://doi.org/10.1161/CIR.0b013e3182160726.
Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298(3):299–308. https://doi.org/10.1001/jama.298.3.299.
Ooi EM, Barrett PH, Chan DC, Watts GF. Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci (London, England: 1979). 2008;114(10):611–24. https://doi.org/10.1042/cs20070308.
• Reiner Z. Hypertriglyceridaemia and risk of coronary artery disease. Nat Rev Cardiol. 2017; https://doi.org/10.1038/nrcardio.2017.31. Thorough review of hypertriglyceridemia and coronary artery disease with good figures.
Kugiyama K, Doi H, Motoyama T, Soejima H, Misumi K, Kawano H, et al. Association of remnant lipoprotein levels with impairment of endothelium-dependent vasomotor function in human coronary arteries. Circulation. 1998;97(25):2519–26.
Ginsberg HN, Brown WV. Apolipoprotein CIII: 42 years old and even more interesting. Arterioscler Thromb Vasc Biol. 2011;31(3):471–3. https://doi.org/10.1161/atvbaha.110.221846.
• Norata GD, Tsimikas S, Pirillo A, Catapano AL. Apolipoprotein C-III: from pathophysiology to pharmacology. Trends Pharmacol Sci. 2015;36(10):675–87. https://doi.org/10.1016/j.tips.2015.07.001. Thorough review on Apolipoprotein C III with good figures.
Kohan AB. Apolipoprotein C-III: a potent modulator of hypertriglyceridemia and cardiovascular disease. Curr Opin Endocrinol Diabetes Obes. 2015;22(2):119–25. https://doi.org/10.1097/med.0000000000000136.
• Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371(1):22–31. https://doi.org/10.1056/NEJMoa1307095. Interesting exome analysis of genes associated with plasma triglyceride levels followed by an evaluation of the association between these genes and risk of coronary heart disease.
• Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371(1):32–41. https://doi.org/10.1056/NEJMoa1308027.. Population study showing an association between loss-of-function mutations in APOC3 and risks of ischemic vascular disease and ischemic heart disease.
Luo M, Peng D. The emerging role of apolipoprotein C-III: beyond effects on triglyceride metabolism. Lipids Health Dis. 2016;15(1):184. https://doi.org/10.1186/s12944-016-0352-y.
Tachmazidou I, Dedoussis G, Southam L, Farmaki AE, Ritchie GR, Xifara DK, et al. A rare functional cardioprotective APOC3 variant has risen in frequency in distinct population isolates. Nat Commun. 2013;4:2872. https://doi.org/10.1038/ncomms3872.
Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science (New York, NY). 2008;322(5908):1702–5. https://doi.org/10.1126/science.1161524.
Wang F, Kohan AB, Dong HH, Yang Q, Xu M, Huesman S, et al. Overexpression of apolipoprotein C-III decreases secretion of dietary triglyceride into lymph. Physiol Rep. 2014;2(3):e00247. https://doi.org/10.1002/phy2.247.
Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Boren J, Catapano AL, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32(11):1345–61. https://doi.org/10.1093/eurheartj/ehr112.
Wang YI, Bettaieb A, Sun C, DeVerse JS, Radecke CE, Mathew S, et al. Triglyceride-rich lipoprotein modulates endothelial vascular cell adhesion molecule (VCAM)-1 expression via differential regulation of endoplasmic reticulum stress. PLoS One. 2013;8(10):e78322. https://doi.org/10.1371/journal.pone.0078322.
Wang L, Gill R, Pedersen TL, Higgins LJ, Newman JW, Rutledge JC. Triglyceride-rich lipoprotein lipolysis releases neutral and oxidized FFAs that induce endothelial cell inflammation. J Lipid Res. 2009;50(2):204–13. https://doi.org/10.1194/jlr.M700505-JLR200.
Eiselein L, Wilson DW, Lame MW, Rutledge JC. Lipolysis products from triglyceride-rich lipoproteins increase endothelial permeability, perturb zonula occludens-1 and F-actin, and induce apoptosis. Am J Physiol Heart Circ Physiol. 2007;292(6):H2745–53. https://doi.org/10.1152/ajpheart.00686.2006.
Kawakami A, Yoshida M. Apolipoprotein CIII links dyslipidemia with atherosclerosis. J Atheroscler Thromb. 2009;16(1):6–11.
