
Abstract
MicroRNAs (miRNAs) are a newly discovered class of noncoding endogenous small RNAs involved in plant growth and development as well as response to environmental stresses. miRNAs have been extensively studied in various plant species, however, only few information are available in cassava, which serves as one of the staple food crops, a biofuel crop, animal feed and industrial raw materials. In this study, the 169 potential cassava miRNAs belonging to 34 miRNA families were identified by computational approach. Interestingly, mes-miR319b was represented as the first putative mirtron demonstrated in cassava. A total of 15 miRNA clusters involving 7 miRNA families, and 12 pairs of sense and antisense strand cassava miRNAs belonging to six different miRNA families were discovered. Prediction of potential miRNA target genes revealed their functions involved in various important plant biological processes. The cis-regulatory elements relevant to drought stress and plant hormone response were identified in the promoter regions of those miRNA genes. The results provided a foundation for further investigation of the functional role of known transcription factors in the regulation of cassava miRNAs. The better understandings of the complexity of miRNA-mediated genes network in cassava would unravel cassava complex biology in storage root development and in coping with environmental stresses, thus providing more insights for future exploitation in cassava improvement.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are evolutionary-conservation non-coding endogenous small RNAs with ~22 nt in length [1]. Recent studies have shown that they negatively regulated their target gene expression at the post-transcriptional and translational levels through mRNAs cleavage or repression of translation, depending on the complementarity between miRNA and their target genes [2–4]. The findings revealed that miRNAs regulated a great number of genes involved in plant growth and development, environmental stress response, signal transduction as well as pathogen invasion [5]. Like their protein-coding counterparts, miRNAs are also transcribed from their own genes [2, 6]. Mature miRNA formation is a multi-step processes. First, miRNA genes are usually transcribed by RNA polymerase II as a long primary transcripts called primary microRNA (pri-miRNA) [7, 8]. Then, the pri-miRNA forms a hairpin-like secondary structure, which subsequently processed into precursor microRNA (pre-miRNA). Unlike in animal, the pre-miRNA in plant is further cleaved into miRNA:miRNA* duplex by Dicer-like1 enzyme (DCL1) [1, 9] and subsequently exported into the cytoplasm via HASTY [10, 11]. Finally, the single-stranded miRNAs are incorporated with ARGONAUTE (AGO) proteins to form a complex known as RNA-induced silencing complex where the regulation of target gene expression occurs [1, 5, 12].
Investigation of plant miRNAs has gained more attention, during last decade, a large number of miRNAs have been widely discovered in a variety of a model plant and non-model plants such as Arabidopsis [13], maize [14], cotton [15], tomato [16], citrus [17], potato [18], Brassica rapa L. [19], and Populus euphratica [20]. Several approaches have been established for identifying miRNAs in various plant species, for instance, genetic screening, direct cloning, computational prediction as well as EST analysis [4]. The low level expression pattern of specific miRNAs in some tissues, stage of development as well as growth conditions can cause difficulty to warranty the good quality of small RNA library. The recently developed deep sequencing technology has great promise to generate an accurate and comprehensive picture of the small RNA transcriptome in different plants, tissues, and at different developmental stages. Meanwhile, such a tool provides the direct way of discovering miRNAs, computational approaches provide a useful complement to wet experiments based on high evolutionary conservation of miRNA among various plant species [13, 21, 22]. The overwhelming EST and GSS sequence databases in various plants are available and facilitated the miRNA prediction using computational approaches [23]. An in silico search of homologs in public databases can greatly help to identify miRNAs in several plants, especially those whose complete genome sequences are unavailable, such as tomato [16], sorghum [24], Vigna unguiculata [25], tobacco [26], and switchgrass [27]. However, only homology search is not enough for identifying miRNAs, therefore other additional criteria were set for distinguish miRNA from other types of small RNA. Predicting the secondary structure of pre-miRNA along with calculating the free energy are necessary for reducing the number of false positive identified miRNAs [13, 16, 22, 28]. Even though computational approach has been demonstrated to be helpful, the experimental validation must be performed to confirm.
In order to investigate the function of miRNAs, the knowledge of miRNA target genes is essential for understanding the comprehensive central role of miRNAs [29]. Because of complementary base-pairing between miRNAs and their target genes, this feature has been widely used to predict hundred of target genes [4, 30]. Much evidence has shown that miRNAs regulate a great amount of genes involved in plant growth and development, environmental stress response, signal transduction as well as pathogen invasion [1, 3, 5, 6, 12, 31].
