
Abstract
A methodology is developed to calculate Cr-evaporation rates from Cr2O3 with a flat planar geometry. Variables include temperature, total pressure, gas velocity, and gas composition. The methodology was applied to solid-oxide, fuel-cell conditions for metallic interconnects and to advanced-steam turbines conditions. The high velocities and pressures of the advanced steam turbine led to evaporation predictions as high as 5.18 × 10−8 kg/m2/s of CrO2(OH)2(g) at 760 °C and 34.5 MPa. This is equivalent to 0.080 mm per year of solid Cr loss. Chromium evaporation is expected to be an important oxidation mechanism with the types of nickel-base alloys proposed for use above 650 °C in advanced-steam boilers and turbines. It is shown that laboratory experiments, with much lower steam velocities and usually much lower total pressure than found in advanced steam turbines, would best reproduce chromium-evaporation behavior with atmospheres that approach either O2 + H2O or air + H2O with 57% H2O.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The oxidation of alloys protected by the formation of Cr2O3 (chromia formers) can undergo scale loss due to reactive evaporation of chromium containing gas species. Water vapor increases the evaporation loss by allowing the formation of CrO2(OH)2(g), which for the same conditions has a higher vapor pressure than CrO3(g). CrO3(g) is the predominate Cr gas specie in dry air or oxygen. This degradation mechanism is of great importance on the cathode-side of solid-oxide, fuel-cell (SOFC) interconnects, where deposition of Cr at triple-phase points can degrade the fuel cell [1–4]. It may also be important as a long-term degradation mechanism in the steam side of advanced-coal-power plants.
A generalized reaction equation for Cr evaporation from Cr2O3 is
For CrO2(OH)2(g), n = 1 and m = 3/4 so Eq. 1 becomes:
Evaporation can change the overall oxidation kinetics from parabolic behavior to linear kinetics or even to breakaway oxidation. Linear kinetics can arise after scale growth from oxidation, which decreases with increasing scale thickness, matches the scale loss from reactive evaporation. The change in scale thickness, x, with time, t, can be described in terms of the parabolic rate constant, k p, and the linear reactive-evaporation rate, k e, as:
At long times or high reactive evaporation rates, a limiting-scale thickness, x L, arises that is given by:
In this case metal loss rates are linear, but still involve diffusion through a protective scale. Rapid metal loss can occur when reactive evaporation of Cr depletes the scale (and sometimes the substrate metal) of Cr [5, 6]. Decreased Cr in the scale or metal can lead to the formation of less-protective oxides, such as Fe–Cr oxides in Fe–Cr base alloys. Unprotective scales can lead to rapid metal loss, or “break-away” oxidation.
A methodology for calculating evaporation rates in a variety of environments is presented, including those found in a SOFC and in a high pressure steam turbine. Experimental results will be used to validate the methodology.
Methodology
Evaporation
One way to determine evaporation rates is to assume that volatility is limited by the transport of the volatile specie through a boundary layer in the gas phase. For flat plate geometry with laminar flow, the evaporation rate can be calculated by Eq. 5: [7, 8]
where Re and Sc are the dimensionless Reynolds and Schmidt numbers, D AB is the gaseous-diffusion coefficient between the Cr gas specie and the solvent gas (m2/s), ρ is the density (kg/m3) of the evaporative specie in the gas, and L is the length (m) in the flow direction of the flat plate. Equation 5 is valid for Sc numbers between 0.6 and 50 [7]. Assuming ideal-gas behavior and a reaction described by Eq. 1, this can be expanded to:
where P i is the partial pressure of gas specie i (atm), M i is the molecular mass (kg/g mol) of gas specie i (in this case i is the Cr-containing gas specie), R 1 is the gas constant (8.20594 × 10−5 m3atm/K g mol), and T is the absolute temperature (K). The dimensionless Reynolds and Schmidt numbers are defined as:
where ρs is the density of the solvent gas (kg/m3), η is the absolute viscosity (kg/m/s) and u is the gas velocity (m/s).
For turbulent flow (Re > 5 × 105), the equation equivalent to Eq. 6 is: [7]
In the case of reaction in Eq. 2, the last term in Eqs. 6 and 9 \( {\left( {P_{{{\text{CrO}}_{2} ({\text{OH}})_{2} }} } \right)} \) is found by:
where \( a_{{{\text{Cr}}_{2} {\text{O}}_{3} }} \) is the activity Cr2O3, \( \Updelta G^{^\circ }_{2} \) is the Gibbs energy of Eq. 2 (J/g mol), and R 2 is the gas constant (8.3146 J/K g mol).