Li H, Han Y, Qi R, Wang Y, Zhang X, Yu M, et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: the effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc Res. 2015;107(4):579–89. https://doi.org/10.1093/cvr/cvv192.
Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114(7):681–7. https://doi.org/10.1161/circulationaha.106.622514.
Kawakami A, Aikawa M, Nitta N, Yoshida M, Libby P, Sacks FM. Apolipoprotein CIII-induced THP-1 cell adhesion to endothelial cells involves pertussis toxin-sensitive G protein- and protein kinase C alpha-mediated nuclear factor-kappaB activation. Arterioscler Thromb Vasc Biol. 2007;27(1):219–25. https://doi.org/10.1161/01.ATV.0000249620.68705.0d.
Zheng C, Azcutia V, Aikawa E, Figueiredo JL, Croce K, Sonoki H, et al. Statins suppress apolipoprotein CIII-induced vascular endothelial cell activation and monocyte adhesion. Eur Heart J. 2013;34(8):615–24. https://doi.org/10.1093/eurheartj/ehs271.
Talayero B, Wang L, Furtado J, Carey VJ, Bray GA, Sacks FM. Obesity favors apolipoprotein E- and C-III-containing high density lipoprotein subfractions associated with risk of heart disease. J Lipid Res. 2014;55(10):2167–77. https://doi.org/10.1194/jlr.M042333.
Xiong X, Liu H, Hua L, Zhao H, Wang D, Li Y. The association of HDL-apoCIII with coronary heart disease and the effect of statin treatment on it. Lipids Health Dis. 2015;14:127. https://doi.org/10.1186/s12944-015-0129-8.
Jensen MK, Rimm EB, Furtado JD, Sacks FM. Apolipoprotein C-III as a potential modulator of the association between HDL-cholesterol and incident coronary heart disease. J Am Heart Assoc. 2012;1(2). https://doi.org/10.1161/jaha.111.000232.
Riwanto M, Rohrer L, Roschitzki B, Besler C, Mocharla P, Mueller M, et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. 2013;127(8):891–904. https://doi.org/10.1161/circulationaha.112.108753.
• Mendivil CO, Rimm EB, Furtado J, Chiuve SE, Sacks FM. Low-density lipoproteins containing apolipoprotein C-III and the risk of coronary heart disease. Circulation. 2011;124(19):2065–72. https://doi.org/10.1161/circulationaha.111.056986. Prospective cohort study of individuals initially free of cardiovascular disease showing a significant higher risk of CHD in patients with LDL that contains APOC-III when compared to LDL that does not contain APOCIII.
Lee SJ, Campos H, Moye LA, Sacks FM. LDL containing apolipoprotein CIII is an independent risk factor for coronary events in diabetic patients. Arterioscler Thromb Vasc Biol. 2003;23(5):853–8. https://doi.org/10.1161/01.atv.0000066131.01313.eb.
Staels B, Vu-Dac N, Kosykh VA, Saladin R, Fruchart JC, Dallongeville J, et al. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest. 1995;95(2):705–12. https://doi.org/10.1172/jci117717.
Maki KC, Bays HE, Dicklin MR, Johnson SL, Shabbout M. Effects of prescription omega-3-acid ethyl esters, coadministered with atorvastatin, on circulating levels of lipoprotein particles, apolipoprotein CIII, and lipoprotein-associated phospholipase A2 mass in men and women with mixed dyslipidemia. J Clin Lipidol. 2011;5(6):483–92. https://doi.org/10.1016/j.jacl.2011.09.001.
Dallinga-Thie GM, Berk P II, Bootsma AH, Jansen H. Atorvastatin decreases apolipoprotein C-III in apolipoprotein B-containing lipoprotein and HDL in type 2 diabetes: a potential mechanism to lower plasma triglycerides. Diabetes Care. 2004;27(6):1358–64.
Chan DC, Watts GF, Ooi EM, Ji J, Johnson AG, Barrett PH. Atorvastatin and fenofibrate have comparable effects on VLDL-apolipoprotein C-III kinetics in men with the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28(10):1831–7. https://doi.org/10.1161/atvbaha.108.170530.
Ooi EM, Watts GF, Chan DC, Chen MM, Nestel PJ, Sviridov D, et al. Dose-dependent effect of rosuvastatin on VLDL-apolipoprotein C-III kinetics in the metabolic syndrome. Diabetes Care. 2008;31(8):1656–61. https://doi.org/10.2337/dc08-0358.