Cassava (Manihot esculenta L.) along with maize, sugarcane, and rice are major sources of energy for millions people in most tropical countries. Apart from its traditional role as a food crop, a growing demand of cassava starch has indicated its applications in diverse industries including for paper, textile, and adhesive as well as an alternative energy resource. Despite the economically importance of cassava, the molecular genetics information especially regarding miRNAs of this plant remains largely unknown. Only a few miRNAs in cassava were reported and most of them were conserved. Identification of comprehensive sets of miRNAs in cassava is a critical step to facilitate our understanding of regulatory mechanisms or networks. Based on computational prediction, therefore, we aimed to extend cassava miRNA study by using all reported plant miRNAs deposited in miRNA database (miRBase) including conserved miRNAs and species-specific miRNAs to search against cassava genome database (Phytozome). A total of 169 cassava miRNAs were identified and their characteristics were investigated. After classification, the results showed that they belonged to 34 miRNA families including those 17 previously reported ones. The new miRNAs identified in this study should enable investigation of the complexity of miRNA-mediated genes network in storage root development and various stress responses in cassava. The knowledge gained from such study should be beneficial in development of cassava variety with desired properties.
Materials and Methods
Data Sets and Softwares
All previously known plant miRNA sequences were downloaded from the miRBase (http://www.mirbase.org) (Release 16.0: Sept 2010) [32, 33] that consist of a total number of 3,070 mature miRNA sequences from Arabidopsis, Brassica, Glycine, Sorghum, Vitis, Solanum, Oryza, Chlamydomonas, and other plant species. The cassava genome database was provided by Phytozome (http://www.phytozome.net/cassava). To predict the secondary structure of pre-miRNA, Zuker RNA folding algorithm software MFOLD 3.1 (http://mfold.bioinfo.rpi.edu/) [34–36] was used for generating RNA secondary structure and calculating the minimum free energy. The miRNA target genes were predicted by using two databases—Phytozome and the web tool psRNA-target (http://bioinfo3.noble.org/psRNATarget/) [37] using the Manihot esculenta (cassava) DFCI Gene index (MAESGI) release 1 as the sequence library for target search.
Identification of Cassava miRNA
A computational prediction was used for predicting potential miRNAs in cassava by using homology search based on miRNA conservation among different plant species [13, 22]. All mature plant miRNA sequences identified so far from other plant species were used in this study. In September 2010, a total of 3,070 sequences had been deposited in miRBase (Release 16.0). In order to avoid redundancy, the repeated sequences from the same miRNA families were removed and the remaining unique 1,367 sequences were left as miRNA reference set to blast against cassava genome. This step significantly reduced the number of query sequences. Then, the output sequences with less than three mismatches when compared with query miRNA sequences, and not less than 18 nucleotides (nt) in length were selected manually for further secondary structure prediction. Precursor sequences of 400 nt were extracted (200 nt upstream and downstream from the blast hits). If the length of a sequence was less than 400 nt, the entire available sequence was used as a miRNA precursor sequence. The extracted sequences were subjected to remove the protein-coding sequences by using BLASTX (http://blast.ncbi.nlm.nih.gov/Blast.cgi). After removing the protein-coding sequences, the remaining sequences were then subjected to secondary structure prediction using MFOLD program. Only sequences that fit the following criteria were designated as potential miRNAs in cassava: (1) mature miRNA should be 18–25 nt in length; (2) the predicted pre-miRNA folded into a perfect or near perfect stem-loop hairpin secondary structure; (3) the potential mature miRNA sequence located on one arm of hairpin structure; (4) no loops or breaks were allowed in the miRNA/miRNA* duplex; (5) 6 nt mismatches were allowed between miRNA/miRNA* duplex; (6) A + U content should be 30–75%; and (7) the predicted pre-miRNA had a high negative minimal free–folding energy (MFE) which obtained from the negative folding free energies (∆G) and MFE index (minimal free-folding energy index, MFEI) in order to distinguish from other small RNAs. The MFEI was calculated from [(MFE/length of the RNA sequence)*100]/(G + C)%. The predicted pre-miRNA should have a higher MFEI than 0.85, in order to distinguish from other small RNAs such as tRNAs, and rRNAs whose MFEI are generally between 0.59 and 0.66 [28]. The size of each pre-miRNA was calculated from the length of predicted secondary hairpin structure of each pre-miRNA. All potential miRNAs were further analyzed for the presence of miRNA clusters and antisense miRNAs.