Each of the parameters in Eqs. 6–10 that require additional commentary will be described.
Diffusion Coefficient, D AB
Estimation of the diffusion coefficient, D AB, between the Cr gas species and the solvent gas is the most tenuous of the parameters. Tucker and Nelken [9] compared several different methods for estimation of D AB and of two recommended choices, the one developed by Fuller et al. [10] is used here because it contains fewer parameters that themselves need to be estimated. After conversion to SI units (but with atm for pressure), the estimation equation is:
Here v i is the diffusion volume of species i (m3/mol) and P T is the total pressure (atm). Diffusion volumes, as given by Fuller et al. [10], were from a fit of Eq. 11 with an extensive list of diffusion-data measurements of various A–B pairs. Table 1 lists M i and v i for several gas species and gas mixtures. Diffusion volumes of Cr-gas species are not available and so were estimated based on a molecule with a radius of 1.6 × 10−10 m, then converted to molar volume. The value of 1.6 × 10−10 m comes from a density-functional-theory estimation of the length of a Cr–O bond in CrO [11]. The values of v i for the mixtures other than air were based on a weighted average of the component v i values.
An additional consideration for supercritical-steam-turbine environments is that D AB can diverge from the inverse-pressure relationship of Eq. 11 at high pressures [12]. As an approximation, Fig. 1 can be used to estimate the reduction in D AB at high pressures. In Fig. 1, PD/(PD)° is the ratio of the pressure-diffusivity product at pressure P to the pressure-diffusivity product at low pressure (both at the same T). T r and P r are the reduced temperature and pressure and are equal to T/T critical and P/P critical. For water T critical is 647.25 K and P critical is 218.25 atm. As an example, consider the conditions of 760 °C and 340 atm. In this case T r is 1.60 and P r is 1.56, which gives a PD/(PD)° value of 0.88. So the value of D AB obtained from Eq. 11 is adjusted by multiplying it by 0.88. This reduction factor is an approximation because this figure is a part of a diagram that was developed for self-diffusivity using Enskog kinetic-theory and fragmentary data [13].

Reduction of D for dense gases. PD/(PD)° is the ratio of the pressure-diffusivity product at pressure P to the pressure-diffusivity product at atmospheric pressure (both at the same T). T r and P r are the reduced temperature and pressure and are equal to T/T critical and P/P critical. Derived from Slattery and Bird [12, 13]
Absolute Viscosity, η
The absolute viscosity of non-polar gases, for example O2, N2, Ar, and air, can be calculated from the following equation based on the Lennard-Jones potential: [7, 8]
where η is in kg/m/s, σ is a characteristic diameter of the molecule in m, and Ωη is the dimensionless collision integral, which is a function of κB T/ε. The term κB T/ε is a viscosity function based on the Lennard-Jones potential, where κB is the Boltzmann constant and −ε is the minimum energy of the Lennard-Jones potential function. Values of σ and ε/κB are given in Table 2 [7, 8]. The value of Ωη can then be found from Table 3 [7, 8].
The absolute viscosity of water, in the temperature range of interest, can be found using Eq. 13, which was obtained from linear portions of absolute-viscosity curves as functions of temperature and pressure [14]. Equation 13 is for the temperature range 811–1,089 K and pressures up to 340 atm where the linear fit has a correlation coefficient (R 2) of 0.994. Absolute viscosities for temperatures below 811 K, closer to the critical point of water, are decidedly non-linear and Eq. 13 should not be used.
For gas mixtures, the absolute viscosity of each component gas was combined using Eqs. 14 and 15, which is the semiempirical formulation of Wilke: [7, 8, 15]
Solvent Gas Density, ρs
The density of the solvent gas is found by assuming ideal gas behavior, which allows Eq. 16:
Where M Ave is the average molecular weight of the solvent–gas mixture.
