
Abstract
Plant micro-RNAs (miRNAs) are a distinct class of non-coding, small regulatory RNA molecules emerging as key regulators of growth, development, and stress responses in plants. Therefore, plant miRNAs are looked upon as one of the most potent tools for crop improvement including generation of stress resilient crops. These small elements regulate the gene expression patterns at post-transcriptional as well as translational stages, and are proved to be concomitant with epigenetic regulations in plants during growth and developmental phases, and stress-mediated modulations/adaptations. Recent advancements in sequencing technologies have delivered the high-resolution clusters of sequence information from various organisms, making the miRNA characterization procedure more reliable, rapid, and promising. A typical miRNA investigation usually incorporates preparation of wide-range small RNA library and high-throughput sequencing, followed by the computational analysis of the obtained sequences using a variety of in silico tools. These in silico tools are proving vital in context of identification of miRNAs, their corresponding targets, and inclusive reports on metabolic networks incorporating the identified miRNAs. Current in silico progressions have made available a wide range of tools and databases for miRNA analysis. Therefore, diverse approaches can be seen amongst the researchers which might vary in terms of specificity of analysis, such as plant-, function-, target-specific analysis of miRNAs. Through this review, we discuss the recent developments and current understandings about the plant miRNAs and present the workflow of plant miRNAs including modern high-precision approaches for isolation, identification, target prediction and confirmation, miRNA-target network analysis, and functional characterization of plant miRNAs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The micro-RNAs (miRNAs) are small (19–24 nt), endogenous, non-coding RNAs abundant in eukaryotic organisms. These tiny molecules are key post-transcriptional and translational regulators of gene expression which interact with their respective target-mRNA transcripts in both plants and animals (Devi et al. 2018; Kumar et al. 2018). Despite the similarities, plant and animal miRNAs differ in their biogenesis, interaction sites, and target recognition (Axtell et al. 2011). For instance, the miRNA-producing loci have distinctive genomic arrangements in animal and plant kingdoms. Furthermore, excision of miRNAs from the respective precursor transcripts varies amongst plants and animals, and in various sub-cellular sections. In animals, the miRNA-mediated repression of the gene expression is attained via translational attenuation through (multiple) binding sites for miRNAs within the 3′ UT region of the target gene (Miller and Waterhouse 2005). On the other hand, almost all plant miRNAs direct the mRNA cleavage to regulate the target gene(s) because of the near-perfect base complementation at single sites in the coding regions. These differences, therefore, emphasize the need for plant-specific tools and databases.
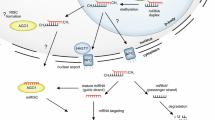
In addition to the vital roles plant miRNAs play in cellular growth and development; there is growing evidence confirming them as master modulators of plant responses to stress factors (Shivaprasad et al. 2012; Ouyang et al. 2014; Shriram et al. 2016; de Vries et al. 2018, 2019; Kumar et al. 2018). Plant miRNAs also contribute in maintaining plant genome stability (Cimini et al. 2019) besides their potential applicability in genome editing (Xu et al. 2019). Owing to this, recent years have seen a sharp increase in investigations related to miRNAs. Plant miRNAs comprise of endogenously conserved entities spread throughout the plant kingdom and are produced via RNAs with imperfectly base-paired hairpin structures. The synthesis of mature plant miRNA involves RNA Pol II-driven miRNA-gene transcription which produces the primary miRNA which is then processed to mature miRNA in two phases (Zhang et al. 2006a); the first phase includes the generation of precursor miRNA from the primary transcripts in the nucleus followed by the formation of mature miRNA from precursor miRNA in the cytoplasm. Both the cleavage steps take place in the nucleus. In this process, the primary miRNAs are converted into the precursor miRNAs in a reaction which is catalyzed by the RNase III like enzyme DICER-LIKE (DCL) along with proteins HYL1 and SERRATE (SE) (Chapman and Carrington 2007; Meyers et al. 2008). The DCL-1 further processes these hairpin-looped structures giving rise to miRNA:miRNA* duplexes (Hannon 2002) that are exported from their site of synthesis by the protein HASTY. These duplexes are then loaded into RNA-induced silencing complex (RISC), after which they unwind by the ARGONAUTE (AGO) proteins (Zhu 2008). Finally, the mature miRNAs lead the RISC to target the complementary mRNAs (reviewed by Shriram et al. 2016; Djami-Tchatchou et al. 2017; Kumar et al. 2018; Treiber et al. 2019).
There are several recent changes in the methods used for identifying plant miRNAs and their respective targets; involved in life processes and responses to environmental stimuli, such as next-generation sequencing (NGS) (Simon et al. 2009), microarray (Yin et al. 2008), RNA ligase-mediated amplification of cDNA ends (RLM-RACE) (Jones-Rhoades et al. 2006), and homology search of public databases (Xu et al. 2019). Small-RNA deep sequencing coupled with degradome analysis has revolutionized this field. In silico tools and techniques are proving fast, efficient, and dependable for assisting the identification and characterization of plant miRNAs (Hannoufa et al. 2018). Wide applications of small RNA-sequencing technology have led to the explosion of miRNA discovery in the last few years. Therefore, miRNA entries in miRBase from different plant species are on a constant rise (Kozomara et al. 2018; Yu et al. 2019). We are presenting herein a review on plant miRNAs, their roles in plant life processes and plant responses to challenging environments, available tools and technologies for their identification and characterization, target prediction, confirmation and validation, and network analysis, with an emphasis on the current status and future challenges.
MiRNAs: emerging regulators of gene expression during plant growth, development, and stress responses
MiRNAs are known to regulate almost all aspects of plant life as they have sufficient diversity to match any RNA entity encoded in the genome, and this regulation is often via mediating the transcription factors (Chen and Rajewsky 2007; Carlsbecker et al. 2010; Huang et al. 2011; Djami-Tchatchou et al. 2017). MiRNAs are known to regulate a broad spectrum of plant developmental processes (reviewed by Singh et al. 2018). Several studies have confirmed the developmental roles of plant miRNAs (Chen 2009; Li and Zhang 2015). High-throughput or deep sequencing (NGS platforms like Illumina) and degradome analysis assisted with advanced in silico or bioinformatics tools and data sets have led to identification of an increasing number of miRNAs involved in plant growth, development, morphogenesis, and responses to external environmental stimuli (Brant and Budak 2018). Eventually, plant miRNAs are emerging tools for crop improvement (Shriram et al. 2016; Budak and Zhang 2017; Khare et al. 2018).
Plant miRNAs have proved their involvement throughout the life-cycle of plant, i.e., from seed germination to seedling establishment, vegetative growth, flowering, and reproduction. Plant seeds exemplify miniature new plants in the form of dormant embryos and miRNAs are reported to be involved in the regulation of seed germination (miR156 and miR172) (Li and Zhang 2015; Das et al. 2018). Similarly, miRs-156, 159, 165, 166, and 319 are involved in regulation of leaf formation (reviwed by Xu et al. 2019). miRNAs are also involved in shoot development including controlling of shoot meristem functions (Liu et al. 2018). Plant miRNAs also play a decisive role in root development (Khan et al. 2011). Almost all aspects of flowering are regulated by the miRNAs (reviewed by Teotia and Tang 2015). All these findings establish that miRNAs are critical for plant cell identity establishment in plant development for accomplishing appropriate morpho-/physiological state (D’Ario et al. 2017).
Similarly, plant miRNAs regulate how plants respond and/or adapt to changing and challenging environments, including countering with abiotic or abiotic stresses (Shivaprasad et al. 2012; de Vries et al. 2015; Khare et al. 2018; Jiang et al. 2018; Xu et al. 2019). Recent reports have confirmed that plant miRNAs re-program the gene expression for modulating the deleterious stress impacts on plants (Brant and Budak 2018).
In spite of the inherently conserved nature, miRNAs (and families thereof) may be specific to plants, development or physiological stage, type of tissues as well as type and exposure to stress condition (Sunkar et al. 2017). Many miRNA candidates are described as stress-responsive in diverse plant species. Such studies were conducted in the context of specific abiotic factors such as salinity (Deng et al. 2015; Eren et al. 2015), drought (Wang et al. 2011; Awasthi et al. 2019), nutrient deficiency (Valdés‐López et al. 2010; Zhao et al. 2012), UV radiations (Jia et al. 2009), heat (Yu et al. 2011; Chen et al. 2012), cold (Zhang et al. 2014; Sun et al. 2015), and heavy metals (Ghosh et al. 2017; Zhou et al. 2019). Likewise, several miRNA candidates, responsive to biotic stresses caused by bacteria (Xie et al. 2017), fungi (Inal et al. 2014), insects (Wu et al. 2017), nematods (Li et al. 2012a), and viruses (Sun et al. 2017), have been reported. Along with in silico analysis, it is helpful to analyze the real-time expression patterns of miRNAs, as it provides actual expression levels. Therefore, miRNAs are analyzed from control as well as stressed plants and differentially expressed miRNAs are identified after normalization and comparison of miRNA expression patterns from plants under stress against their control counterparts.
All such investigations have resulted into deciphered miRNomes of many plants, responsive against specific biotic or abiotic stress factors. However, there is a recent emphasis on identifying miRNAs responsive to multiple stresses, to help develop climate resilient crops (Xu et al. 2019). Though deeper insights are needed on the regulation of expression levels of miRNAs as well as their targets, where in silico tools might be of great support.
Approaches for identification of miRNAs from small RNA transcriptome
Owing to the importance of miRNA in plant growth and adaptability to stressful conditions, several new methods and databases are being developed for identifying miRNAs and further necessary analyses (Backofen et al. 2017). Initially, the miRNA identification process used to involve direct cloning and sequencing; however, the small sizes of miRNAs, their multiple occurrences in genome, and methylation status were the factors making the process tedious (Prabu and Mandal 2010). Progressions in cloning and computational techniques boosted the miRNA identification procedure (Tripathi et al. 2015). The current pipelines for identification of miRNAs are a blend of interdisciplinary paths involving biological and bioinformatics approaches (Akhtar et al. 2016).
Current approaches of miRNA analysis revolve around getting an exact sequence of the miRNA transcriptome, and the revolutionized NGS strategy is acting as a backbone for this. The founding methods in nucleotide sequencing were Sanger’s dideoxy synthesis method (Sanger and Coulson 1975; Sanger et al. 1977) and Maxam–Gilbert’s chemical cleavage method (Maxam and Gilbert 1980). Despite being considered relatively slow, the modified, automated, and commercialized Sanger’s approach is still widely used for many applications. These modifications include incorporation of fluorescent dyes for detection, use of in silico tools for investigating high-throughput data, and efficient use of thermal-cycle sequences which reduces the quantity of input genomic material to be sequenced besides increasing the accuracy and efficiency (Slatko et al. 2018). After automation of Sanger’s method, the requirement of cost-effectiveness for large genome sequencing triggered the evolution of creative methodologies which are referred as second-generation sequencing techniques. We can classify these second-generation methods in two major groups, the sequencing by hybridization, and the sequencing by synthesis.
The Ion Torrent™ Technology directly translates the chemical encoded information into the digital one on a semiconductor chip (Rothberg et al. 2011). The process uses the pH fluctuation during release of hydrogen at the DNA extension event, which are recorded in form of voltage spike. Ion Torrent™ supports a wide array of applications in sequencing including de novo/targeted DNA/RNA sequencing, transcriptome sequencing, and small RNA and miRNA sequencing (Furey 2012).
