
Abstract
The oxidation kinetics of a Cr3Si-Cr7C3/SiC/SiC-coated C/SiC were comparatively investigated in dry and wet air at 1300°C under 1 atm. After oxidation for 10 h, Cr2O3 and SiO2 were produced in both oxidations. However, a larger amount of SiO2 formed during the wet air oxidation. After the dry air oxidation, the surface of Cr3Si-Cr7C3/SiC/SiC coating became relatively loose with many pores. After the wet air oxidation, the surface of Cr3Si-Cr7C3/SiC/SiC coating became undulating and loose with notable damage at the position of bulges. The Cr3Si-Cr7C3/SiC/SiC-coated C/SiC showed paralinear weight gains in both oxidation environments. Although the initial weight gains within the first 1 h in the wet air oxidation condition were higher, the gradual weight gains thereafter were lower. The parabolic rate constant for oxide formation and the linear rate constant for oxide volatilization in the wet air oxidation condition were higher than those in the dry air oxidation condition.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Continuous carbon fiber-reinforced silicon carbide (C/SiC) composites are one of the most promising thermostructural materials for high-temperature heat exchangers, hot gas filters, turbine engines, spacecraft reentry thermal protection systems, and ultra-lightweight mirrors, etc.1,2,3,4,–5 Most of these applications operate under high-temperature oxidizing environments, even containing water vapor, such as combustor liners, turbine vanes and thrusters for propulsion.
The oxidation of SiC in air is passive up to 1700°C and the formed SiO2 film has a low oxygen diffusion coefficient. Chemical vapor-deposited (CVD) SiC has been predominantly used as the coating material to protect thermal structural composites from oxidation.6,7,8,9,–10 However, below the deposition temperature (usually 1000°C), oxidizing agents can diffuse inward through microcracks mainly produced by mismatch in the coefficient of thermal expansion (CTE) between the composite and the coating, leading to oxidation of the carbon phases in the composite.7,8 Earlier work has demonstrated that, compared to a SiC/SiC/SiC coating, a Cr3Si-Cr7C3/SiC/SiC coating showed enhanced oxidation protection because of the formation of a protective Cr2O3 layer below 1000°C and a SiO2 layer at high temperatures.11 It has been confirmed that oxidation of SiC and Cr2O3 formers, such as chromium, chromium carbides and chromium silicides in dry air are different from those in wet air.12,13,14,15,–16 Research has confirmed that water vapor strongly enhances the oxidation rate of SiC.12,13 At temperatures from 1100°C to 1300°C, oxide growth of iron-chromium-aluminum alloys (FeCrAl) in dry air was faster than that in wet air.14 It has been well acknowledged that water vapor can not only accelerate formation of Cr2O3 but also enhance its vaporization.17,18,19,–20 It has been revealed that Cr2O3 evaporates linearly with time in N2-O2-H2O atmospheres, and that the evaporation rate increases with temperature.19 Therefore, above 1000°C, the oxidation behavior of the Cr3Si-Cr7C3/SiC/SiC coating in wet air might differ from that in dry air. Unfortunately, reports on the comparisons of the oxidation of the Cr3Si-Cr7C3/SiC/SiC coating above 1000°C in dry and wet air, especially on the oxidation kinetics and oxidation rate, are lacking.
The present work deals with the high-temperature oxidation behavior of a Cr3Si-Cr7C3/SiC/SiC coating investigated in dry air and in 18 vol.% O2 + 72 vol.% N2 +10 vol.% H2O under 1 atm with a flow rate of 3.0 cm s−1 at 1300°C. Much of the analysis and discussion are mainly focused on the differences in the oxidation kinetics, micromorphologies of the coating, the parabolic rate constant for oxide formation, and the linear rate constant for oxide volatilization.
Experimental Procedure
Preparation of Samples
A two-dimensional C/SiC composite prepared by low-pressure chemical vapor infiltration was used as the substrate. The preform was laid up with polyacrylonitrile (PAN)-based carbon fiber clothes (T300™). After deposition of a pyrolytic carbon (PyC) interface layer using butane, the preform was deposited with a SiC matrix using methyltrichlorosilane (MTS). The deposition conditions for the PyC interface layer were as follows: temperature 960°C, pressure 5 kPa, time 20 h, Ar flow 200 mL min−1, and C4H10 flow 15 mL min−1. The deposition conditions for the SiC matrix were as follows: temperature 1000°C, pressure 5 kPa, time 120 h, H2 flow 350 mL min−1, Ar flow 350 mL min−1, and the molar ratio of H2 to MTS was 10:1. The as-received composite was cut and machined to obtain substrates with a size of 3.0 mm × 4.0 mm × 30 mm.