Gibbs Energy, ΔG
The two primary Cr-gas species for reactive evaporation are CrO3(g) in either dry conditions or moist conditions at higher temperatures, and CrO2(OH)2(g) in moist conditions at most of the temperatures of interest here. Equation 2 describes the evaporation reaction for CrO2(OH)2(g). It is necessary to know the Gibbs energy of formation for each of the products and reactants in Eq. 2 to obtain the ΔG of the reaction used in Eq. 10. The general form for ΔG of Eq. 1 is given by:
The ΔG f for CrO2(OH)2(g) is not well established. Opila [16] has reviewed the literature and has found that using data based on Glusko [17] results in much lower calculated partial pressures of CrO2(OH)2(g) than using data based on Ebbinghaus [18]. Glusko [17] is the source of CrO2(OH)2(g) data for the ITVAN [19] and HSC [20] thermodynamics programs. The experimental data of Gindorf et al. [21] lie between that predicted by Glusko [17] and Ebbinghaus [18] (in terms of \( {\text{log}}\,P_{{\text{CrO}}_{2} ({\text{OH}})_{2}} \)). The Glusko [17] and Ebbinghaus [18] data sources form lower and upper bounds for predicting evaporation losses. Ebbinghaus [18] used estimates of molecular parameters to formulate thermodynamic information. Gindorf [21] used transpiration experiments to measure the partial pressure of CrO2(OH)2(g). It is unclear [16] how the Glusko [17] data was generated. Table 4 shows ΔG f values for compounds and species of interest.
The partial pressures of CrO3(g) and CrO2(OH)2(g) over pure Cr2O3 (activity of 1) were found for conditions of atmospheric pressure, \( P_{{{\text{O}}_{2} }} = 0.20 \), and \( P_{{{\text{H}}_{2} {\text{O}}}} = 0.03 \) (air plus 3% H2O) and are shown in Fig. 2a. Figure 2b and c also show partial pressures of CrO3(g) and CrO2(OH)2(g) over pure Cr2O3, but in these cases for H2O with 180 ppb dissolved O2 (DO). Figure 2b is at atmospheric pressure and Fig. 2c is at 300 atm.

Partial pressures of CrO3(g) and CrO2(OH)2(g) over pure Cr2O3 (activity of 1) for (a) 3% H2O in air at atmospheric pressure, (b) steam with 180 ppb DO at atmospheric pressure, and (c) steam with 180 ppb DO at 300 atm
Since the formation of CrO2(OH)2(g) reduces the total moles of gas, higher pressures increase its partial pressure (Fig. 2b and c). Conversely the formation of CrO3(g) increases the total moles of gas, so higher pressures decrease its partial pressure (Fig. 2b and c).
Experimental Comparison
Experiments best suited to verify the methodology have situations where a steady-state scale thickness (Eq. 4) is quickly established and mass loss due to reactive evaporation of Cr2O3 can be found from mass-change-with-time measurements. Several such tests are described below. Otherwise it would be necessary to separate the effects from scale growth from oxidation and scale thinning from evaporation by the integration of Eq. 3, from which it can be difficult to obtain reliable k p and k e values.
Cyclic-oxidation experiments on Haynes 230 (UNS NO6230) [22] and Inconel 625 (UNS NO6625) were conducted in air in the presence of steam at atmospheric pressure. The compositions of these alloys are given in Table 5. This was designed to examine the adhesion and spallation behavior of protective oxides. The tests consisted of 1-h cycles of heating and cooling (55 min in the furnace and 5 min out of the furnace) in a tube furnace equipped with a programmable slide to raise and lower the samples, Fig. 3. Periodically (between cycles) the samples were removed for mass measurements and then returned for more exposure. The suspension of the samples as shown in Fig. 3 allowed the passage of the gas steam to flow unimpeded across the samples. Water was metered into the bottom of the furnace along with compressed air. Two total gas flow rates were used with rates of 1.9 × 10−3 m/s (38% water vapor and air, by volume) and 7.6 × 10−3 m/s (37% water vapor and air, by volume). The exposure temperature for these tests was 760 °C. There was no evidence of scale spallation during these tests. In similar tests on certain other alloys, for example with TP347HFG, there was evidence of scale spallation from visible scale debris from handling during mass measurements.

Cyclic-oxidation apparatus for testing in atmospheric pressure steam/air mixtures
Note that the reactive-evaporation rates calculated from the preceding methodology are on a CrO2(OH)2(g) basis and the experimental mass losses were from chromia scale evaporation and so are on a Cr2O3(s) basis. To compare the two rates on the same Cr2O3 basis, the following conversion was used:
Results from these tests are shown in Fig. 4 in comparison with predicted slopes from reactive evaporation of Cr2O3(s) to CrO2(OH)2(g) using the Gindorf [21] data for CrO2(OH)2(g). The agreement is close, suggesting that the reactive-evaporation methodology is validated for this case.