However, by far, the most prominent player in sequencing is Illumina. This approach is a bridge amplification strategy in which nucleic acid template undergoes repeated amplification on a solid support to create millions of sequence clusters. Currently, wide range of models available for Illumina platform including MiniSeq, MiSeq, NextSeq, NovaSeq and HiSeq provide a differential range of outcomes. If we compare the wide array of sequencing strategies, it can be depicted that currently the Illumina platform seems a best option for its high resolution (Slatko et al. 2018). Illumina HiSeq can provide 600 Gb of sequence yield per run which is significantly higher than the Ion Torrent, PacBio, as well as other sequencing platforms. The comparison between next-generation strategies may also provide the insights regarding the cost-effectiveness and higher accuracy and efficiency of Illumina HiSeq compared to the rest next generation sequencing platforms (Liu et al. 2011; Quail et al. 2012). Sequencing only small RNA transcriptome is proving more suitable because of their specificity towards small RNA transcripts including miRNAs. Therefore, the small RNA-sequencing strategy combined with the depth offered by Illumina NGS has emerged as a method of choice to identify novel miRNA candidates.
Advanced NGS platforms have made the objectives including identification of miRNA fractions, miRNA expression levels, prediction of miRNA precursors, variants, miRNA targets, and other characters achievable with good precision (Motameny et al. 2010; Conesa et al. 2016). The efficacy levels in miRNA studies are increased because of the integrative applications of NGS (Ma et al. 2015). Increased sensitivity and cost-effectiveness of NGS has led to large miRNA data from plants. Strand-specific non-coding RNA transcripts even with low abundance can be analyzed using NGS and different bioinformatics approaches (Turner et al. 2012; Li et al. 2013a; Wu et al. 2014).
Channeling for exact recognition of conserved/novel miRNAs is intricate but a necessary task. The in silico techniques used to identify miRNAs can be grouped into two categories (Kleftogiannis et al. 2013), the comparative approach and the machine learning approaches. Comparative approach incorporates mainly the flow-connected programing, while machine learning approach focuses on predictive modeling constructed from a set of known information. For both the approaches, two common processes are applicable for miRNA identification (a) similarity-based search against existing databases and (b) secondary structure folding (Numnark and Suwannik 2019).
Approaches based on deep-sequencing and available in silico tools are being explored to decipher plant miRNAs involved in biological processes and phenomena. If we consider the current available data of miRNAs from various plants, several miRNAs seem conserved (Zhang et al. 2006a, b). Based on this, many computational approaches are being developed for miRNA identification (Patanun et al. 2013). In silico tools have been exploited for predicting and/or identifying miRNAs from plants including Arabidopsis, Oryza sativa, Zea mays, Lycopersicon esculantum, Setaria italica, Glycine max, Brassica napus, Vitis vinifera, and Gossypium hirsutum (reviewed by Banerjee et al. 2016).
Pre-processing of raw sequence data
A typical workflow for miRNA work comprises the generation of raw sequences from small RNA libraries, alignment of the generated raw data with available data sets, miRNA identification, and prediction of secondary structures of recognized miRNA candidates, miRNA-target prediction, and the putative functional assignment (Liao et al. 2018). Specific in silico tools have been developed for all these steps (Chen et al. 2018). The primary step for miRNA analysis is mapping of the raw sequencing data on the reference genome. However, the raw data also involve adapter (used during library preparation) sequences which can hinder the correct processing and further downstream analysis of actual small RNA data. Hence, the adapter sequences need to be eliminated from the raw data to get the pure transcriptome sequence devoid of any artificial adapter sequence (Lindgreen 2012). Trimming of adapters was first implemented for small RNA sequencing where target fragments typically ranged between 18 and 30 nucleotides, with read length requirement of 36 nt (He et al. 2014). The trimming process differs from removal of contaminant sequences. Trimming-off the adapters is usually associated with next-generation sequencing protocols, where the specific adapter sequences are synthesized by reagent venders. As the pattern of adapters is known, semi-global or end-space free alignment is modeled for adapter removal. These alignments are usually performed via Smith–Waterman or Needleman–Wunsch algorithms (Jiang et al. 2014). Based on such algorithms, some of the tools developed initially involve cutadapt (Martin 2011) and Btrim (Kong 2011) (Table 1a). Trimmomatic (v0.38) (https://www.usadellab.org/cms/?page=trimmomatic, Bolger et al. 2014) is trimming tool which analyze paired- or single-ended NGS data. Depending on the Illumina pipeline used, this tool works with FASTQ (uncompressed or zipped) format. The cutadapt (v1.18) (https://cutadapt.readthedocs.io/en/stable/, Martin 2011) is another tool which can find and eliminate adapter sequences, primers, and polyA tails from high-throughput sequence reads. This tool supports paired-end reads as well as color-space data. To process the short-read sequences generated through NGS, FASTX-Toolkit (https://hannonlab.cshl.edu/fastx_toolkit/, Gordon 2011) is also an option developed by Hannon Lab Public Galaxy, which is a collection of available tools useful in processing of FASTA/FASTQ files. One of the available tools from this collection is FASTQ/A Trimmer, which works for shortening the reads by removing barcodes/noise. For actual removal of adapters/linkers sequences, FASTQ/A Clipper tool is available. The development of newer and more effective tools for serving the same purpose has taken place with the advent of new in silico tools with improved accuracy and efficiency. Some of these tools are SeqPurge (https://github.com/imgag/ngs-bits, Sturm et al. 2016), a very sensitive tool for adapter trimming with a capacity to remove very short adapter sequence (even one base) AdapterRemoval v2 (https://github.com/MikkelSchubert/adapterremoval/, Schubert et al. 2016). All these in silico tools act as filters for complete removal of adapter sequences providing clear and true sequence reads without any partial or complete contamination with adapter sequences.
After getting high-quality reads, the next step includes the mapping of the reads to known databases for identification of known miRNAs and identification of other non-coding/repetitive small RNAs. The mapping of reads for identification of miRNA/other nc-small RNAs/repetitive small RNAs is achieved via exploring the databases Rfam and RepBase (Table 1b). The Rfam database (https://rfam.xfam.org/, Kalvari et al. 2018) is a collection of RNA families with multiple sequence alignments, covariance models, and consensus secondary structures. These Rfam families are frequently assembled via external sources and comprise three broad functional classes, the nc-RNA genes, self-splicing RNAs, and cis-regulatory elements. The first Rfam database was published in 2003 (Griffiths-Jones et al. 2003) as a collection of multiple sequence alignment and covariance models representing families of non-coding RNAs with 25 such families, the current version 14.0 (2019), incorporates information from 3016 families. RepBase (https://www.girinst.org/about/repbase.html, Bao et al. 2015) is also available for repetitive sequences from eukaryotic species and is considered as a reference standard for annotation of repetitive genomic data, having more than 44,000 sequences from over hundred model organisms. This database is necessary for repetitive sequence masking tools such as RepeatMasker (Smit et al. 2015), REPET (Flutre et al. 2011), and CENSOR (Kohany et al. 2006). Application of such repetitive sequence masking process omits the repeats present in the raw sequence. Therefore, after clearing the adapter sequences and repetitive sequences, the raw sequence can now be considered as high-throughput, non-repetitive, small RNA reads, ready to be compared with the known miRNA databases. The processed raw reads can then be subjected for sequence alignment using general purpose aligners such as SOAP2 (Li et al. 2009) or Bowtie2 (Langmead and Salzberg 2012) (Table 1c).
Following alignment, the obtained miRNA counts are required to be normalized for eliminating the data variation. The normalization procedure compares raw sequence counts against known common denominator (McCormick et al. 2011). An effective normalization minimizes the technical and experimental bias without a noise to provide an authentic biological variation across the samples. There are different approaches proposed for miRNA sequence normalization including linear/non-linear scaling and quantile normalization (Garmire and Subramaniam 2012; Dillies et al. 2013). Several normalization methods have been developed including counts per million based normalization (cpm), total count scaling, upper quartile scaling (UQ), Trimmed mean of M-values (TMM), linear regression, quantile-based normalization, and cyclic loess normalization (Tam et al. 2015). One of the widely used methods in NGS for length normalization is reads per kilobase per million mapped reads (RPKM). ERPKM is an upgraded form of RPKM as it follows an effective read length (Li et al. 2015). Expression of raw abundance values in form of transcripts per million (TPM) is also a way to obtain normalized biological expression abundance. It represents the number of specific transcripts found per million transcripts, apparently developed to produce more equivalent results across the sample sequences than RPKM (Wagner et al. 2012). Although the accuracy of miRNA-abundance quantification is affected with respect to the normalization technique, the downstream impacts of application of different aligners for mapping the reads to a reference are unknown. Moreover, the normalization process will not ensure the data without any inconsistencies caused due to original alignment (Tam et al. 2015). The housekeeping genes, the conventional normalizers, display considerable variation with different biological conditions (Lee et al. 2002). Therefore, the global normalization approaches discussed above are preferred due to their greater stability achieved by exploiting the abundance measures of large number of entities. Nevertheless, the availability of large number of profiled and detected miRNAs have provided stable global normalization approach for miRNA sequences (Tiberio et al. 2015). For comprehensive miRNA profiling, the application of suitable aligner followed by use of appropriate miRNA count normalizer is highly recommended.
Once the data are normalized, numerous analysis can be performed on it, most of which falls under differential expression analysis category. Methods for identification of statistically significant, differentially expressed genes may vary as per the number of biological replicates under consideration (McCormick et al. 2011). There are different methods implemented for differential expression analysis. For instance, Bioconductor (https://bioconductor.org/) offers a tool as edgeR which identifies variable gene expression patterns in biological replicates via over dispersed Poisson model (Robinson et al. 2009). This tool requires raw sequence counts as well the total library counts as an input, and hence, pre-normalized data are not required. DESeq2 is yet another impressive tool provided by Bioconductor which uses negative binomial generalized linear model for testing differential gene expression. This tool also requires no pre-normalized data as DESeq2 model internally corrects the library size (Love et al. 2014). For differential expression analysis, another approach is provided by Fahlgren et al. (2009), which is an adaptation of significance analysis of microarrays (SAM) to sequencing data, called as SAM-Seq. In this approach, the differential expression score is calculated amongst the samples by considering the average gene abundance in every replicate for sRNA under consideration along with the standard deviations. It also minimizes the data variation via integrating a small, positive constant.
Only the correct quantification of differential miRNA expression along with statistical significance will provide a clear scenario of miRNA-driven biological differences in the samples (compared as control vs test). The information regarding miRNA:mRNA interaction will be meaningless if miRNA-abundance changes between control and test are not evaluated correctly. Therefore, application of tools such as DESeq2 or edgeR is must for better results.
Searching hits in miRNA databases
After pre-processing, the conserved miRNA candidates from the raw sequences are then compared with already established databases for miRNAs (Table 1d). Amongst the miRNA databases, the miRBase (v22, https://www.mirbase.org/index.shtml, Kozomara et al. 2018) is the preferred miRNA database which delivers integrated interfaces to an inclusive miRNA sequence data, annotation, and predicted target genes. The database initially published as ‘The microRNA Registry’ (Griffiths-Jones 2004) with 506 miRNA entries from six organisms, at present, (v22) has miRNAs from 271 organisms representing 38,589 hairpin precursors along with 48,860 mature miRNAs. These data are collected from 1493 small RNA sequence data sets and have mapped 5.5 billion reads to miRNA sequences. Each entry in this database denotes a predicted hairpin segment of miRNA transcript (referred as ‘mir’) along with location and mature miRNA sequence (referred as ‘miR’). Retrieval of this information is possible via providing name, keywords, annotations, or references for particular miRNA. This database is extensively applied in most of the miRNA-related investigations and considered as a central repository for miRNA sequences, nomenclature annotation, and target prediction.