Prediction of Potential Cassava miRNA Target Genes
The target genes of miRNAs could be predicted according to their perfect or nearly perfect complementarity between them and their target genes through homology algorithm [29]. Cassava genome provided by cassava genome database in Phytozome has not only served for identifying potential miRNA in cassava, but also served for predicting potential cassava miRNA target genes. All cassava miRNAs were used to search against cassava genome database. All of the blast results matching the query miRNA sequence with appropriate direction were further searched against BLASTX in order to annotate their functions. The web tool psRNA-target, an update version of web-based miRU, was applied for predicting cassava miRNA targets [37]. The analysis parameters were set as default. Briefly, the following criteria were set for predicting the potential cassava miRNA target genes: (1) not more than four mismatches between identified miRNA and target mRNA; (2) no mismatches were allowed between positions 10th, 11th because this site was believed as a cleavage site; (3) one mismatch was allowed between position 2nd and 12th and up to three mismatches between position 12th and 25th; and (4) not more than two consecutive mismatches.
Transcriptional Regulatory Motif Analysis of Potential Cassava miRNA Gene Promoter
In this study, we have focused on the regulations of miRNA gene expression in order to understand the complexity of miRNA-mediated genes network. Like protein-coding genes, miRNAs were transcribed by RNA Polymerase II [7, 38–40]. Therefore, cis-acting element on miRNA gene promoter could be analyzed by using tools designed for protein-coding genes such as PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) [41] and PLACE (http://www.dna.affrc.go.jp/htdocs/PLACE/) [42]. Because of easy to understand and user-friendly features, PlantCARE was used for searching known cis-regulatory elements in miRNA gene promoters. The 1,000 bp upstream promoter regions from the 5′-end of the entire hairpin structure of miRNA precursor sequence of each predicted miRNA genes were downloaded for cis-regulatory elements analysis.
Field Cultivation and Preparation of Plant Materials and RNA Extraction
Cassava cultivar Kasetsart 50 (KU50) was planted at the Rayong Field Crops Research Center, Ministry of Agriculture and Cooperatives, Rayong, Thailand. Fibrous roots (4 weeks old) and storage roots (8 weeks old) were collected from six healthy plantlets after planting. The roots were defined as follows: fibrous roots (less than 0.5 cm diameter) and storage roots (more than 1 cm in diameter). Concert™ plant RNA reagent (Invitrogen, Carlsbad, CA) was used for root RNA extraction. The RNA quality and quantity were analyzed by spectrophotometer, gel electrophoresis.
First-Strand cDNA Synthesis and Analysis of Gene Expression by Real-Time Quantitative PCR
Following isolation of total RNAs and DNase I treatment (Ambion), 100 ng of total RNA was used as template for the first-strand cDNA synthesis. First-strand cDNA synthesis was performed using Ncode™ miRNA first-strand cDNA synthesis and quantitative real-time PCR (qRT-PCR) Kits (Invitrogen). All miRNAs in the sample are polyadenylated using poly A polymerase and ATP. Following polyadenylation, Superscript™ III RT and a specially designed Universal RT Primer are used to synthesize cDNA from the tailed miRNA population.
To amplify miRNA159, sequence specific primer (5′-CCGGAGTTCGTTTGGATTGAAGGGA-3′) was used in conjunction with 3′ primers complementary to the adapter sequence (5′-GTAGAAGAGCGCCAACCGGATTAGTGAT-3′). This miR159 specific primer design leaves ~7 “unconstrained” nt at the 3′ end of the specific target miRNA. This portion of the amplified fragment is not determined by either of the primers and is used to confirm the identity of the amplified miRNA, by cloning and sequencing the PCR product. The adaptors were designed to be non-homologous to any known genes, as determined by Blast homology searches in order to minimize non-specific PCR amplification. Cassava cDNA samples from two types of cassava root system were used for real-time PCR employing the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and were performed on an Applied Biosystems 7500 instrument (Applied Biosystems). The relative quantity of miR159 transcripts, with the U6 gene as internal standard, was automatically calculated as 2−∆∆Ct by the ABI-7500 software. The Ct (cycle threshold) data were determined using default threshold settings. All first-strand cDNA syntheses and real-time PCR amplifications were performed in triplicate for each gene and time treatment. SE calculation and ANOVA were used for statistical and significance analyses, respectively, taking into account all three-by-three replicates.