Cyclic oxidation of Haynes 230 (UNS NO6230) [22] and Inconel 625 (UNS NO6625) at 760 °C in moist air. Triangle data points for 1.9 × 10−3 m/s and plus data points for 7.6 × 10−4 m/s. Straight solid lines are the predicted slopes (on a Cr2O3 basis) from reactive evaporation of Cr2O3(s) to CrO2(OH)2(g) using the Gindorf [21] data for CrO2(OH)2(g)
Figure 5 is a back-scattered electron micrograph of Haynes 230 (UNS NO6230) after exposure at 760 °C in moist air for 2,000 cycles. It shows a very thin oxide scale, approximately 1 μm thick. Aluminum was internally oxidized.

Micrograph using back-scattered electrons of Haynes 230 (UNS NO6230) after exposure at 760 °C in moist air for 2,000 cycles [22]. The scale is predominately Cr2O3. The bright second phase is W-rich
Table 6 shows a comparison of the experimental slopes (after 200 h for the 1.9 × 10−3 m/s data and after 24 h with the 7.7 × 10−3 m/s data) and the predicted reactive evaporation rates using the three sets of ΔG f data for CrO2(OH)2(g). The evaporation model results using Gindorf ΔG values are in good to excellent agreement with experimental values.
Table 6 also compares experimental results from Asteman et al. [7] for 304L (UNS S30403, composition in Table 5) at 600 °C in 10% H2O in O2. In this case evaporation was evident from 72 to 168 h of exposure. Once again there is good agreement between experiment and evaporation rates calculated using the Gindorf [21] data for CrO2(OH)2(g).
For the applications and experimental designs that follow, the Gindorf [21] data for CrO2(OH)2(g) is used.
Asteman et al. [6] also showed breakaway oxidation for 304L at 600 °C in 40% H2O in O2. For a gas mixture of H2O and O2, the equilibrium constant in Eq. 2 can be used to show that the partial pressure of CrO2(OH)2(g) is at a maximum with \( P_{{{\text{H}}_{2} {\text{O}}}} = \frac{4} {7}P_{{{\text{O}}_{2} }} \)(∼57% H2O). So increasing the water content in Asteman et al. [6] from 10% to 40% increased the evaporation rate, which led to breakaway oxidation due to a depletion of Cr in the scale.
Solid-oxide, Fuel-cell Environments
The environment of the cathode of a SOFC consists of ambient air—thus air containing some fraction of water vapor. For SOFC with metallic interconnects, Cr-gas species from the interconnect can deposit to form Cr2O3 at the triple points at the porous cathode and electrolyte interface (the triple point refers to locations at the interface where the electrolyte, cathode, and gas phases meet). Cell-degradation results from this poisoning of the triple points by Cr2O3. The methodology of Cr evaporation using Eq. 6. for laminar-gas flow should work well in SOFC environments.
Mitigation
Some of the efforts to mitigate Cr-evaporation act to reduce the activity of Cr2O3 in the scale, which reduces the partial pressure of CrO2(OH)2(g) as seen through the equilibrium constant in Eq. 17. The formation of MnCr2O4 spinel should reduce Cr evaporation by a factor of 55 at 700 °C and by a factor of 35 at 800 °C [23]. In this case the reactive-evaporation reaction is:
This strategy is inherent in interconnect alloys that contain relatively large amounts of Mn, such as Crofer 22 APU (DIN EN X1CrTiLa22) with 0.3–0.8 wt% Mn and stainless steel 430 (UNS S43000) with 1.0 wt% Mn (max). The MnCr2O4 spinel has been reported [24–26] on the outer scale for Crofer 22 APU after exposure in moist air at 800 °C.
Other mitigation techniques involve perovskite coatings such as La0.9Sr0.1CrO3 (LSC), La0.9Ca0.1CrO3 (LCaC), and La0.8Sr0.2MnO3 (LSM) [21, 27]. Some, like LSC and LCaC aim to reduce Cr evaporation by lowering the activity of Cr in the oxide. Others, like LSM, seek to eliminate Cr evaporation by removing it from the outer surface of the interconnect. However, even with LSM, Cr diffuses through the coating and subsequently can evaporate [21].