Apart from the miRBase, there are some plant-specific miRNA databases (Table 1e). For instance, at Université du Québec à Montréal, Laboratoire Bioinformatique, a new wheat miRNA database as Wheat MicroRNA Portal (WMP) has been developed (https://wheat.bioinfo.uqam.ca/index.php, Remita et al. 2016). This encompasses ten small RNA libraries obtained from the plant samples subjected to different abiotic stresses with a compilation of published wheat miRNAs (total 5036). The miRNAs are available from aerial and root tissues of wheat under specific conditions including developmental phases (vegetative/reproductive), salinity, cold, and aluminum stresses. Another web resource, Soybean knowledge base (SoyKB) (https://soykb.org/, Joshi et al. 2017) is based on translational genomics of soybean. It is a comprehensive web resource specific to soybean, which also provides data regarding miRNAs and sRNAs. This database allows soybean-specific miRNA search against query in form of miRNA ID, target gene or sequence. Similarly, Fei et al. (2010) developed a tomato functional genomics database (TFGD) (https://ted.bti.cornell.edu/cgi-bin/TFGD/sRNA/miRNA.cgi) which incorporates tomato expression database along with metabolite profiles and large-scale data sets of tomato small RNAs. The TFGD harbors a list of putative miRNAs from tomato and the miRNAs can be searched on the basis of miRNA ID or name from this database along with the target prediction tool. The sorghum-specific multidimensional network analysis database, referred to as Sorghum Functional Genomics Database (SorghumFDB) (https://structuralbiology.cau.edu.cn/sorghum/miRNA.php), is developed at Zhen Su’s lab, China Agriculture University (Tian et al. 2016). One feature of this database includes integration of miRNAs from other miRNA databases such as miRBase, PMRD, and PNRD. The list provides miRNAs, respective chromosome number with base-position and nature of strand (positive or negative). The Cereal Small RNAs Database (CSRDB) (https://sundarlab.ucdavis.edu/smrnas/, Johnson et al. 2006) encompasses miRNA knowledge base consisting exhaustive list of miRNAs present in rice and maize. This database encompasses small RNAs from mitochondria as well as chloroplast. Li et al. (2012a, b) have designed SoMART (Solanaceae miRNA/tasiRNA Analysis Resource and Tools) webserver (https://bakerlab.berkeley.edu/somart-webserver-mirna-sirna-analysis), a suite of providing the databases and computational tools for identification and analysis of small RNAs from Solanaceae family members. This web server can be used for identification of miRNAs targeting the gene-of-interest, detection of miRNA/siRNA cleavage transcripts, identification of miRNA progenitors, and target confirmation. Along with all these specific web-based resources, the whole-genome databases, available for many plants now can provide search tools for identification of specific sequences including miRNAs.
Besides, there are some other miRNA identification pipelines incorporating miRNA-related data from more than one plant (Table 1d). Plant miRNA database (PMRD) (https://bioinformatics.cau.edu.cn/PMRD/, Zhang et al. 2010) is a database integrating available evidence regarding miRNA sequences, their target genes, secondary structures, and expression profiles along with the genome browsers, an updated version was released in 2014 as plant non-coding RNA database (PNDB) (https://structuralbiology.cau.edu.cn/PNRD/, Yi et al. 2015). Current version of PNRD consists of 28,214 non-coding RNA entries from 166 species. This database also contains functional search and analytical tools such as non-coding RNA keyword search, the functional search based on the literature, target gene search along with miRNA prediction toolkit, coding potential calculator toolkit, and BLAST tool. The miRNEST2.0 (https://rhesus.amu.edu.pl/mirnest/copy/, Szcześniak and Makałowska 2014) is an integrative miRNA resources from several plant species for browsing different miRNA candidates, besides in silico tools for deep-sequence predictions and miRNA genes. For identification and quantification of the known/novel miRNAs from raw small RNA sequences, An et al. (2012) developed an integrated miRNA identification tool known as miRDeep* (https://www.australianprostatecentre.org/research/software/mirdeep-star). It is a user-friendly graphic interphase which accepts FASTQ or SAM/BAM formats for miRNA analysis. It is principally a java-coded in silico application. There are development of variants for this database, one of the latest of such kind is miRDeep-P2 (https://sourceforge.net/projects/mirdp2/, Kuang et al. 2018). Even though there are continuous progressions in the development of in silico analytical tools, there is always a challenge of minimizing the false-positive predictions as well as analytical time. The current version of miRDeep-P2 provides superiority in speed and precision because of the updated annotation criteria and new scoring system, and, therefore, may serve as a method of choice.
Assigning appropriate name to the newly identified miRNA member is essential. The current miRNA nomenclature system implies primary forms (as pri-miRNA), miRNA precursors (as pre-miRNA) along with name of gene or corresponding number assigned. The miRNA annotation ending with -3p and -5p signifies the 3′ and 5′ arm mature sequence of that miRNA (Desvignes et al. 2015). The perfect nomenclature is vital in miRNA-related investigations as the exact annotation of identified miRNAs ensures comparability of the results obtained.
Secondary structure prediction for miRNAs
As miRNAs are established as key modulators of plant responses to environmental stress and pathogen invasions/disease environment, prediction of new potential miRNA species involved during such an interaction is of high interest. The ab initio predictions are usually helpful in this, as they use intrinsic properties of an actual sequence for prediction (Tav et al. 2016). These ab initio tools predict the miRNA precursors having characteristic hairpin structure, which plays a crucial role during biogenesis of mature miRNAs. Hence, prediction of secondary structure by computational tools is always an area of interest for researchers (Li et al. 2010). These secondary structures may suggest the presence of lower loop/bulge or unstructured region, actual miRNA sequence, upper junction region, and upper/terminal loop region, giving rise to the hairpin or stem-loop with single- or double-stranded regions (Meyers et al. 2010). Presence of bulge at particular position (upper, middle, or lower regions of hairpin) is a characteristic feature of the predicted secondary structure.
Lower stem region is crucial in pri-/pre-miRNA processing as initial cleavage takes place at ~ 15 nucleotide position from the single-stranded (unpaired) lower stem to form a mature miRNA. The presence or absence of a big bulge in this region decides the fate of the initial cleavage (Bologna et al. 2013). Thus, this bulge above the unpaired region at lower region of hairpin describes an optimum assembly certifying very precise and effective processing of plant miRNA precursors (Meyers et al. 2010). On the other hand, bulge or terminal loop in an uppermost region of hairpin along with upper junction region is important in miRNA processing as these are the sites for second Dicer-mediated cleavage (Mateos et al. 2010; Werner et al. 2010). Owing to the importance, the input miRNA sequences, hence, are subjected to the search and analysis for bulges/internal loops. In most cases, the compact hairpin precursor displaying minimum-free energy is finalized from each input sequence (Miskiewicz et al. 2017). Hence, the presence or absence of loops at upper/middle/lower position and minimum-free energy requirements are critical for optimum secondary structure predictions.
The process of miRNA secondary structure prediction is boosted by the development of several in silico tools presented in Table 1f. The miRFinder (https://www.bioinformatics.org/mirfinder/, Huang et al. 2007) is a pre-miRNA prediction tool which estimates the secondary structure by an initial search of hairpin-candidate sequences, leaving the non-robust structures. This tool represents the output using symbols to show a state of paired/unpaired, insertion/deletion, and a bulging in the hairpin. On a similar line, ab initio approach for miRNA prediction in the genome is also implemented in another in silico tool, the miRNAFold (https://evryrna.ibisc.univ-evry.fr/miRNAFold, Tav et al. 2016). It is dedicated to identification of miRNA precursor in a highly sensitive and fast way which initiates the searching by approximation of miRNA hairpin assembly. It is followed by reconstituting the pre-miRNA structures and allows a considerable reduction in theoretical complexity and search time. This tool gives output in form of well-presented structure with sequence positions and hairpin region marked with different colors (stem: green, loops/bulges: brown, terminal loop: blue). Another webserver frequently applied for secondary structure predictions is mfold (https://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form, Zuker 2003). This server provides easy access to nucleic acid folding and hybridization. It uses universally available web graphical user interphases (GUIs). The folding event of sequences is provided with detailed outputs as energy dot plots, single-stranded frequency plots, and structure plots with reliability information. This and one more web server are amalgamated for providing a new service as UNAFold (https://unafold.rna.albany.edu/, Markham and Zuker 2008), an expansion of existing algorithms and software to gain enhanced analysis. Lorenz et al. (2011) developed The ViennaRNA Package (https://www.tbi.univie.ac.at/RNA/). This software comprises of C-code library and many programs for predicting and comparing RNA secondary structures, where the energy minimization is the method of choice for secondary structure prediction. Three dynamic algorithms are provided under this package, first the minimal free algorithm to get structure, second, the partition function algorithm for calculating the base pair probabilities (thermodynamic ensemble), and third being sub-optimal folding algorithm to generate sub-optimal structures for provided energy. This package also provides an algorithm for inverse folding (designing sequence with pre-determined secondary structure). Similarly, RNAstructure (v6.1) (https://rna.urmc.rochester.edu/RNAstructure.html, Mathews 2014) is also a complete package for secondary structure prediction and investigation. Along with algorithms for structural predictions, this facility also provides probabilities for base pairing, which predicts bimolecular structures and equilibrium binding affinity of nucleotide to a structured RNA target (Mathews 2014).
As discussed earlier, huge raw small RNA sequence data are generated during the miRNA analysis and there is a need for separating real miRNA precursors from non-miRNA hairpins. To achieve this sorting, so far the machine learning approaches seem most promising. Though machine learning requires the use of training data, which is tricky because of the inequality in positive (no. of miRNA) and negative (number of non-miRNA) data leading lesser efficiency.
Secondary structure of miRNAs is crucial intermediary levels providing the vital information about miRNAs which condenses dominating part of folding energy. These structures are evolutionarily well conserved and frequently serve as an explanation of respective experimental findings. As discussed above various in silico tools are available for secondary structure predictions of miRNAs. Besides, predicting the probable position of plant miRNA is also important and can be obtained using programs like MaturePred (https://39.106.10.28/maturepred/, Xuan et al. 2011) which works on machine learning methods. This web server predicts the start position of miRNA for its pre-miRNA candidature. For an optimistic progression in miRNA analysis, there is a persistent necessity of new methods for positional predictions of miRNAs for aiding discovery of new pre-miRNA species (Szwacka et al. 2018).
mRNA–miRNA interaction analysis
The modulation of gene expression through miRNAs revolves around and depends on the alteration of translation efficiency and/or mRNA targets’ stability. It is assumed that wide share of total protein-encoding RNAs are subjected under miRNA-mediated control, but identification of such miRNA–mRNA interactions necessitates intricate procedures or prior knowledge of target mRNAs (Afonso-Grunz and Müller 2015). However, experimental evidence for these miRNA–mRNA interactions is infrequent and in silico analysis based on sequence complementarity, suffers from a high degree of false-positive outcomes. Hence, a comprehensive analysis of interactions of small non-coding RNAs and mRNAs is needed for better knowledge and prediction of multifaceted dynamics of miRNA-mediated gene regulation (Afonso-Grunz and Müller 2015). Recent approaches show the application of cross-linking and ARGONAUTE immuno-precipitation coupled with high-throughput CLIP-Seq and degradome sequencing, also known as parallel analysis of RNA ends (PARE) for target isolation. These methods have curtailed the frequency of false-positive results, besides reducing the search time. The improved frequency of application of CLIP-Seq and PARE, however, urges the need of integrated databases and tools to assist the analysis and annotation of these interactive miRNA/mRNA entities, and available in silico tools for this are presented in Table 1g. starBase (https://starbase.sysu.edu.cn/), a database to explore miRNA–mRNA interaction maps generated from CLIP-Seq and/or PARE has been developed by Yang et al. (2010). This database helps in inclusive annotation and searching of miRNA and respective targets. It also contains information regarding Ago-binding and cleavage sites. As an extension of this database, starBase v2.0 is launched by Li et al. (2013b) for understanding the regulatory interactions between multiple classes of RNAs. This updated version provides complete and experimentally proven information about miRNA–mRNA interactions.