The obtained composite substrates were initially coated with two layers of SiC by CVD, then an additional Cr3Si-Cr7C3 coating was prepared using a powder immersion reaction-assisted coating (PIRAC) method. The conditions for the CVD SiC deposition were the same as that for the SiC matrix, except that the deposition time was 30 h per cycle. In the PIRAC processing, samples with two layers of SiC coatings were inside sealed into a Cr-rich stainless steel container packed with Cr powders. They were then sealed into an additional Cr-rich stainless steel container with small amounts of titanium and chromium powder, acting as getters for N2 and O2, respectively. They were finally annealed at 1000°C for 2 h to form a Cr3Si-Cr7C3 coating via the reaction of Cr and SiC.11 The prepared coating is referred to as the Cr3Si-Cr7C3/SiC/SiC coating. A schematic of the sample fabrication processes is shown in Fig. 1.

Schematic of samples fabrication processes
Oxidation Tests
Oxidation tests were conducted in dry air (composed of 22 vol.% O2 and 78% vol.% N2) and wet air at 1300°C under 1 atm. The wet air oxidation tests were carried out in 18 vol.% O2 + 72.% vol.% N2 +10 vol.% H2O atmosphere. Deionized water was used to generate water vapor. In each test, three specimens were placed into an alumina tube with a purity of 99.99% at the desired temperature, and then the mixed gas flow was introduced into the alumina tube. The velocity of the mixed gas was 3.0 cm s−1 to minimize the volatility of the SiO2 scale formed on the SiC coating. The masses of the specimens before and after oxidation were measured using an electronic analytical balance (resolution: 0.01 mg).
Characterization of Phase Composition and Microstructure
Phase composition and microstructure of the samples were characterized using x-ray diffraction (XRD; BRUKER, D8 ADVANCE A25 X) and scanning electron microscopy (SEM; Hitachi S4800) equipped with energy-dispersive x-ray spectroscopy (EDS), respectively. XRD analysis was operated at 40 kV and 40 mA. Step scans were taken in the range of 2θ = 20°–80° with a 0.02-step, 0.1°/s scan speed and a 2-s exposure. The Rietveld method was adopted to quantitative analysis using DIFFRAC TOPAS 5.0.
Results and Discussion
Figure 2 shows the XRD patterns of the samples before and after oxidation for 10 h. It showed that the PIRAC-prepared Cr3Si-Cr7C3 outer layer was mainly composed of Cr3Si and Cr7C3 along with a small amount of Cr3C2. After oxidation at 1300°C for 10 h, Cr2O3 and SiO2 were detected for the both oxidations. Quantitative analysis on XRD data showed the weight ratios of Cr2O3 to SiO2 in the wet air oxidation condition and the dry air oxidation condition were 13.7 and 29.6, respectively. Distinct lower weight ratios of Cr2O3 to SiO2 in the wet air oxidation condition indicated that there was a larger amount of SiO2 formed, suggesting that the wet air oxidation condition enhanced the formation of SiO2.

XRD patterns of the samples before and after oxidation at 1300°C for 10 h
Figure 3 shows the surface morphologies of the Cr3Si-Cr7C3/SiC/SiC-coated samples before and after oxidation for 10 h. It can be seen that the surface morphologies of the oxidized samples were different from each other and from that of the as-received coating. The as-received Cr3Si-Cr7C3 outer layer exhibited a uniform folded ridge morphology, as shown in Fig. 3a. After the dry air oxidation, the surface showed a relatively loose morphology with many pores. After the wet air oxidation, the surface showed an undulating and loose morphology with notable damage at the position of the bulges, as shown in Fig. 3c.