Supercritical-steam-turbine Environments
Development of advanced-steam turbines is underway in much of the world to improve the efficiency of power generation from coal. While much of the alloy development involves improving high-temperature creep strength, steam oxidation resistance is also of importance. Current US Department of Energy research programs are aimed at 60% efficiency from coal generation, which would require increasing the operating conditions to as high as 760 °C and 37.9 MPa (374 atm) for the high-pressure (HP) turbine. Current technology limits operation to about 620 °C. Research is also being directed at intermediate temperatures of 650, 700 and 732 °C. Above 650 °C, it is expected that nickel-base alloys will be required based on creep strength limitations of ferritic and austenitic stainless steels.
Since candidate alloys for this application are all chromia formers, reactive evaporation could be an important degradation mechanism. Representative environments for current and advanced-steam turbines were chosen as: temperatures of 540, 600, 680, 720, 740, and 760 °C, pressures of 163, 197, 306, and 340 atm, steam velocity of 300 m/s (calculated from 60 Hz, 3,600 revolutions per minute, and 0.8 m rotor + blade radius), and characteristic length of 0.05 m. This is turbulent flow, so Eq. 9 was used. The values used for the partial pressure of oxygen were based off of oxygenated feedwater that is typical of once-through supercritical power plants, i.e., dissolved oxygen (DO) of 50–150 ppb and a pH of 8.0–8.5 controlled with ammonia additions [14]. By the time the feedwater enters the boiler, most of the DO has been removed to less than 1 ppb [28]. However, at high temperatures, water undergoes dissociation to O2 and H2 to levels above 1 ppb. To estimate the DO at temperature and pressure, the program FactSage [29] was used to first determine the amount of NH3 required for a pH of 8.25 at 25 °C—34.5 ppb. This agreed well with the reported [14] 20–65 ppb NH3 used for pH control to 8.0–8.5. Next FactSage was used to find the value of \( P_{{{\text{O}}_{2} }} \) for each temperature and pressure combination from water with 34.5 ppb NH3. A minimum of 1 ppb of DO was used for cases where the dissociation pressure of O2 was less than 1 ppb. Output from FactSage included the fugacities of H2O and O2, so these were used in place of \( P_{{{\text{H}}_{2} {\text{O}}}} \) and \( P_{{{\text{O}}_{2} }} \) in Eq. 9. The use of fugacities made only a minor difference because the fugacity adjustments tended to cancel each other out in Eq. 9. Results are shown in Table 7 and Fig. 6.

Predicted evaporation rates in supercritical steam turbine conditions with DO set by the greater of the dissociation of water or 1 ppb, 300 m/s flow rate, and a characteristic length of 0.05 m. Region “A” is typical for conditions in current power plants, “B” is for current advanced power plants, and “C” is the DOE target conditions (see Table 7)
These predicted rates are large compared to the experimental tests at atmospheric pressure and low gas velocities (Table 6). The highest value in Table 7 (for 760 °C and 340 atm) of 5.18 × 10−8 kg/m2/s is equivalent to 0.080 mm per year of solid Cr loss (assumes a metal density of 9 g/cm3 and a conversion to a Cr basis in a manner similar to Eq. 18). This is a large value for metal loss for a component expected to operate many years, and it may be larger if the scale loses enough Cr to become non-protective. Current state-of-the-art steam turbines operate at approximately 600 °C and 31 MPa (306 atm), with a predicted evaporation rate of 1.90 × 10−9 kg/m2/s. Typical subcritical-steam-power plants operate at 538 °C and 16.5 MPa (163 atm), with a predicted evaporation rate of about 4.2 × 10−10 kg/m2/s. These later two cases should have lower evaporation rates in practice because the ferritic–martensitic steels used usually form Fe–Cr spinel outer scales instead of chromia scales. This lowers the activity of chromia in the scale, which lowers the partial pressure of CrO2(OH)2(g) (Eq. 17) and thus lower the evaporation rate.