Results and Discussion
Identification of Potential Cassava miRNAs
In plant kingdom, a substantial number of miRNAs are conserved in different plant species, in lineages from mosses and gymnosperms to flowering plants. Such homologous miRNA families typically have conserved and essential regulatory functions across many plants. In 2009, Zeng et al. [43] studied species- and condition-specific miRNA expression patterns in Euphorbiaceous plant. The four agri-economically valuable Euphorbiaceous species, castor bean, cassava, rubber tree and jatropha were studied by combining computational prediction and some experimental validation. They predicted 85 conserved miRNAs in 23 families in castor bean and experimentally verified and characterized 58 miRNAs in at least one of four Euphorbiaceous species during normal seedling development. According to their method, 20 conserved miRNA families in cassava showed the different miRNA expression profiling during cold and drought stress. Recently, Amiteye et al. [44] computationally predicted 17 well-conserved miRNA families in cassava by using Arabidopsis mature miRNA sequence to search against incomplete sequences of cassava genome. Of the 17 identified miRNA families, only two miRNA families have not been verified their existence in previous report by Zeng et al.
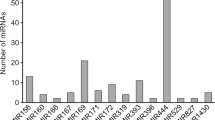
In this study, the total of 1,367 mature miRNA sequences from all previously known plant miRNA sequences, after removing redundancy sequences, were subjected to blast analysis against cassava genome database, then removed the protein-coding sequences and subsequently predicted the hairpin-loop secondary structure. The 169 potential cassava miRNAs belonging to 34 miRNA families were identified in this study (Fig. 1, Supplement Fig. S1 and Supplement Table S1), along with 17 previously identified cassava miRNAs by Amiteye et al. [44]; miR156, miR157, miR159, miR160, miR162, miR166, miR167, miR168, miR171, miR172, miR390, miR394, miR397, miR398, miR399, miR408, and miR414. Of these 34 miRNA families, the newly identified miR 169 family in cassava was the largest family with 29 members, similar to what have been observed in tobacco [26]. The 3 families, miR 166, miR156/157, and miR171 were found to contain 10, 11 and 12 members per family, respectively (Fig. 2). The other 30 families contained fewer than ten members. The rest contained only one or two members per family. The majority of identified potential cassava miRNAs were 21 nt in length (75.15%) followed by 20 nt (14.2%), 22 nt (7.69%), 23 nt (2.37%), and 19 nt (0.59%), respectively (Fig. 3). Of the 169 identified cassava miRNAs, 103 (60.95%) were found to be located on the 5′ arm of the hairpin secondary structure. The remaining 66 (39.05%) located on the 3′ arm. Moreover, we found that miRNAs belonging to the same miRNA family were not required to be located on the same arm of the pre-miRNA. As shown here by in silico prdiction, miR168a was located on the 3′ arm, while the miR168b was located on the 5′ arm. In order to analyze whether both mature miRNAs are present in cassava, the thermodynamic of 5′ and 3′ arm of the pre-miRNA have to be further analyzed. Most of the mature miRNA sequences in cassava began with the base uracil (U), which was consistent with previously results in other plants [19, 20, 45], due to the high affinity of AGO proteins to bind with U base in the 5′ terminus of mature miRNA sequences [46]. The average length distribution of predicted pre-miRNAs is consistent with previous results in other plant species. The average length of a potential cassava pre-miRNA sequences was 116.2 ± 35 nt, as found in other plant species. The mes-miR168a exhibited the shortest precursor length of 47 nt, whereas mes-miR319g exhibited the longest precursor length of 225 nt (Fig. 3). The nt composition of the newly identified potential cassava miRNA precursor sequences had an average A + U content of 54.65 ± 5% and G + C content of 45.35 ± 5%. The average minimal folding free energy (MFE) of the putative cassava pre-miRNAs was −52.69 ± 15.2% (Table 1).

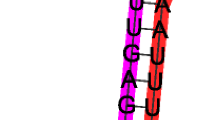
Predicted secondary hairpin structure for some cassava miRNAs identified in this study. Mature miRNA sequences are labeled in red (Color figure online)

Number of cassava miRNAs per family identified in this study

Size distribution of mature miRNA (a) and pre-miRNA (b)
Due to the fact that the hairpin-loop secondary structure is not enough for distinguish miRNA from other types of coding or non-coding RNAs, the MFEI has been calculated in order to distinguish miRNA from other RNAs precisely [13, 22, 28]. The plant pre-miRNA should have a MFEI higher than 0.85, while other types of RNAs, mRNAs, tRNAs, and rRNAs have a lower MFEI, with 0.62–0.66, 0.64, and 0.59, respectively. In this study, we found a wide range of MFEI from 0.48 to 1.49 with an average of 1.02 ± 0.16 (Table 1), suggesting that most of the predicted miRNA could be present as real mature miRNAs in cassava. It should be noted that miR473 and miR477 shared the same pre-miRNA sequences. This phenomenon was also discovered in miR159 and miR319 in V. unguiculata [25]. The mature miR473 and miR477 sequences were different in a few bases at the 5′ and 3′ terminus. It is interesting to elucidate the biogenesis of these 2 mature miRNAs in cassava.