Considering the significance of information on miRNA–mRNA interactions, several such interactions have been experimentally discovered and confirmed. However, plenty of such knowledge is available in fragmented form across thousands of manuscripts and raw sequence data sets. Hence, to index the published (and experimentally authenticated) miRNA–gene interaction events, DIANA-TasBase (v7.0) (https://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index) was generated by Vlachos et al. (2014). This database enables researchers to recognize negative/positive experimental outcomes along with implemented methods and experimental conditions. This version also delivers advanced information such as binding site locations (experimental and in silico), primer sequences for cloning.
Some resources are plant specific as well; for instance, a rice miRNA:mRNA interaction resource (ARMOUR) (https://armour.icgeb.trieste.it/login) was launched recently (Sanan-Mishra et al. 2018). This database comprises experimentally validated expression patterns from seven different indica rice cultivars under variable environmental conditions and developmental phases. It is flexible in terms of query as it uses different means like recognized gene–gene ontology/KEGG identifiers, besides permitting to analyze fly-fold-change.
Target prediction for miRNAs
Experimental evidence has suggested that many TFs, and development-related genes, are target mRNAs for the miRNAs. Interestingly, most of the algorithms developed initially for target prediction were animal miRNA-centric (Lim et al. 2003; Li et al. 2010), but there are variations between plant and animal miRNAs. For instance, to accomplish a miRNA–mRNA interaction in animal system, loose complementarity with target at approximately first 8 nucleotides of the miRNA is necessary, while a plant system requires complete mature miRNA sequence in near-perfect alignment with its respective target mRNA. Also, inhibition of expression of target gene(s) is achieved by animal miRNAs at the translational level, while plant miRNAs achieve the same via direct cleavage of target transcript (Lim et al. 2003; Li et al. 2010) along with some reports of translational repression (Ouyang et al. 2014). Animal miRNAs identify the 3′-UTR region of the corresponding target mRNA, but plant miRNA requires no such preference (Lim et al. 2003; Li et al. 2010; Dai and Zhao 2011). These dissimilarities demanded the need to develop separate recognition process for miRNA targets in plants (Dai and Zhao 2011). The stable state of miRNA–mRNA duplex, which depends on the complementarity between target sites and respective miRNAs, is a decisive factor during target gene analysis. Another important measure to filter out the miRNA target is the conserved nature of the target sequence. Orthologous search for such conserved region may facilitate the target prediction process. Multiple occurrences of the target sequence are also considered while targeting prediction (Dai et al. 2010).
A region of first ~ 6–8 nucleotides within the structure of a mature miRNA initiating at 5′ and counting towards 3′ is known as a seed region, a highly conserved and crucial region which binds to the complementary target in the 3′ UTR. A slight change in nucleotide sequence of seed region may result into the miRNA dysfunctioning and subsequent variation in the expression pattern of respective miRNA target (Borel and Antonarakis 2008). Overall, a higher degree of conservation is found in seed region than others (Lewis et al. 2003). The characterization of conserved nature of these seed sites at 3′ UTR indicates the fact that single miRNA might regulate several genes (Lewis et al. 2003; Brennecke et al. 2005; Krek et al. 2005).
The stability of miRNA:mRNA interaction is measured in terms of free energy (also referred as hybridization energy), which is calculated by considering high and low free energies of terminal loop region and stem region, respectively (Yue et al. 2009). Following the transcription process, the mRNA is usually in a secondary structure form which may hinder the miRNAs’ accession to its binding site on target-mRNA (Mahen et al. 2010). The site accessibility is granted to miRNA after initial binding of miRNA to short, accessible region on mRNA, followed by unfolding of secondary mRNA structure and complete miRNA:mRNA hybridization (Long et al. 2007). Therefore, to check the probability of mRNA being target of a particular miRNA, the energy essential for making site available to miRNA is considered. Collectively, free energy of the system indicates its stability and correctness in the target prediction. As, direct quantification of the free energy is tough, the variations in free energy levels during the interaction are considered. The negative difference in free energy results in a system with increased stability (Peterson et al. 2014).
Continuous developments in in silico tools for miRNA-target predictions have highlighted the major biological information, namely, seed region complementarity and conservation, free energy, and site accessibility, as factors to be considered for prediction of miRNA targets (Du and Zamore 2005). Along with these, physicochemical properties of miRNA and its respective targets’ interaction should be given a due consideration. The accessibility of miRNA-target site is an effective structural feature for target predictions (Dai et al. 2010). Keeping these benchmarks in considerations, various in silico tools have been developed for target predictions (Table 1h).
Dai and Zhao (2011) developed an analytical server psRNATarget (https://plantgrn.noble.org/psRNATarget/) for predicting the plant miRNA targets. This server provides two important analytical utilities, namely, a reverse complementary matching of small RNAs with their target transcripts via the proven scoring system and unpaired energy calculation-mediated target-site accessibility evaluations. The current updated version of psRNATarget (2017 release, Dai et al. 2018) provides enhanced analytical efficiency via the development of a new set of scoring systems. Both canonical and non-canonical target search is possible via a customizable process in this server. It is compelling, clear, and user-friendly interphase which presents the data concisely and clearly. Another tool for plant miRNA prediction, Target-align (https://www.leonxie.com/targetAlign.php, Xie and Zhang 2010) is developed using Smith–Waterman-like algorithm. This dynamic program contains complementarity based score matrix system to trace the optimal local alignments. In this tool, maximum mismatches/consecutive mismatches are used for clarifying optimal local alignments. This tool can also identify multi-targets and non-cleaved targets. Another web-based integrated in silico system, miRU (https://bioinfo3.noble.org/miRU.htm, Zhang 2005), is used for plant miRNA-target prediction, in case several sequences needs to be analyzed. After providing a mature miRNA sequence, possible complementary target sites with acceptable target mismatches are searched within the system. Evolutionary conservation is also one of the considered criteria for target predictions. The TAPIR (https://bioinformatics.psb.ugent.be/webtools/tapir, Bonnet et al. 2010) webserver for plant miRNA-target prediction uses two different search engines, the FASTA and RNAhybrid, providing more precision to give rich output along with free energy calculations and ratios. This server also provides possibilities for probable target mimics. This feature is helpful as target mimics representing large loop bearing miRNA-target duplexes which are undetectable by the traditional methods. TarHunter (https://github.com/XMaBio, Ma et al. 2017) is one more tool for miRNA-target prediction which takes target mimics into account. Since, endogenous target mimics along with non-canonical targets play a crucial role for particular miRNAs, this tool has a mandatory filter for cross-speciation to find out probable mimics, which ultimately improves the precision of the target prediction.
Another tool for plant miRNA and target identification, C-mii (https://www.biotec.or.th/isl/c-mii), was developed by Numnark et al. (2012). It is a ready-made software package which provides some distinct features. It is user friendly and all in one database with easy graphical user interphase for the user. It also provides a comprehensive view of identified miRNA/targets with info-graphics and grouping information and supports multi-project as well as multi-thread management. Kakrana et al. (2014) developed a small RNA-PARE target analyzer (sPARTA) (https://github.com/atulkakrana/sPARTA/releases) which utilizes high-throughput sequence data to profile a genome-wide cleavage product. A built-in parallelized genome-wide target prediction module, called as miRferno, is also merged with this analyzer. This multi-core server achieves two-dimensional parallelization for maintenance of low-memory footprints for complete genome analysis. Another bioinformatics pipeline referred as miRTour (https://bio2server.bioinfo.uni-plovdiv.bg/miRTour/, Milev et al. 2011) presents an automated steps for miRNA similarity searches, pre-miRNA selection, target prediction, and annotation. The experimentally authenticated interactions between the miRNAs and their respective targets have been compiled in miRTarBase (https://mirtarbase.mbc.nctu.edu.tw/php/index.php, Chou et al. 2017). The 2018 release of this database contains 422,517 curated interactions between miRNA and their targets from 4076 miRNAs and 23,054 target genes, compiled from 8500 reports. This version also provides downloadable information about target sites validated by reporter assays.
Interestingly, Srivastava et al. (2014) compared the performance of different plant miRNA-target prediction tools. The authors assessed the performance of eleven computational tools used for genome-wide identification of targets in Arabidopsis and other plants using optimized score cutoffs. Targetfinder was found most proficient with 89% precision (exactness in prediction) and 97% ‘recall’ (sensitivity) in prediction of true-positive targets in Arabidopsis. Overall, authors concluded that Targetfinder alone or with psRNATarget/Tapirhybrid proved best for the prediction of miRNA targets (Srivastava et al. 2014).
Gene ontology and pathway analysis of identified miRNA targets
Gene ontology of the identified targets is preferred to get a good understanding of the functions of identified miRNAs and their targets, followed by the analysis of respective pathways in which the target genes are involved. In silico tools for such analysis are summarized in Table 1i. The Gene Ontology resource (https://geneontology.org/, Gene Ontology Consortium 2014) is a comprehensive and widely used knowledge portal. This knowledge base is constructed in ontology form by stratifying classes of gene functions having specific relation with each other. The gene ontology facilitates systematized and accurate vocabularies and classification of domains in the considered biological process, cellular mechanisms, and gene functions. Another tool designed on a similar line (enabling gene ontology) is Balst2GO (https://www.blast2go.com/, Conesa et al. 2005). This tool enables ontology-based data mining from raw sequences without requiring the previous annotations. It combines similarity-based searches for annotation with statistical analysis, with highlighted visualization on direct graphical representations. Apart from these two plug-ins, BiNGO (A Biological Network Gene Ontology tool, https://www.psb.ugent.be/cbd/papers/BiNGO/Home.html, Maere et al. 2005) (v3.0.3), also a Java-based tool, defines that which gene ontology categories are statistically over-represented in the biological network being analyzed. This app maps the pre-dominant functions of considered gene/s on ontological hierarchy and outputs these maps on interaction networks. The WEGO (Web Gene Ontology Annotation Plot, https://wego.genomics.org.cn/, Ye et al. 2018) (v2.0) is simple and useful tool for visualization, comparison and graphical representation of gene ontology annotation results. This tool analyzes multiple files at one operation with harboring data for nine different organisms. The agriGO (https://systemsbiology.cau.edu.cn/agriGOv2/, Tian et al. 2017) platform specifically focuses on gene ontology enrichment analysis of crop species. The current version of agriGO accumulates information from 394 species and 865 datatypes. The agriGO provides the user a high computational efficiency because of the batch analysis, p-value distribution, and greater user-friendliness. Additional visualization features are add-ons for this platform including a cross comparison of singular enrichment analysis (SEACOMPARE), direct acyclic graph (DAG), and scatter plots. ClueGO (https://www.ici.upmc.fr/cluego/cluegoDownload.shtml, Bindea et al. 2009) is yet another tool for interpreting the gene ontology as well as pathway annotation networks. The main feature of this tool includes the integration of gene ontology terms and KEGG/BioCarta pathways which results in the creation of functionally organized GO/pathway term network. Gene set enrichment analysis (GSEA) is one of the preferred ways for interpreting genes via calculating the overlaps with already defined gene sets. Using this method, Yi et al. (2013) developed a webserver named PlantGSEA (https://structuralbiology.cau.edu.cn/PlantGSEA) which focuses on plants. This server provides 20,290 defined gene sets from various resources. A rapid genome-wide functional annotation of large set of sequences via fast orthology assignment can be accomplished through a tool, known as eggNOG-mapper (https://eggnog-mapper.embl.de/, Huerta-Cepas et al. 2017). This tool validates the ontology predictions against BLAST and InterProScan, two widely used homology-based approaches. Functional annotation in eggNOG-mapper is done using pre-computed clusters and phylogenies from the eggNOG database. This tool is rapid, with higher proteome coverage and precision in ontology prediction than the other tools like BLAST and InterProScan (Huerta-Cepas et al. 2017).