In a recent review of the current understanding in steam oxidation in power plants, Saunders and McCartney [30] consider and then reject Cr evaporation as an important oxidation mechanism (except for its possible role inside pores and fissures in Cr-migration). At 627 °C, 440 ppb DO and 300 atm, a \( P_{{{\text{CrO}}_{2} ({\text{OH}})_{2} }} \) of about 10−11 atm was reported [30] and was considered too small to be of importance. This value is much lower than is found using Eq. 17 and the Gibbs energy of formation data in Table 4, where values of 1.8 × 10−8 atm (Glusko), 2.5 × 10−7 atm (Gindorf), and 4.8 × 10−6 atm (Glusko) are obtained. The evaporation methodology, using the Gindorf \( \Updelta G_{{f,{\text{CrO}}_{2} ({\text{OH}})_{2} }} \), predicts an evaporation rate of 2.3 × 10−7 kg/m2/s (equivalent to about 0.34 mm per year of solid Cr) for these conditions in a steam turbine. Note that these conditions probably assume a too large of a value of DO, which lead to quite high evaporation rate predictions. An estimate from Table 7, with a DO of 1 ppb, predicts about 2 × 10−9 kg/m2/s (equivalent to about 0.003 mm per year of solid Cr). For the types of materials in current power plants (and the focus of Saunders and McCarney [30]), this evaporation rate would be even lower as the assumption of a pure Cr2O3 scale would not be met—and indeed may be of little importance. These materials include T22, P91, and P92 ferritic steels and 300-series austenitic steels. Scale thicknesses are much larger (i.e., 60–100 μm at 600 °C and 120–240 μm at 650 °C for P92 after 1,000 h [30]) for these materials than for the nickel-base superalloys considered for higher temperatures (i.e., ∼1 μm for Haynes 230 at 760 °C in moist air for 2,000 hourly cycles—Fig. 5. [21]). Equation 4 shows that the eventual steady-state-scale thickness is related to the relative sizes of k p and k e. So evaporation will be of much greater relative importance in describing oxidation mechanisms in nickel-base superalloys with slower-growing-chromia scales.
Mitigation
Mitigation of these predicted high evaporation rates can be speculated upon. One way could be to reduce the activity of Cr in the oxide scale in similar ways as used in SOFC interconnect development. However, other properties of the alloy would have to be maintained. For example, Mn-alloy additions generally reduce high-temperature creep strength in 9–12% Cr ferritic alloys [31]. The adherence of perovskite coatings is not established, and would need to be evaluated.
Turbine-blade coatings might be effective. These could consist of either a coating to lower Cr-oxide activity (as discussed with SOFC coatings) or a thermal-barrier coating (TBC). Porous TBCs would in effect increase the width of the diffusion boundary layer and thus reduce the evaporation rate. When combined with internal-blade cooling, a TBC would also decrease the surface temperature. This would reduce the evaporation rate and also reduce the overall oxidation rate. There is much experience with TBC in gas turbines, but these add significant expense in both capital and maintenance costs.
Finally, the presence of Cr evaporation taking place in the superheater tubes prior to the turbine may to some degree saturate the steam with CrO2(OH)2(g), thereby reducing the driving force for evaporation. The lower steam velocity in the superheater tubes (10–25 m/s is typical [14].) will result in lower evaporation rates than in the steam turbine (∼300 m/s), but there is considerable length of superheater tubing at the high temperature and pressure of the HP turbine that could allow a build up of CrO2(OH)2(g). Evaporation in the superheater may move evaporation issues upstream and reduce them in the turbine.
Laboratory Experimentation
Laboratory corrosion tests generally seek to mimic the process environment as closely as possible. In cases where this is difficult, then one seeks to establish conditions where the corrosion mechanisms are the same. For steam turbines, laboratory tests with the same combination of temperature, pressure, gas velocities, and steam chemistry are extremely difficult and expensive. Therefore tests sacrifice one or more of the conditions—usually pressure and or gas velocity.
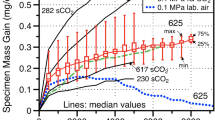
For examining the effects of Cr-evaporation as a corrosion mechanism, laboratory tests may be best served with much-higher-oxygen partial pressures so as to increase the evaporation rate. A comparison of the evaporation rates from Tables 6 and 7 shows that even with air and water vapor mixtures, experimental tests (Table 6) fail to achieve the predicted evaporation rates at high pressures and gas flows (Table 7) by several orders of magnitude. As discussed earlier, for O2 + H2O mixtures, a \( P_{{{\text{H}}_{2} {\text{O}}}} \) equal to \( \frac{4} {7}P_{{{\text{O}}_{2} }} \) (∼57% H2O) should maximize the evaporation rate. The same holds true (albeit at a lower evaporation rate) for air + H2O mixtures with the maximum also at 57% H2O. Laboratory tests in steam at atmospheric pressure will have extremely small evaporation rates due to the low partial pressure of oxygen. This is all illustrated in Fig. 7 for predictions made at 760 °C. In Fig. 7 the advanced steam turbine points are from the A, B, and C data in Table 7 (point C at 760 °C is the direct comparison, points A and B are at lower temperatures). The representative laboratory curves are as a function of the partial pressure of O2 in either O2 + H2O or air + H2O atmospheres. The laboratory curves were all calculated at atmospheric pressure, u = 0.02 m/s and L = 0.02 m. The right-hand-side of the laboratory curves drop sharply as \( P_{{{\text{H}}_{2} {\text{O}}}} \) approaches zero. The right-hand-side of the laboratory curves are limits. Reactive evaporation in drier O2 or drier air would switch at that point from CrO2(OH)2(g) being the dominate gas specie to CrO3(g), and would not drop further with less H2O.