Nagashima K, Lopez C, Donovan D, Ngai C, Fontanez N, Bensadoun A, et al. Effects of the PPARgamma agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J Clin Invest. 2005;115(5):1323–32. https://doi.org/10.1172/jci23219.
• Graham MJ, Lee RG, Bell TA 3rd, Fu W, Mullick AE, Alexander VJ, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112(11):1479–90. https://doi.org/10.1161/circresaha.111.300367.
•• Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, et al. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med. 2015;373(5):438–47. https://doi.org/10.1056/NEJMoa1400283.. Randomized, double-blind, placebo-controlled, phase 2 study of volanesorsen in patients with hypertriglyceridemia.
• Yang X, Lee SR, Choi YS, Alexander VJ, Digenio A, Yang Q, et al. Reduction in lipoprotein-associated apoC-III levels following volanesorsen therapy: phase 2 randomized trial results. J Lipid Res. 2016;57(4):706–13. https://doi.org/10.1194/jlr.M066399. Study with antisense inhibitor of APOC3 synthesis in patients with the FCS and LPL deficiency suggesting that APOC3 also strongly regulates the metabolism of TRL through LPL-independent pathways.
•• Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371(23):2200–6. https://doi.org/10.1056/NEJMoa1400284.. Randomized, double blind phase 3 study with volanesorsen in FCS.
• Digenio A, Dunbar RL, Alexander VJ, Hompesch M, Morrow L, Lee RG, et al. Antisense-mediated lowering of plasma apolipoprotein C-III by volanesorsen improves dyslipidemia and insulin sensitivity in type 2 diabetes. Diabetes Care. 2016;39(8):1408–15. https://doi.org/10.2337/dc16-0126. Randomized, double-blind, placebo-controlled trial of volanesorsen in patients with diabetes.
• Stegemann C, Pechlaner R, Willeit P, Langley SR, Mangino M, Mayr U, et al. Lipidomics profiling and risk of cardiovascular disease in the prospective population-based Bruneck study. Circulation. 2014;129(18):1821–31. https://doi.org/10.1161/circulationaha.113.002500. Study applying mass spectrometry-based lipidomics profiling to the Bruneck study population.
•• Gaudet D, Digenio A, Alexander VJ, Arca M, Jones AF, Stroes E et al. The APPROACH study: a randomized, double-blind, placebo-controlled, phase 3 study of volanesorsen administered subcutaneously to patients with familial chylomicronemia syndrome (FCS). J Clin Lipidol. 2017;11(3):814–5. https://doi.org/10.1016/j.jacl.2017.04.071. Randomized, double blind phase 3 study with volanesorsen in FCS.
Gouni-Berthold I, Alexander V, Digenio A, DuFour R, Steinhagen-Thiessen E, Martin S et al. Apolipoprotein C-III inhibition with volanesorsen in patients with hypertriglyceridemia (COMPASS): a randomized, double-blind, placebo-controlled trial. J Clin Lipidol. 2017;11(3):794–5. https://doi.org/10.1016/j.jacl.2017.04.038.
Dewey FE, Gusarova V, O’Dushlaine C, Gottesman O, Trejos J, Hunt C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374(12):1123–33. https://doi.org/10.1056/NEJMoa1510926.
Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, Konig IR, et al. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med. 2016;374(12):1134–44. https://doi.org/10.1056/NEJMoa1507652.
Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119(1):70–9. https://doi.org/10.1172/jci37118.
Helgadottir A, Gretarsdottir S, Thorleifsson G, Hjartarson E, Sigurdsson A, Magnusdottir A, et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet. 2016;48(6):634–9. https://doi.org/10.1038/ng.3561.
Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017; https://doi.org/10.1056/NEJMoa1612790.
Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518(7537):102–6. https://doi.org/10.1038/nature13917.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Natalia A. Rocha, Cara East, Jun Zhang, and Peter A. McCullough declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Nonstatin Drugs
Rights and permissions
About this article

Cite this article
Rocha, N.A., East, C., Zhang, J. et al. ApoCIII as a Cardiovascular Risk Factor and Modulation by the Novel Lipid-Lowering Agent Volanesorsen. Curr Atheroscler Rep 19, 62 (2017). https://doi.org/10.1007/s11883-017-0697-3
Published:
DOI: https://doi.org/10.1007/s11883-017-0697-3