In modern plant science studies, long with GO, MapMan (https://mapman.gabipd.org/, Thimm et al. 2004) is also a widely used ontology specifically for a plant. MapMan was initially tailored only for Arabidopsis, but later was extended to other plants such as maize, tomato, potato, and medicago (Klie and Nikoloski 2012). It is a user-driven tool which presents large data sets as a diagrammatic representation of metabolic pathways. In this tool, different SCAVENGER modules assign the hierarchical classes of measured parameters (such as transcripts or metabolites), whereas IMAGEANNOTATOR module uses this classified information to organize a diagrammatic display of experimental data as per the user’s need. As both MapMan and GO offer alternative ways for ontology-driven analysis of raw sequence data, the choice of tool totally depends on the requirements of the researcher. The comparative account on the choice between MapMan and GO for plant gene function prediction is provided excellently by Klie and Nikoloski (2012) and the readers are advised to refer it.
Cytoscape (https://cytoscape.org/cy3.html, Su et al. 2014) is one of the most prevalent, open-source in silico tool implemented for visual investigation of biological networks incorporating genes, proteins, and different interactions. It provides the researchers with a versatile and interactive conception of complex metabolic inter-connections, along with experimental details and annotations. It offers significant functionality of loading, visualizing, searching, filtering, and saving the network data. This function is further improved because of the availability of hundreds of apps to address particular needs, e.g., GeneMANIA (Montojo et al. 2014) to construct a network of composite gene–gene functional interactions or KEGGscape (Nishida et al. 2014) for replicating hand-drawn pathway diagrams with details in Cytoscape). Kyoto Encyclopedia of Gene and Genomes (KEGG, https://www.genome.jp/kegg/, Kanehisa and Goto 2000) is considered as the main knowledge base which provides a systematic analysis of gene function and helps in linking the genomic interactions with a high degree of functional information.
Functional characterization and validation of plant miRNAs
One of the easiest approaches to validate the assigned target for a particular miRNA is profiling of the expression patterns and probable target(s) of the miRNAs by quantitative PCR (qPCR). In a typical state, when miRNA expression increases the quantity for their respective target decreases and vice versa. However, this approach is not useful for studying translational repression. Additionally, only expression patterns cannot be considered to understand the regulatory mechanisms between transcript regulated by many miRNAs or single miRNA regulating many transcripts (Devi et al. 2018). The cloning-based approach such as RLM-RACE and PolyA polymerase-mediated (PPM)-RACE can be implemented for recognition of direct cleavage products of miRNA targets (Wang and Fang 2015). These approaches can separate the cleavage events by different miRNAs. The cleaved transcripts are added either by oligonucleotide (for RLM-RACE) or by poly-A tail (for PPM RACE), followed by reverse transcriptase-mediated transcription, cloning, and then, the products are sequenced. A cleavage product of miRNA targets can also be detected via PARE, as described in previous sections. This high-throughput analytical approach is also referred to as degradome sequencing in which initial processing is equivalent to that of the RLM RACE (German et al. 2009). The exact target-miRNA interaction can be methodically validated using HITS-CLIP approach (Chi et al. 2009) and photoactivable ribonucleoside-enhanced cross-linking and immuno-precipitation (PAR-CLIP) (Hafner et al. 2010). Both these approaches principally involve linking of miRNA target to AGO protein followed by precipitation of epitope-tagged AGO and sequencing (Thomson et al. 2011; Hafner et al. 2012). For validation of miRNA targets in transgenics, reporter gene assays utilizing luciferase or green fluorescent proteins can be used. Luciferase activities from different species are also utilized where luciferase from one species possesses miRNA binding site which is subsequently cleaved and another counterpart devoid of the cleavage site, hence acting as an internal control (Moyle et al. 2017). These methods functionally validate the miRNA-target pairs that can be placed appropriately in the gene-interactive networks to attain the comprehensive knowledge of the miRNA-mediated regulatory event. A workflow for plant identification and characterization of miRNA from plants, their target prediction and confirmation, and functional validation is presented in Fig. 1.

A workflow for plant miRNA identification, characterization, target prediction and confirmation, and their functional validation
Conclusion and future perspectives
Plant miRNA research has witnessed an upward swing in the past few years, owing to the key roles played by these non-coding RNA species in plant life processes and their adjustments to the challenging environmental conditions. However, the research community should expand its base and should identify miRNAs from the non-model and lesser-explored plants. Similarly, genome-wide expression analysis of plant miRNA families is needed.
Technological advancements, particularly in high-throughput small RNA sequencing and degradome analysis along with advent of modern computational or in silico tools and databases useful for identifying the miRNAs and their respective targets, have revolutionized this field. The NGS approaches available at reduced costs with high precision are enabling researchers to fast and reliable miRNA discovery from the plants even if their genome sequences are unavailable. Computational tools and algorithms thereof are playing significant roles in increasing the precision in identification of miRNAs and their targets. The interplay between miRNAs and their targets is still a field to be explored fully. However, experimental approaches coupled with the use of bioinformatics tools and databases have the potential to unfold the mysteries of these tiny but powerful molecules and their roles in plant cellular events.
Author contributions statement
XZ and VK conceived the idea. XZ, VK and TK authored the manuscript. All three authors contributed substantially.
References
Afonso-Grunz F, Müller S (2015) Principles of miRNA–mRNA interactions: beyond sequence complementarity. Cell Mol Life Sci 72:3127–3141. https://doi.org/10.1007/s00018-015-1922-2
Akhtar MM, Micolucci L, Islam MS, Olivieri F, Procopio AD (2016) Bioinformatics tools for microRNA dissection. Nucleic Acids Res 44:24–44. https://doi.org/10.1093/nar/gkv1221
An J, Lai J, Lehman ML, Nelson CC (2012) miRDeep*: an integrated application tool for miRNA identification from RNA sequencing data. Nucleic Acids Res 41:727–737. https://doi.org/10.1093/nar/gks1187
Awasthi JP, Chandra T, Mishra S, Parmar S, Shaw BP, Nilawe PD, Chauhan NK, Sahoo S, Panda SK (2019) Identification and characterization of drought responsive miRNAs in a drought tolerant upland rice cultivar KMJ 1–12-3. Plant Physiol Biochem 137:62–74. https://doi.org/10.1016/j.plaphy.2019.01.029
Axtell MJ, Westholm JO, Lai EC (2011) Vive la différence: biogenesis and evolution of microRNAs in plants and animals. Genome Biol 12:221
Backofen R, Engelhardt J, Erxleben A, Fallmann J, Grüning B, Ohler U, Rajewsky N, Stadler PF (2017) RNA-bioinformatics: tools, services and databases for the analysis of RNA-based regulation. J Biotechnol 261:76–84. https://doi.org/10.1016/j.jbiotec.2017.05.019
Banerjee A, Roychoudhury A, Krishnamoorthi S (2016) Emerging techniques to decipher microRNAs (miRNAs) and their regulatory role in conferring abiotic stress tolerance of plants. Plant Biotechnol Rep 10:185–205. https://doi.org/10.1007/s11816-016-0401-z
Bao W, Kojima KK, Kohany O (2015) Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile Dna 6:11. https://doi.org/10.1186/s13100-015-0041-9
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z, Galon J (2009) ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25:1091–1093. https://doi.org/10.1093/bioinformatics/btp101
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bologna NG, Schapire AL, Zhai J, Chorostecki U, Boisbouvier J, Meyers BC, Palatnik JF (2013) Multiple RNA recognition patterns during microRNA biogenesis in plants. Genome Res 23:1675–1689. https://doi.org/10.1101/gr.153387.112
Bonnet E, He Y, Billiau K, Van de Peer Y (2010) TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26:1566–1568. https://doi.org/10.1093/bioinformatics/btq233
Borel C, Antonarakis SE (2008) Functional genetic variation of human miRNAs and phenotypic consequences. Mamm Genome 19:503–509. https://doi.org/10.1007/s00335-008-9137-6
Brant EJ, Budak H (2018) Plant small non-coding RNAs and their roles in biotic stresses. Front Plant Sci 9:1038. https://doi.org/10.3389/fpls.2018.01038
Brennecke J, Stark A, Russell RB, Cohen SM (2005) Principles of microRNA-target recognition. PLoS Biol 3:e85. https://doi.org/10.1371/journal.pbio.0030085
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190. https://doi.org/10.1126/science.1159151
Budak H, Zhang B (2017) MicroRNAs in model and complex organisms. Funct Integr Genom 17:121. https://doi.org/10.1007/s10142-017-0544-1
Carlsbecker A, Lee JY, Roberts CJ, Dettmer J, Lehesranta S, Zhou J, Lindgren O, Moreno-Risueno MA, Vatén A, Thitamadee S, Campilho A (2010) Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature 465:316–321. https://doi.org/10.1038/nature08977
Chapman EJ, Carrington JC (2007) Specialization and evolution of endogenous small RNA pathways. Nat Rev Genet 8:884. https://doi.org/10.1038/nrg2179
Chen X (2009) Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol 25:21–44. https://doi.org/10.1146/annurev.cellbio.042308.113417
Chen K, Rajewsky N (2007) The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8:93–103. https://doi.org/10.1038/nrg1990
Chen L, Ren Y, Zhang Y, Xu J, Sun F, Zhang Z, Wang Y (2012) Genome-wide identification and expression analysis of heat-responsive and novel microRNAs in Populus tomentosa. Gene 504:160–165. https://doi.org/10.1016/j.gene.2012.05.034
Chen L, Heikkinen L, Wang C, Yang Y, Sun H, Wong G (2018) Trends in the development of miRNA bioinformatics tools. Brief Bioinform 2018:1–17. https://doi.org/10.1093/bib/bby054
Chi SW, Zang JB, Mele A, Darnell RB (2009) Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 460:479–486. https://doi.org/10.1038/nature08170
Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, Chiew MY (2017) miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 46:D296–D302. https://doi.org/10.1093/nar/gkx1067
Cimini S, Gualtieri C, Macovei A, Balestrazzi A, De Gara L, Locato V (2019) Redox balance-DDR-miRNA triangle: relevance in genome stability and stress responses in plants. Front Plant Sci 10:989. https://doi.org/10.3389/fpls.2019.00989
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. https://doi.org/10.1093/bioinformatics/bti610
Conesa A, Madrigal P, Tarazona S, Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ, Elo LL, Zhang X, Mortazavi A (2016) A survey of best practices for RNA-seq data analysis. Genome Biol 17:13. https://doi.org/10.1186/s13059-016-0881-8
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159. https://doi.org/10.1093/nar/gkr319
Dai X, Zhuang Z, Zhao PX (2010) Computational analysis of miRNA targets in plants: current status and challenges. Brief Bioinform 12:115–121. https://doi.org/10.1093/bib/bbq065
Dai X, Zhuang Z, Zhao PX (2018) psRNATarget: a plant small RNA target analysis server (2017 release). Nucleic Acids Res 46:W49–W54. https://doi.org/10.1093/nar/gky316
D'Ario M, Griffiths-Jones S, Kim M (2017) Small RNAs: big impact on plant development. Trends Plant Sci 22:1056–1068. https://doi.org/10.1016/J.TPLANTS.2017.09.009
Das SS, Yadav S, Singh A, Gautam V, Sarkar AK, Nandi AK, Karmakar P, Majee M, Sanan-Mishra N (2018) Expression dynamics of miRNAs and their targets in seed germination conditions reveals miRNA-ta-siRNA crosstalk as regulator of seed germination. Sci Rep 8:1233. https://doi.org/10.1038/s41598-017-18823-8
de Vries S, Kloesges T, Rose LE (2015) Evolutionarily dynamic, but robust, targeting of resistance genes by the miR482/2118 gene family in the Solanaceae. Genome Biol Evol 7:3307–3321. https://doi.org/10.1093/gbe/evv225
de Vries S, Kukuk A, von Dahlen JK, Schnake A, Kloesges T, Rose LE (2018) Expression profiling across wild and cultivated tomatoes supports the relevance of early miR482/2118 suppression for Phytophthora resistance. Proc Royal Soc B Biol Sci 285(1873):20172560
de Vries S, de Vries J, Rose LE (2019) The elaboration of miRNA regulation and gene regulatory networks in plant–microbe interactions. Genes 10(4):310
Deng P, Wang L, Cui L, Feng K, Liu F, Du X, Tong W, Nie X, Ji W, Weining S (2015) Global identification of microRNAs and their targets in barley under salinity stress. PLoS ONE 10:e0137990. https://doi.org/10.1371/journal.pone.0137990
Desvignes T, Batzel P, Berezikov E, Eilbeck K, Eppig JT, McAndrews MS, Singer A, Postlethwait JH (2015) miRNA nomenclature: a view incorporating genetic origins, biosynthetic pathways, and sequence variants. Trends Genet 31:613–626. https://doi.org/10.1016/j.tig.2015.09.002
Devi K, Dey KK, Singh S, Mishra SK, Modi MK, Sen P (2018) Identification and validation of plant miRNA from NGS data—an experimental approach. Brief Funct Genom 18:13–22. https://doi.org/10.1093/bfgp/ely034
Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, Keime C, Marot G, Castel D, Estelle J, Guernec G (2013) A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform 14:671–683. https://doi.org/10.1093/bib/bbs046
Djami-Tchatchou AT, Sanan-Mishra N, Ntushelo K, Dubery IA (2017) Functional roles of microRNAs in agronomically important plants—potential as targets for crop improvement and protection. Front Plant Sci 8:378. https://doi.org/10.3389/fpls.2017.00378
Du T, Zamore PD (2005) microPrimer: the biogenesis and function of microRNA. Development 132:4645–4652. https://doi.org/10.1242/dev.02070
Eren H, Pekmezci MY, Okay S, Turktas M, Inal B, Ilhan E, Atak M, Erayman M, Unver T (2015) Hexaploid wheat (Triticum aestivum) root miRNome analysis in response to salt stress. Ann Appl Biol 167:208–216. https://doi.org/10.1111/aab.12219
Fahlgren N, Sullivan CM, Kasschau KD, Chapman EJ, Cumbie JS, Montgomery TA, Gilbert SD, Dasenko M, Backman TW, Givan SA, Carrington JC (2009) Computational and analytical framework for small RNA profiling by high-throughput sequencing. RNA 15:992–1002. https://doi.org/10.1261/rna.1473809
Fei Z, Joung JG, Tang X, Zheng Y, Huang M, Lee JM, McQuinn R, Tieman DM, Alba R, Klee HJ, Giovannoni JJ (2010) Tomato functional genomics database: a comprehensive resource and analysis package for tomato functional genomics. Nucleic Acids Res 39:D1156–D1163. https://doi.org/10.1093/nar/gkq991
Flutre T, Duprat E, Feuillet C, Quesneville H (2011) Considering transposable element diversification in de novo annotation approaches. PLoS ONE 6:e16526. https://doi.org/10.1371/journal.pone.0016526
Furey TS (2012) ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet 13:840–852. https://doi.org/10.1038/nrg3306
Garmire LX, Subramaniam S (2012) Evaluation of normalization methods in mammalian microRNA-Seq data. RNA 18:1279–88. doi: 10.1261/rna.030916.111
Gene Ontology Consortium (2014) Gene ontology consortium: going forward. Nucleic Acids Res 43:D1049–D1056. https://doi.org/10.1093/nar/gku1179
German MA, Luo S, Schroth G, Meyers BC, Green PJ (2009) Construction of parallel analysis of RNA ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat Protoc 4:356. https://doi.org/10.1038/nprot.2009.8
Ghosh S, Singh K, Shaw AK, Azahar I, Adhikari S, Ghosh U, Basu U, Roy S, Saha S, Sherpa AR, Hossain Z (2017) Insights into the miRNA-mediated response of maize leaf to arsenate stress. Env Exp Bot 137:96–109. https://doi.org/10.1016/j.envexpbot.2017.01.015
Gordon, A. 2011. FASTX‐Toolkit. Computer program distributed by the author, website https://hannonlab.cshl.edu/fastx_toolkit/index.html. Accessed 1 May 2019
Griffiths-Jones S (2004) The microRNA registry. Nucleic Acids Res 32:D109–D111. https://doi.org/10.1093/nar/gkh023
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR (2003) Rfam: an RNA family database. Nucleic Acids Res 31:439–441. https://doi.org/10.1093/nar/gkg006
Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, Ulrich A (2010) Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141:129–141. https://doi.org/10.1016/j.cell.2010.03.009
Hafner M, Lianoglou S, Tuschl T, Betel D (2012) Genome-wide identification of miRNA targets by PAR-CLIP. Methods 58:94–105. https://doi.org/10.1016/j.ymeth.2012.08.006
Hannon GJ (2002) RNA interference. Nature 418:244–251. https://doi.org/10.1038/418244a
Hannoufa A, Matthews C, Feyissa BA, Gruber MY, Arshad M (2018) Progress toward deep sequencing-based discovery of stress-related microRNA in plants and available bioinformatics tools. In: Progress in Botany. Springer, Berlin, Heidelberg. doi: 10.1007/124_2018_25
He HH, Meyer CA, Hu SS, Chen MW, Zang C, Liu Y, Rao PK, Fei T, Xu H, Long H, Liu XS, Brown M (2014) Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods 11:73–78. https://doi.org/10.1038/nmeth.2762
Huang TH, Fan B, Rothschild MF, Hu ZL, Li K, Zhao SH (2007) MiRFinder: an improved approach and software implementation for genome-wide fast microRNA precursor scans. BMC Bioinform 8:341. https://doi.org/10.1186/1471-2105-8-341
Huang Y, Shen XJ, Zou Q, Wang SP, Tang SM, Zhang GZ (2011) Biological functions of microRNAs: a review. J Physiol Biochem 67:129–139. https://doi.org/10.1007/s13105-010-0050-6
Huerta-Cepas J, Forslund K, Coelho LP, Szklarczyk D, Jensen LJ, von Mering C, Bork P (2017) Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol 34:2115–2122. https://doi.org/10.1093/molbev/msx148
Inal B, Türktaş M, Eren H, Ilhan E, Okay S, Atak M, Erayman M, Unver T (2014) Genome-wide fungal stress responsive miRNA expression in wheat. Planta 240:1287–1298. https://doi.org/10.1007/s00425-014-2153-8
Jia X, Ren L, Chen QJ, Li R, Tang G (2009) UV-B-responsive microRNAs in Populus tremula. J Plant Physiol 166:2046–2057. https://doi.org/10.1016/j.jplph.2009.06.011
Jiang H, Lei R, Ding SW, Zhu S (2014) Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform 15:182. https://doi.org/10.1186/1471-2105-15-182
Jiang N, Meng J, Cui J, Sun G, Luan Y (2018) Function identification of miR482b, a negative regulator during tomato resistance to Phytophthora infestans. Hort Res 5(1):9
Johnson C, Bowman L, Adai AT, Vance V, Sundaresan V (2006) CSRDB: a small RNA integrated database and browser resource for cereals. Nucleic Acids Res 35:D829–D833. https://doi.org/10.1093/nar/gkl991
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53. https://doi.org/10.1146/annurev.arplant.57.032905.105218
Joshi T, Wang J, Zhang H, Chen S, Zeng S, Xu B, Xu D (2017) The evolution of soybean knowledge base (SoyKB). In: van Dijk A (eds) Plant Genomics Databases Methods in Molecular Biology, vol 1533. Humana Press, New York, 10.1007/978-1-4939-6658-5_7
Jung JH, Park CM (2006) MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 225:1327–1338. https://doi.org/10.1007/s00425-006-0439-1
Kakrana A, Hammond R, Patel P, Nakano M, Meyers BC (2014) sPARTA: a parallelized pipeline for integrated analysis of plant miRNA and cleaved mRNA data sets, including new miRNA target-identification software. Nucleic Acids Res 42:e139–e139. https://doi.org/10.1093/nar/gku693
Kalvari I, Nawrocki EP, Argasinska J, Quinones ON, Finn RD, Bateman A, Petrov AI (2018) Non-Coding RNA Analysis Using the Rfam Database. Curr Protoc Bioinform 62:51. https://doi.org/10.1002/cpbi.51
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. https://doi.org/10.1093/nar/28.1.27
Khan GA, Declerck M, Sorin C, Hartmann C, Crespi M, Lelandais-Brière C (2011) MicroRNAs as regulators of root development and architecture. Plant Mol Biol 77:47–58. https://doi.org/10.1007/s11103-011-9793-x
Khare T, Shriram V, Kumar V (2018) RNAi Technology: the role in development of abiotic stress-tolerant crops. In: Wani S. (eds) Biochemical, physiological and molecular avenues for combating abiotic stress tolerance in plants. Academic Press. 10.1016?C2016-0-04133-5
Kleftogiannis D, Aigli Korfiati KT, Likothanassis S, Tsakalidis A, Mavroudi S (2013) Where we stand, where we are moving: surveying computational techniques for identifying miRNA genes and uncovering their regulatory role. Biomed Inf 46:563–573. https://doi.org/10.1016/j.jbi.2013.02.002
Klie S, Nikoloski Z (2012) The choice between MapMan and Gene Ontology for automated gene function prediction in plant science. Front Genet 3:115. https://doi.org/10.3389/fgene.2012.00115
Kohany O, Gentles AJ, Hankus L, Jurka J (2006) Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform 7:474. https://doi.org/10.1186/1471-2105-7-474
Kong Y (2011) Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics 98:152–153. https://doi.org/10.1016/j.ygeno.2011.05.009
Kozomara A, Birgaoanu M, Griffiths-Jones S (2018) miRBase: from microRNA sequences to function. Nucleic Acids Res 47(D1):D155–D162. https://doi.org/10.1093/nar/gky1141
Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N (2005) Combinatorial microRNA target predictions. Nat Genet 37:495–500. https://doi.org/10.1038/ng1536
Kuang Z, Wang Y, Li L, Yang X (2018) miRDeep-P2: accurate and fast analysis of the microRNA transcriptome in plants. Bioinformatics. https://doi.org/10.1093/bioinformatics/bty972
Kumar V, Khare T, Shriram V, Wani SH (2018) Plant small RNAs: the essential epigenetic regulators of gene expression for salt-stress responses and tolerance. Plant Cell Rep 37:61–75. https://doi.org/10.1007/s00299-017-2210-4
Langmead B, Salzberg S (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923
Lee PD, Sladek R, Greenwood CM, Hudson TJ (2002) Control genes and variability: absence of ubiquitous reference transcripts in diverse mammalian expression studies. Genome Res 12:292–297
Lee WC, Lu SH, Lu MH, Yang CJ, Wu SH, Chen HM (2015) Asymmetric bulges and mismatches determine 20-nt microRNA formation in plants. RNA Biol 12:1054–1066. https://doi.org/10.1080/15476286.2015.1079682
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB (2003) Prediction of mammalian microRNA targets. Cell 115:787–798. https://doi.org/10.1016/S0092-8674(03)01018-3
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li C, Zhang B (2015) MicroRNAs in control of plant development. J Cell Physiol 231:303–313. https://doi.org/10.1002/jcp.25125
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967. https://doi.org/10.1093/bioinformatics/btp336
Li L, Xu J, Yang D, Tan X, Wang H (2010) Computational approaches for microRNA studies: a review. Mamm Genome 21:1–12. https://doi.org/10.1007/s00335-009-9241-2
Li F, Orban R, Baker B (2012a) SoMART: a web server for plant miRNA, tasiRNA and target gene analysis. Plant J 70:891–901. https://doi.org/10.1111/j.1365-313X.2012.04922.x
Li X, Wang X, Zhang S, Liu D, Duan Y, Dong W (2012b) Identification of soybean microRNAs involved in soybean cyst nematode infection by deep sequencing. PLoS ONE 7:e39650. https://doi.org/10.1371/journal.pone.0039650
Li JH, Liu S, Zhou H, Qu LH, Yang JH (2013a) starBase v2. 0: decoding miRNA-ceRNA, miRNA-ncRNA and protein–RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42:D92–D97. https://doi.org/10.1093/nar/gkt1248
Li S, Liberman LM, Mukherjee N, Benfey PN, Ohler U (2013b) Integrated detection of natural antisense transcripts using strand-specific RNA sequencing data. Genome Res 23:1730–1739. https://doi.org/10.1101/gr.149310.112
Li P, Piao Y, Shon HS, Ryu KH (2015) Comparing the normalization methods for the differential analysis of Illumina high-throughput RNA-Seq data. BMC Bioinform 16:347. https://doi.org/10.1186/s12859-015-0778-7
Liao P, Li S, Cui X, Zheng Y (2018) A comprehensive review of web-based resources of non-coding RNAs for plant science research. Int J Biol Sci 14:819–832. https://doi.org/10.7150/ijbs.24593
Lim LP, Lau NC, Weinstein EG, Abdelhakim A, Yekta S, Rhoades MW, Burge CB, Bartel DP (2003) The microRNAs of Caenorhabditis elegans. Genes Dev 17:991–1008. https://doi.org/10.1101/gad.1074403
Lindgreen S (2012) AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res Notes 5:337. https://doi.org/10.1186/1756-0500-5-337
Liu J, Jennings SF, Tong W, Hong H (2011) Next generation sequencing for profiling expression of miRNAs: technical progress and applications in drug development. J Biomed Sci Eng 4:666. https://doi.org/10.4236/jbise.2011.410083
Liu H, Yu H, Tang G, Huang T (2018) Small but powerful: function of microRNAs in plant development. Plant Cell Rep 37:515–528. https://doi.org/10.1007/s00299-017-2246-5
Long D, Lee R, Williams P, Chan CY, Ambros V, Ding Y (2007) Potent effect of target structure on microRNA function. Nat Struct Mol Biol 14:287–294. https://doi.org/10.1038/nsmb1226
Lorenz R, Bernhart SH, Zu Siederdissen CH, Tafer H, Flamm C, Stadler PF, Hofacker IL (2011) ViennaRNA Package 2.0. Algorithms Mol Biol 6:26. https://doi.org/10.1186/1748-7188-6-26
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. https://doi.org/10.1186/s13059-014-0550-8
Ma X, Tang Z, Qin J, Meng Y (2015) The use of high-throughput sequencing methods for plant microRNA research. RNA Biol 12:709–719. https://doi.org/10.1080/15476286.2015.1053686
Ma X, Liu C, Gu L, Mo B, Cao X, Chen X (2017) TarHunter, a tool for predicting conserved microRNA targets and target mimics in plants. Bioinformatics 34:1574–1576. https://doi.org/10.1093/bioinformatics/btx797
Maere S, Heymans K, Kuiper M (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21:3448–3449. https://doi.org/10.1093/bioinformatics/bti551
Mahen EM, Watson PY, Cottrell JW, Fedor MJ (2010) mRNA secondary structures fold sequentially but exchange rapidly in vivo. PLoS Biol 8:e1000307. https://doi.org/10.1371/journal.pbio.1000307
Markham NR, Zuker M (2008) UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol 453:3–31. https://doi.org/10.1007/978-1-60327-429-6_1
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet 17:10–12. https://doi.org/10.14806/ej.17.1.200
Mateos JL, Bologna NG, Chorostecki U, Palatnik JF (2010) Identification of microRNA processing determinants by random mutagenesis of Arabidopsis MIR172a precursor. Curr Biol 20:49–54. https://doi.org/10.1016/j.cub.2009.10.072
Mathews DH (2014) RNA secondary structure analysis using RNAstructure. Curr Protoc Bioinform 46:12–16. https://doi.org/10.1002/0471250953.bi1206s46
Maxam AM, Gilbert W (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol 65:499–560
McCormick KP, Willmann MR, Meyers BC (2011) Experimental design, preprocessing, normalization and differential expression analysis of small RNA sequencing experiments. Silence 2:2. https://doi.org/10.1186/1758-907X-2-2
Meng Y, Ma X, Chen D, Wu P, Chen M (2010) MicroRNA-mediated signaling involved in plant root development. Biochem Biophys Res Commun 393:345–349. https://doi.org/10.1016/j.bbrc.2010.01.129
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S (2008) Criteria for annotation of plant MicroRNAs. Plant Cell 20:3186–3190. https://doi.org/10.1105/tpc.108.064311
Meyers BC, Simon SA, Zhai J (2010) MicroRNA processing: battle of the bulge. Curr Biol 20:R68–70. https://doi.org/10.1016/j.cub.2009.12.008
Milev I, Yahubyan G, Minkov I, Baev V (2011) miRTour: plant miRNA and target prediction tool. Bioinformation 6:248
Millar AA, Waterhouse PM (2005) Plant and animal microRNAs: similarities and differences. Funct Integr Genom 5:129–135. https://doi.org/10.1007/s10142-005-0145-2
Miskiewicz J, Tomczyk K, Mickiewicz A, Sarzynska J, Szachniuk M (2017) Bioinformatics study of structural patterns in plant microRNA precursors. BioMed Res Int 2017:6783010. https://doi.org/10.1155/2017/6783010
Montojo J, Zuberi K, Rodriguez H, Bader GD, Morris Q (2014) GeneMANIA: fast gene network construction and function prediction for Cytoscape. F1000Research. doi: 10.12688/f1000research.4572.1
Motameny S, Wolters S, Nürnberg P, Schumacher B (2010) Next generation sequencing of miRNAs–strategies, resources and methods. Genes 1:70–84. https://doi.org/10.3390/genes1010070
Moyle RL, Carvalhais LC, Pretorius LS, Nowak E, Subramaniam G, Dalton-Morgan J, Schenk PM (2017) An optimized transient dual luciferase assay for quantifying microRNA directed repression of targeted sequences. Front Plant Sci 8:1631. https://doi.org/10.3389/fpls.2017.01631
Nishida K, Ono K, Kanaya S, Takahashi K (2014) KEGGscape: a Cytoscape app for pathway data integration. F1000Research. https://doi.org/10.12688/f1000research.4524.1
Numnark S, Suwannik W (2019) An emerging technique for reducing the response time in plant miRNA identification. Comput Biol Chem 78:382–388. https://doi.org/10.1016/j.compbiolchem.2018.12.019
Numnark S, Mhuantong W, Ingsriswang S, Wichadakul D (2012) C-mii: a tool for plant miRNA and target identification. In: BMC genomics (Vol. 13, No. 7, p. S16). BioMed Central. doi: 10.1186/1471–2164–13-S7-S16
Ouyang S, Park G, Atamian HS, Han CS, Stajich JE, Kaloshian I, Borkovich KA (2014) MicroRNAs suppress NB domain genes in tomato that confer resistance to Fusarium oxysporum. PLoS Pathog 10(10):e1004464
Patanun O, Lertpanyasampatha M, Sojikul P, Viboonjun U, Narangajavana J (2013) Computational identification of MicroRNAs and their targets in Cassava (Manihot esculenta Crantz.). Mol Biotechnol 53:257–269. https://doi.org/10.1007/s12033-012-9521-z
Peterson SM, Thompson JA, Ufkin ML, Sathyanarayana P, Liaw L, Congdon CB (2014) Common features of microRNA target prediction tools. Front Genet 5:23. https://doi.org/10.3389/fgene.2014.00023
Prabu G, Mandal A (2010) Computational identification of miRNAs and their target genes from expressed sequence tags of tea (Camellia sinensis). Genom Proteom Bioinform 8:113. https://doi.org/10.1016/S1672-0229(10)60012-5
Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y (2012) A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genom 13:341. https://doi.org/10.1186/1471-2164-13-341
Remita MA, Lord E, Agharbaoui Z, Leclercq M, Badawi MA, Sarhan F, Diallo AB (2016) A novel comprehensive wheat miRNA database, including related bioinformatics software. Curr Plant Biol 7:31–33. https://doi.org/10.1016/j.cpb.2016.10.003
Robinson MD, McCarthy DJ, Smyth GK (2009) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. https://doi.org/10.1093/bioinformatics/btp616
Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M et al (2011) An integrated semiconductor device enabling non-optical genome sequencing. Nature 475:348–352. https://doi.org/10.1038/nature10242
Sanan-Mishra N, Tripathi A, Goswami K, Shukla RN, Vasudevan M, Goswami H (2018) ARMOUR–A Rice miRNA: mRNA Interaction Resource. Front Plant Sci. https://doi.org/10.3389/fpls.2018.00602
Sanger F, Coulson AR (1975) A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol 94:441–448
Sanger F, Nicklen S, Coulson AR (1977) DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA 74:5463–5467
Schubert M, Lindgreen S, Orlando L (2016) AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res Notes 9:88. https://doi.org/10.1186/s13104-016-1900-2
Shivaprasad PV, Chen HM, Patel K, Bond DM, Santos BA, Baulcombe DC (2012) A microRNA superfamily regulates nucleotide binding site–leucine-rich repeats and other mRNAs. Plant Cell 24(3):859–874
Shriram V, Kumar V, Devarumath RM, Khare TS, Wani SH (2016) MicroRNAs as potential targets for abiotic stress tolerance in plants. Front Plant Sci 7:8173389–8173817. https://doi.org/10.3389/fpls.2016.00817
Simon SA, Zhai J, Nandety RS, McCormick KP, Zeng J, Mejia D, Meyers BC (2009) Short-read sequencing technologies for transcriptional analyses. Annu Rev Plant Biol 60:305–333. https://doi.org/10.1146/annurev.arplant.043008.092032