Predicted evaporation rates for steam turbines (A, B, and C) compared with atmospheric pressure laboratory tests (760 °C, u = 0.02 m/s, L = 0.02 m) as a function of the partial pressure of O2 for tests in either O2 + H2O or air + H2O. “A” is typical for conditions in current power plants, “B” is for current advanced power plants, and “C” is the DOE target conditions (see Table 7)
Efforts to improve laboratory tests for higher evaporation rates would include testing in either O2 + H2O or air + H2O at 57% H2O, increasing the gas velocity (k e is proportional to u 1/2), increasing the sample size (k e is proportional to L 1/2), or increasing the total pressure (moving the reaction of Eq. 2 to the right).
Conclusions
A methodology was developed to calculate Cr evaporation rates from Cr2O3 with a flat, planar geometry. As part of this calculation, the interdiffusion coefficient, absolute viscosity, and the Gibbs energy of reaction were determined. The major variables include temperature, total pressure, gas velocity, and gas composition. Experimental verification was done at atmospheric pressure in moist air and moist oxygen. It was concluded that the Gindorf [21] data for \( \Updelta G_{{f,{\text{CrO}}_{2} ({\text{OH}})_{2} }} \) gave a close match with observed evaporation rates, and so was used for further calculations.
The methodology was applied to SOFC conditions for metallic interconnects. Current mitigation techniques for reducing Cr poisoning of the SOFC with Mn additions to the alloy and perovskite coatings were discussed in terms of the methodology.
The methodology was also applied to advanced-steam-turbines conditions. The high velocities and pressures of the advanced steam turbine led to evaporation predictions as high as 5.18 × 10−8 kg/m2/s at 760 °C and 34.5 MPa. This is equivalent to 0.080 mm per year of solid Cr loss. Should this Cr loss be too large to maintain sufficient Cr for a protective-oxide scale, then much higher oxidation rates could result. Chromium evaporation is expected to be an important oxidation mechanism with the types of nickel-base alloys proposed for use above 650 °C in advanced-steam boilers and turbines. Chromium evaporation is of less importance for the ferritic and austenitic alloys used in current steam boilers and turbines due to their relatively large oxidation rates with respect to evaporation rates.
Possible mitigation techniques were discussed, including those used in SOFC metallic interconnects (adding Mn to the alloy and perovskite coatings), thermal barrier coatings (TBC) with or without internal cooling, and partial or full saturation of the steam with CrO2(OH)2(g) from Cr evaporation in the superheater. Of these, Mn additions are limited by reductions in creep strength, and TBC by increased capital and maintenance costs. The partial or full saturation of the steam with CrO2(OH)2(g) from Cr evaporation in the superheater may significantly reduce turbine-blade evaporation, but also move evaporation issues upstream into the superheater.
It was shown that laboratory experiments, with much lower steam velocities and usually much lower total pressure than found in advanced steam turbines, would best reproduce chromium-evaporation behavior with atmospheres that approach either O2 + H2O or air + H2O at 57% H2O instead of with oxygenated steam.
References
K. Hilpert, D. Das, M. Miller, D. H. Peck, and R. Weiß, Journal of the Electrochemical Society 143, 3642 (1996).
S. P. Jiang, J. P. Zhang, and J. Föger, Journal of the Electrochemical Society 147, 3195 (2000).
S. P. Jiang, J. P. Zhang, L. Apateanu, and K. Föger, Journal of the Electrochemical Society 147, 4013 (2000).
F. Chen, E. Sun, J. Yamanis, J. Hawkes, J. Smeggil, S. Warrier, and J.-W. Kim. in Fuel Cells and Energy Storage Systems: Materials, Processing, Manufacturing and Power Management Technologies, Proceedings of Materials Science and Technology (MS&T) 2006: Materials and Systems (2006), Vol. I, p. 303.