Surface morphologies of the Cr3Si-Cr7C3/SiC/SiC coated samples before and after oxidation for 10 h: (a) as-received, (b) dry air, and (c) wet air
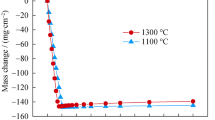
Figure 4 shows the weight changes during oxidation in the two environments. It can be seen from the figure that the samples showed nonlinear weight gains for both oxidation conditions, with a very rapid weight gains within the first 1 h followed by gradual weight gains thereafter. However, it should be noted that, during the wet air oxidation, the initial rapid weight gains are slightly higher, while the following gradual weight gains are much less. When the oxidation is over 1.75 h, the weight gains induced by the dry air oxidation are larger than those induced by the wet oxidation. The above results are different to those of iron-chromium-aluminum alloys (FeCrAl), of which the oxidation weight gains in dry air condition were faster than in wet air condition.14

The oxidation-induced weight changes in the two environments
Figure 5 shows the cross-section morphologies of the Cr3Si-Cr7C3/SiC/SiC-coated samples before and after oxidation for 10 h. It was observed that the PIRAC-prepared Cr3Si-Cr7C3 outer layer was homogeneous and well-bonded to the CVD SiC/SiC coatings. After dry air oxidation for 10 h, the outer layer showed a double-layered structure, although the outer layer was homogeneous and well-bonded to the CVD SiC/SiC coatings. After wet air oxidation for 10 h, the outer layer was rough and loose, indicating apparently vaporization. However, it should be noted that there is scarcely discernible oxidation under the coating, as shown in Fig. 5d, indicating that oxidation is taking place on the surface during the two oxidations.

Cross-section morphologies of the Cr3Si-Cr7C3/SiC/SiC coated samples before and after oxidation for 10 h: (a) as-received, (b) dry air, (c) wet air, (d) view of sub-coating region of (c)
In the present work, possible reactions for the coatings are as follows: 11,15,19,20,21,22,23,24,25,–26
At high temperatures, oxidation of Cr7C3 led to the formation of Cr2O3 and CO according to reaction (1).22 Oxidation of Cr3Si led to the formation of Cr2O3 and SiO2, according to reaction (2).23,24 On the other hand, in the present work, the formed Cr2O3 may volatilize due to further reacting with oxygen to form gaseous CrO3, according to reaction (3)25,26 or water vapor to form gaseous Cr(OH)x, according to the reaction (4),19,20 Oxidation of SiC led to the formation of SiO2 and CO, according to reactions (5) and (6).15
If there is no oxide volatilization, the oxidation-induced weight gains should be parabolic oxidation, as in Eq. 7:27
where x is the weight gain per unit surface area induced by oxide formation, kp is the parabolic oxidation rate constant and t is the time.
Figure 6 shows the log–log plot of oxidation-induced weight gains and time for both oxidation conditions. From the results, during the dry air oxidation, the weight gains, ∆wgain, and time, t, can be described by:

The log–log plot of weight gains and time during oxidation
As the exponent of 0.4284 is lower than 0.5 for parabolic kinetics, it is suggested that there is oxide volatilization during the air oxidation. In the same way, there is oxide volatilization during the wet air oxidation. In these cases, the weight gains versus time should follow a paralinear model, as in Eq. 926:
where kl is the linear rate constant for oxide volatilization, x is the weight gain per unit surface area induced by oxide formation, kp is the parabolic oxidation rate constant and t is the time.
Figure 7 shows the comparison of the fitted weight gains, which were calculated according to Eq. 9, and the measured weight gains. It seems that the weight gains can be well-fitted by the paralinear model for both oxidation conditions. In the air oxidation condition, the calculated parabolic rate constant for oxide formation and the linear rate constant for oxide volatilization were 0.03918 mg2(cm−4 h−1) and 0.02016 mg(cm−2 h−1), respectively. It has been reported that the calculated parabolic rate constant and the linear rate constant of Ni-30Cr at 1300°C in dry air oxidation condition were 0.0882 mg2(cm−4 h−1) and 0.045 mg (cm−2 h−1), respectively.25 As the SiO2 layer reduced the inward diffusion of O and the outward diffusion of Cr,28 oxide scale growth of the Cr3Si-Cr7C3/SiC/SiC should be lower than that of Ni-30Cr. Moreover, at 1300°C in dry air oxidation condition, SiO2 is more stable than Cr2O3.15 Therefore, oxidation at 1300°C in dry air, the calculated parabolic rate constant and the linear rate constant of the Cr3Si-Cr7C3/SiC/SiC are all lower than those of the Ni-30Cr.