Singh A, Gautam V, Singh S, Das SS, Verma S, Mishra V, Mukherjee S, Sarkar AK (2018) Plant small RNAs: advancement in the understanding of biogenesis and role in plant development. Planta 248:545–558. https://doi.org/10.1007/s00425-018-2927-5
Slatko BE, Gardner AF, Ausubel FM (2018) Overview of next generation sequencing technologies. Curr Protoc Mol Biol 122:e59. https://doi.org/10.1002/cpmb.59
Smit AFA, Hubley R, Green P (2015) RepeatMasker Open-4.0. 2013–2015. https://www.repeatmasker.org. Accessed 1 May 2019
Srivastava PK, Moturu TR, Pandey P, Baldwin IT, Pandey SP (2014) A comparison of performance of plant miRNA target prediction tools and the characterization of features for genome-wide target prediction. BMC Genom 15:348. https://doi.org/10.1186/1471-2164-15-348
Sturm M, Schroeder C, Bauer P (2016) SeqPurge: highly-sensitive adapter trimming for paired-end NGS data. BMC Bioinform 17:208. https://doi.org/10.1186/s12859-016-1069-7
Su G, Morris JH, Demchak B, Bader GD (2014) Biological network exploration with Cytoscape 3. Curr Protoc Bioinform 47:8–13. https://doi.org/10.1002/0471250953.bi0813s47
Sun G (2012) MicroRNAs and their diverse functions in plants. Plant Mol Biol 80:17–36. https://doi.org/10.1007/s11103-011-9817-6
Sun X, Fan G, Su L, Wang W, Liang Z, Li S, Xin H (2015) Identification of cold-inducible microRNAs in grapevine. Front Plant Sci 6:595. https://doi.org/10.3389/fpls.2015.00595
Sun Y, Niu X, Fan M (2017) Genome-wide identification of cucumber green mottle mosaic virus-responsive microRNAs in watermelon. Arch Virol 162:2591–2602. https://doi.org/10.1007/s00705-017-3401-6
Sunkar R, Maheswari M, Chakraborty S (2017) Small RNAs: regulators of plant development and climate resilience. Indian J Plant Physiol 22:369–370. https://doi.org/10.1007/s40502-017-0349-1
Szcześniak MW, Makałowska I (2014) miRNEST 2.0: a database of plant and animal microRNAs. Nucleic Acids Res 42:D74–D77. https://doi.org/10.1093/nar/gkt1156
Szwacka M, Pawełkowicz M, Skarzyńska A, Osipowski P, Wojcieszek M, Przybecki Z, Pląder W (2018) Biological significance, computational analysis, and applications of plant microRNAs. Acta Physiol Plant 40:146. https://doi.org/10.1007/s11738-018-2718-4
Tam S, Tsao MS, McPherson JD (2015) Optimization of miRNA-seq data preprocessing. Brief Bioinform 16:950–963. https://doi.org/10.1093/bib/bbv019
Tav C, Tempel S, Poligny L, Tahi F (2016) miRNAFold: a web server for fast miRNA precursor prediction in genomes. Nucleic Acids Res 44(W1):W181–W184. https://doi.org/10.1093/nar/gkw459
Teotia S, Tang G (2015) To bloom or not to bloom: role of microRNAs in plant flowering. Mol Plant 8:359–377. https://doi.org/10.1016/j.molp.2014.12.018
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M (2004) MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J 37:914–939. https://doi.org/10.1111/j.1365-313X.2004.02016.x
Thomson DW, Bracken CP, Goodall GJ (2011) Experimental strategies for microRNA target identification. Nucleic Acids Res 39:6845–6853. https://doi.org/10.1093/nar/gkr330
Tian T, You Q, Zhang L, Yi X, Yan H, Xu W, Su Z (2016) SorghumFDB: sorghum functional genomics database with multidimensional network analysis. Database. https://doi.org/10.1093/database/baw099
Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, Xu W, Su Z (2017) agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 45:W122–W129. https://doi.org/10.1093/nar/gkx382
Tiberio P, Callari M, Angeloni V, Daidone MG, Appierto V (2015) Challenges in using circulating miRNAs as cancer biomarkers. BioMed Res Int 2015:731479. https://doi.org/10.1155/2015/731479
Treiber T, Treiber N, Meister G (2019) Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol 20:5–20. https://doi.org/10.1038/s41580-018-0059-1
Tripathi A, Goswami K, Sanan-Mishra N (2015) Role of bioinformatics in establishing microRNAs as modulators of abiotic stress responses: the new revolution. Front Physiol 6:286. https://doi.org/10.3389/fphys.2015.00286
Turner M, Yu O, Subramanian S (2012) Genome organization and characteristics of soybean microRNAs. BMC Genom 13:169. https://doi.org/10.1186/1471-2164-13-169
Valdés-López O, Yang SS, Aparicio-Fabre R, Graham PH, Reyes JL, Vance CP, Hernández G (2010) MicroRNA expression profile in common bean (Phaseolus vulgaris) under nutrient deficiency stresses and manganese toxicity. New Phytol 187:805–818. https://doi.org/10.1111/j.1469-8137.2010.03320.x
Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL, Maniou S, Karathanou K, Kalfakakou D, Fevgas A (2014) DIANA-TarBase v7. 0: indexing more than half a million experimentally supported miRNA: mRNA interactions. Nucleic Acids Res 43:D153–D159. https://doi.org/10.1093/nar/gku1215
Wagner GP, Kin K, Lynch VJ (2012) Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci 131:281–285. https://doi.org/10.1007/s12064-012-0162-3
Wang C, Fang J (2015) RLM-RACE, PPM-RACE, and qRT-PCR: an integrated strategy to accurately validate miRNA target genes. In: Rederstorff M (eds) Small non-coding RNAs methods in molecular biology, vol 1296. Humana Press, New York, 10.10007/978-1-4939-2547-6_16
Wang T, Chen L, Zhao M, Tian Q, Zhang WH (2011) Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genom 12:367. https://doi.org/10.1186/1471-2164-12-367
Werner S, Wollmann H, Schneeberger K, Weigel D (2010) Structure determinants for accurate processing of miR172a in Arabidopsis thaliana. Curr Biol 20:42–48. https://doi.org/10.1016/j.cub.2009.10.073
Wu J, Wang D, Liu Y, Wang L, Qiao X, Zhang S (2014) Identification of miRNAs involved in pear fruit development and quality. BMC Genom 15:953. https://doi.org/10.1186/1471-2164-15-953
Wu Y, Lv W, Hu L, Rao W, Zeng Y, Zhu L, He Y, He G (2017) Identification and analysis of brown planthopper-responsive microRNAs in resistant and susceptible rice plants. Sci Rep 7:8712. https://doi.org/10.1038/s41598-017-09143-y
Xie F, Zhang B (2010) Target-align: a tool for plant microRNA target identification. Bioinformatics 26:3002–3003. https://doi.org/10.1093/bioinformatics/btq568
Xie S, Jiang H, Xu Z, Xu Q, Cheng B (2017) Small RNA profiling reveals important roles for miRNAs in Arabidopsis response to Bacillus velezensis FZB42. Gene 629:9–15. https://doi.org/10.1016/j.gene.2017.07.064
Xu J, Hou QM, Khare T, Verma SK, Kumar V (2019) Exploring miRNAs for developing climate-resilient crops: a perspective review. Sci Total Environ 653:91–104. https://doi.org/10.1016/j.scitotenv.2018.10.340
Xuan P, Guo M, Huang Y, Li W, Huang Y (2011) MaturePred: efficient identification of microRNAs within novel plant pre-miRNAs. PLoS ONE 6:e27422. https://doi.org/10.1371/journal.pone.0027422
Xue C, Li F, He T, Liu GP, Li Y, Zhang X (2005) Classification of real and pseudo microRNA precursors using local structure-sequence features and support vector machine. BMC Bioinform 6:310. https://doi.org/10.1186/1471-2105-6-310
Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH (2010) starBase: a database for exploring microRNA–mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic Acids Res 39:D202–D209. https://doi.org/10.1093/nar/gkq1056
Ye J, Zhang Y, Cui H, Liu J, Wu Y, Cheng Y, Xu H, Huang X, Li S, Zhou A, Bolund L, Chen Q, Wang J, Yang H, Fang L, Shi C (2018) WEGO 2.0: a web tool for analyzing and plotting GO annotations, 2018 update. Nucleic Acids Res 46:W71–W75. https://doi.org/10.1093/nar/gky400
Yi X, Du Z, Su Z (2013) PlantGSEA: a gene set enrichment analysis toolkit for plant community. Nucleic Acids Res 41:W98–W103. https://doi.org/10.1093/nar/gkt281
Yi X, Zhang Z, Ling Y, Xu W, Su Z (2015) PNRD: a plant non-coding RNA database. Nucleic Acids Res 43:D982–D989. https://doi.org/10.1093/nar/gku1162
Yin JQ, Zhao RC, Morris KV (2008) Profiling microRNA expression with microarrays. Trends Biotechnol 26:70–76. https://doi.org/10.1016/j.tibtech.2007.11.007
Yu X, Wang H, Lu Y, de Ruiter M, Cariaso M, Prins M, van Tunen A, He Y (2011) Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J Exp Bot 63:1025–1038. https://doi.org/10.1093/jxb/err337
Yu D, Wan Y, Ito H, Ma X, Xie T, Wang T, Shao C, Meng Y (2019) PmiRDiscVali: an integrated pipeline for plant microRNA discovery and validation. BMC Genom 20:133. https://doi.org/10.1186/s12864-019-5478-7
Yue D, Liu H, Huang Y (2009) Survey of computational algorithms for MicroRNA target prediction. Curr Genom 10:478–492. https://doi.org/10.2174/138920209789208219
Zhai J, Jeong DH, De Paoli E, Park S, Rosen BD, Li Y, González AJ, Yan Z, Kitto SL, Grusak MA, Jackson SA (2011) MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev 25(23):2540–2553
Zhang Y (2005) miRU: an automated plant miRNA target prediction server. Nucleic Acids Res 33:W701–W704. https://doi.org/10.1093/nar/gki383
Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA (2006a) Conservation and divergence of plant microRNA genes. Plant J 46:243–259. https://doi.org/10.1111/j.1365-313X.2006.02697.x
Zhang B, Pan X, Cobb GP, Anderson TA (2006b) Plant microRNA: a small regulatory molecule with big impact. Dev Biol 289:3–16. https://doi.org/10.1016/j.ydbio.2005.10.036
Zhang Z, Yu J, Li D, Zhang Z, Liu F, Zhou X, Wang T, Ling Y, Su Z (2010) PMRD: plant microRNA database. Nucl Acids Res 38:D806–D813. https://doi.org/10.1093/nar/gkp818
Zhang XN, Li X, Liu JH (2014) Identification of conserved and novel cold-responsive microRNAs in trifoliate orange (Poncirus trifoliata (L.) Raf.) using high-throughput sequencing. Plant Mol Biol Rep 32:328–341. https://doi.org/10.1007/s11105-013-0649-1
Zhao M, Tai H, Sun S, Zhang F, Xu Y, Li WX (2012) Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS ONE 7:e29669. https://doi.org/10.1371/journal.pone.0029669
Zhou M, Zheng S, Liu R, Lu L, Zhang C, Zhang L, Yant L, Wu Y (2019) The genome-wide impact of cadmium on microRNA and mRNA expression in contrasting Cd responsive wheat genotypes. BMC Genom 20:615. https://doi.org/10.1186/s12864-019-5939-z
Zhu JK (2008) Reconstituting plant miRNA biogenesis. Proc Natl Acad Sci USA 105:9851–9852. https://doi.org/10.1073/pnas.0805207105
Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucl Acids Res 31:3406–3415. https://doi.org/10.1093/nar/gkg595
Acknowledgements
XZ acknowledges the financial support from Yangtze Normal University (Z19074) and VK from the Science and Engineering Research Board, Government of India (EMR/2016/003896).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Przemyslaw Wojtaszek.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, X., Khare, T. & Kumar, V. Recent trends and advances in identification and functional characterization of plant miRNAs. Acta Physiol Plant 42, 25 (2020). https://doi.org/10.1007/s11738-020-3013-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-020-3013-8