H. Asteman, J.-E. Svensson, L.-G. Johansson, and M. Norell, Oxidation of Metals 52, 95 (1999).
H. Asteman, J.-E. Svensson, M. Norell, and L.-G. Johansson, Oxidation of Metals 54, 11 (2000).
D. R. Gaskell, An Introduction to Transport Phenomena in Materials Engineering (Macmillan Publishing, New York, NY, 1992), p. 78 (chapter 2) and p. 569 (chapter 11).
G. H. Geiger and D. R. Poirier, Transport Phenomena in Metallurgy (Addison-Wesley Publishing, Reading, MA, 1973), p. 7 (chapter 1), p. 463 (chapter 13) and p. 529 (chapter 15).
W. A. Tucker and L. H. Nelken, in Handbook of Chemical Property Estimation Methods. W. J. Lyman, W. F. Reehl, and D. H. Rosenblatt, eds. (American Chemical Society, Washington DC, 1990), p. 17.9.
E. N. Fuller, P. D. Schettler, and J. C. Giddings, Industrial and Engineering Chemistry 58, 19 (1966).
S. Veliah, K.-H. Xiang, R. Pandey, J. M. Recio, and J. M. Newsam, Journal of Physical Chemistry B 102, 1126 (1988).
R. B. Bird, W. E. Stewart, and E. D. Lightfoot, Transport Phenomena (John Wiley & Sons, New York, NY, 1960), p. 504 (chapter 16).
J. C. Slattery and R. B. Bird, American Institute of Chemical Engineers Journal 4, 137 (1958).
S. C. Stultz and J. B. Kitto, eds., in Steam, 40th edn. (Babcock & Wilcox, Barberton, OH, 1992), p. 3.8, 42.
C. R. Wilke, Journal of Chemical Physics 18, 517 (1950).
E. J. Opila, Materials Science Forum 461–464, 765 (2004).
Glusko Thermocenter of the Russian Academy of Sciences—Izhorskaya 13/19, 127412 (IVTAN Association, Moscow Russia, 1994).
B. B. Ebbinghaus, Combustion and Flame 93, 119 (1993).
V. S. Yungman, V. A. Medvedev, I. V. Veits, and G. A. Bergman. IVTANTHERMO—A Thermodynamic Database and Software System for the Computer (CRC Press and Begell House, Boca Raton FL, 1993).
A. Roine, HSC Chemistry 5.11 (Outokumpu Research Oy, Pori Finland, 2002).
C. Gindorf, K. Hilpert, and L. Singheiser, in Solid Oxide Fuel Cells (SOFC VII), Proceedings, H. Yokokawa and S. C. Singhal, eds. (Electrochemical Society, Pennington, NJ, 2001), Vol. 2001-16, p. 793.
G. R. Holcomb, M. Ziomek-Moroz, and D. E. Alman, in Proceedings of the 23rd Annual Pittsburgh Coal Conference (2006).
G. R. Holcomb and D. E. Alman, Scripta Materialia 54, 1821 (2006).
J. Hammer, S. Laney, W. Jackson, F. Pettit, and G. Meier, in SECA Annual Workshop and Core Technology Program Peer Review (2005).
Z. Yang, M. S. Walker, P. Singh, J. W. Stevenson, and T. Norby, Journal of the Electrochemical Society 151, B669 (2004).
J. W. Stevenson, Z. G. Yang, P. Singh, and G. H. Meier, in 1st International Conference on Fuel Cell Development and Deployment, Storrs (CT) (2004).
Y. Larring and T. Norby, Journal of the Electrochemical Society 147, 3251 (2000).
S. C. Kung, Private Communication (The Babcock & Wilcox Company, Barberton, OH, 2007).
C. W. Bale, A. D. Pelton, W. T. Thompson, G. Eriksson, K. Hack, P. Chartrand, S. Decterov, J. Melançon, and S. Petersen, Factsage 5.5, Thermfact and GTT-Technologies (2007).
S. R. J. Saunders and L. N. McCartney, Materials Science Forum (2006), Vols. 522–523, p. 119.
F. Masuyama, ISIJ International 41, 612 (2001).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Holcomb, G.R. Calculation of Reactive-evaporation Rates of Chromia. Oxid Met 69, 163–180 (2008). https://doi.org/10.1007/s11085-008-9091-4
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11085-008-9091-4