Comparison of the fitted weight gains and the measured weight gains
In the wet air oxidation condition, the calculated parabolic rate constant for oxide formation and the linear rate constant for oxide volatilization were 0.07194 mg2(cm−4 h−1) and 0.1248 mg (cm−2 h−1), respectively. High parabolic rate constant for oxide formation in the wet air oxidation condition indicates that oxidation of the coating in the wet air oxidation condition is higher than that in the dry air oxidation condition, and thus there is a higher weight gain rate at the initial oxidation. The calculated linear rate constant for oxide volatilization for the wet air oxidation condition was much higher than that in the air oxidation condition, indicating that water vapor enhances the oxide volatilization and makes the weight gain more gradual. Based on these calculation results and the surface morphology shown in Fig. 3c, and the pores and dilapidation observed on the oxidized surface, it can be concluded that oxide volatilization occurred. The combination the surface morphology and the fine gap observed on the cross-section, it can be concluded that there are gases formed at the oxide/Cr3Si-Cr7C3 interface.18,21
It has been well-confirmed that water vapor can enhance oxidizing agent diffusion in chromia and silica, and thus enhance the oxidation rate and formation of chromia and silica.13,17,18,29,30,–31 Therefore, the rate of weight gain at the initial stage in the wet air oxidation condition was higher than that in the dry air oxidation condition, which was similar to the parabolic rate constant for oxide formation. On the other hand, water vapor can significantly enhance the evaporation of chromia by the formation of gaseous Cr(OH)x.19,20 Moreover, gaseous Cr(OH)x will simultaneously be formed in the oxide films due to the transport of OH− ions (ionic radius of OH−: 95 × 10−12 m) through the Cr2O3 scale being much faster than that of the O2− ionic (ionic radius of O2−: 140 × 10−12 m).29,32 For the same conditions, gaseous Cr(OH)x has a higher vapor pressure than CrO3(g).33 Consequently, the linear rate constant for oxide volatilization in the wet air oxidation condition was much higher than that in the air oxidation condition, while the oxidation kinetics in the wet oxidation condition showed apparently higher gradual weight gains after the initial rapid weight gains. Meanwhile, the oxidation kinetics in the dry air and the wet air oxidation conditions at 1300°C of Cr3Si-Cr7C3/SiC/SiC are opposite to those of iron-chromium-aluminum alloys (FeCrAl). It has been demonstrated that, at low gas velocity and low pressure, silica volatilization and the corresponding steady-state recession rate are negligible.34 Therefore, the weight ratio of Cr2O3 to SiO2 in the ratio of the intensity of the SiO2 peaks to that of the SiC peaks for the wet air oxidation condition was smaller than that for in the dry air oxidation condition.
During oxidation at 1300°C, Cr2O3 and SiO2 coexisted in the oxide film, which was solid-state. Therefore, evaporation of the external portion of the Cr2O3 scale resulted in the air-oxidized surface showed a relatively rough loose morphology with many pores, as shown in Fig. 3b. During the wet oxidation, the water vapor resulted in viscous silica being formed,13,35,36 which resulted in the release of the Cr(OH)x formed in the oxide films and at the oxide/Cr3Si-Cr7C3 interface being baffled. As a result, the escape of gases induced local bulging and damage to the oxides film, as shown in Fig. 3c.
Conclusions
Oxidation kinetics of a Cr3Si-Cr7C3/SiC/SiC-coated C/SiC composite was comparatively investigated in dry air and wet air.
After oxidation at 1300°C for 10 h, the produced phases were Cr2O3 and SiO2 for the two oxidation conditions. However, a larger amount of SiO2 formed in the wet air oxidation condition. Both oxidations made the surface of Cr3Si-Cr7C3/SiC/SiC coating become relatively loose with many pores. After the wet air oxidation, enhanced evaporation of gases caused the surface of the Cr3Si-Cr7C3/SiC/SiC coating to showed notable damage at the position of bulges.
The Cr3Si-Cr7C3/SiC/SiC-coated C/SiC composite showed paralinear weight gains during both oxidation environments. Although the rapid weight gains within the first 1 h in the wet air oxidation condition were higher, the gradual weight gains thereafter were lower.
In the dry air oxidation condition, the calculated parabolic rate constant for oxide formation and the linear rate constant for oxide volatilization were 0.03918 mg2 (cm−4 h−1) and 0.02016 mg (cm−4 h−1), respectively. In the wet air oxidation condition, the calculated parabolic rate constant for oxide formation and the linear rate constant for oxide volatilization were 0.07194 mg2(cm−4 h−1) and 0.1248 mg (cm−4 h−1), respectively.
References
R. Naslain and F. Christin, MRS Bull. 28, 654 (2003).
S. Schmidt, S. Beyer, H. Knabe, H. Immich, R. Meistring, and A. Gessler, Acta Astronaut. 55, 409 (2004).
J. Swain, K. Gaurav, D. Singh, P.K. Sen, and S.K. Bohidar, Inter. J. Innov. Res. Sci. Technol. 1, 290 (2014).
A. Sommers, Q. Wang, X. Han, C. T’Joen, Y. Park, and A. Jacobi, Appl. Therm. Eng. 30, 1277 (2010).
W. Krenkel, Inter. J. App. Ceram. Technol. 1, 188 (2004).
J.R. Strife and J.E. Sheehan, Ceram. Bull. 67, 369 (1998).
F.J. Buchanan and J.A. Little, Corros. Sci. 35, 1243 (1993).
W. Li, Y. Xiang, B.F. Zhang, S. Wang, J. Liu, Q.G. Lin, and J. Yang, Corros. Sci. 74, 149 (2013).
S.J. Wu, L.F. Cheng, L.T. Zhang, and Y.D. Xu, Surf. Coat. Technol. 200, 4489 (2006).
L. Li, H.J. Li, H.J. Lin, L. Zhuang, S.L. Wang, T. Feng, X.Y. Yao, and Q.G. Fu, Surf. Coat. Technol. 302, 56 (2016).
S.J. Wu, Y.G. Wang, Q. Guo, B. Guo, and L. Luo, Mater. Sci. Eng., A 644, 268 (2015).
E.J. Opila, J. Am. Ceram. Soc. 82, 625 (1999).
S.J. Wu, L.F. Cheng, and L.T. Zhang, Corros. Sci. 66, 111 (2013).
R.B. Rebak, V.K. Gupta, and M. Larsen, JOM 70, 1484 (2018).
E.J. Opila, N.S. Jacobson, D.L. Myers, and E.H. Copland, JOM 58, 22 (2006).
S.J. Wu, B. Guo, T.B. Li, and D.M. Gui, Constr. Build. Mater. 81, 11 (2015).
N.K. Othman, J.Q. Zhang, and D.J. Young, Corros. Sci. 52, 2827 (2010).
Y.P. Jacob, V.A.C. Haanappel, M.F. Stroosnijder, H. Buscail, P. Fielitz, and G. Borchardt, Corros. Sci. 44, 2027 (2002).
A. Yamauchi, K. Kurokawa, and H. Takahashi, Oxid. Met. 59, 517 (2003).
H. Asteman, J.E. Svensson, and L.G. Johansson, Corros. Sci. 44, 2635 (2002).
N.K. Othman, N. Othman, J. Zhang, and D.J. Young, Corros. Sci. 51, 3039 (2009).
Y.N. Kok and P.E.H. Hovsepian, Surf. Coat. Technol. 201, 3596 (2006).
J.H. Ma, Y.L. Gu, L. Shi, L.Y. Chen, Z.H. Yang, and Y.T. Qian, J. Alloy. Compd. 375, 249 (2004).
S.V. Raj, Mater. Sci. Eng., A 192–193, 583 (1995).
P. Berthod, Oxid. Met. 64, 235 (2005).
C.S. Tedmon, J. Electrochem. Soc. 113, 766 (1967).
C. Wagner, J. Electrochem. Soc. 99, 369 (1952).
P. Tunthawiroon, Y.P. Li, N. Tang, Y. Koizumi, and A. Chiba, Corros. Sci. 95, 88 (2015).
J. Ehlers, D.J. Young, E.J. Smaardijk, A.K. Tyagi, H.J. Penkalla, L. Singheiser, and W.J. Quadakkers, Corros. Sci. 48, 3428 (2006).
J. Zurek, D.J. Young, E. Essuman, M. Hänsel, H.J. Penkalla, L. Niewolak, and W.J. Quadakkers, Mater. Sci. Eng., A 477, 259 (2008).
B.E. Deal and A.S. Grove, J. Appl. Phys. 36, 3770 (1965).
H. Nickel, Y. Wouters, M. Thiele, and W.J. Quadakkers, J. Anal. Chem. 361, 540 (1998).
G.R. Holcomb, Oxid. Met. 69, 163 (2008).
E.J. Opila, J. Am. Ceram. Soc. 86, 1238 (2003).
B.B. Karki and L. Stixrude, Phys. Rev. Lett. 104, 215901 (2015).
S.J. Wu, L.F. Cheng, J. Zhang, L.T. Zhang, X.G. Luan, H. Mei, and P. Fang, Mater. Sci. Eng., A 435–436, 412 (2006).
Acknowledgements
The authors gratefully acknowledge the financial support from the fund of the Creative Research Foundation of Science and Technology on Thermostructural Composite Materials Laboratory (6142911020105).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, S., Ma, S., Chen, Y. et al. Paralinear Oxidation of Cr-Si-C-Coated C/SiC at 1300°C in Wet and Dry Air Environments. JOM 72, 361–367 (2020). https://doi.org/10.1007/s11837-019-03841-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11837-019-03841-w