miRNA Clusters in Cassava
The polycistronic miRNAs transcribed from the same pri-miRNA in the same direction were defined as miRNA clusters. The first clustering of miRNA genes have been identified widely in animal. In the past, cluster miRNAs have not been found much in plant. For analysis of miRNA clustering, both upstream and downstream sequences less than 10 kb were considered as clustered miRNAs. A total of 15 clusters were predicted. Our analysis revealed that 31 cassava miRNA genes that classified into seven families were identified in these 15 clusters (Table 2; Fig. 4). The miR 169 families possess the most clustered miRNAs including the mes-miR169q–miR169r cluster, the mes-miR169s–miR169t cluster, the mes-miR169u–miR169v cluster, the mes-miR169w–miR169x cluster, and the mes-miR169y–miR169z cluster. The miR 473 family contained three clusters, while the miR 394 family and miR 477 family contained two clusters for each family. The miR 166 family and miR 167 family contained only one cluster for each family. Based on literature review, we discovered the cluster of miR 394, miR 473, and miR 477 for the first time. It should be noted that the mes-miR394a–miR394c cluster and the mes-miR394b–miR394d cluster were only separated by 62 and 57 nt, on different arm of pre-miRNA strands, respectively. Considering these short separations on different arm of pre-miRNA strands, further experiment is required to verify whether each miRNA cluster produces the naturally existing mature miRNAs in cassava.

miR166j-k are clustered together in a cassava sequence no. 5979. This sequence containing the 2 miRNAs encoded within the cluster. (a) Shadowed sequences represent pre-miRNAs; underlined sequences represent the mature miRNAs. (b) The secondary structure of miR166j-k; underlined sequences represent the mature miRNAs
Antisense miRNAs in Cassava
Another class of miRNAs, antisense miRNAs originated from both sense and antisense strand of the miRNA genomic loci have been discovered in several plant such as tobacco [26], switchgrass [27], and soybean [47]. The pre-miRNA of sense/antisense miRNA are separately transcribed from its own DNA template strands, thus they may be involved in different function in plant. In this study, we identified 12 pairs of sense/antisense strand cassava miRNAs belonging to six different miRNA families (Table 3). The miR169 family exhibited the greatest number of sense/antisense miRNA pairs with the four composed of mes-miR169p/mes-miR169r, mes-miR169q/mes-miR169t, mes-miR169v/mes-miR169w, and mes-miR169x/mes-miR169u. The three pairs of sense/antisense miRNA were found in the miR396 family composed of mes-miR396a/mes-miR396e, mes-miR396c/mes-miR396d (see example in Fig. 5), and mes-miR396g/mes-miR396h, while another two pairs of that were found in the miR399 family composed of mes-miR399a/mes-miR399e, and mes-miR399c/mes-miR399d. All of the miR171, miR172, and miR395 family contained only one pair of sense/antisense miRNA which were mes-miR171c/mes-miR171k, mes-miR172d/mes-miR172e, and mes-miR395a/mes-miR395b, respectively.

Example of the sense/antisense miRNA pair of mes-miR396c and mes-miR396d identified in cassava. The pre-miRNA of sense mes-miR396d (above)/antisense mes-miR396c (below) are separately transcribed from its own DNA template strands. Mature miRNA sequences are labeled in red (Color figure online)
Mirtron in Cassava
Most miRNA loci are encoded by independent transcription units, which are transcribed by RNA polymerase II, however, recent studies revealed that some miRNAs are located in the introns of the mRNA encoding host genes, named mirtrons [48]. Although several studies have been discovered mirtrons widely spread in animals [48, 49], only a few numbers of mirtrons have been identified in rice and poplar [7]. In this study, we discovered one putative mirtron in cassava, mes-miR319b. The precursor of this miRNA lies within an intron of an unknown protein-coding gene. A comparison of conserved miRNA sequences with annotated cassava introns did not reveal other mirtrons. This may be due to the imperfect annotation of the cassava genome, or perhaps because the average size of cassava intron is an unsuitable mimic of pre-miRNAs. In addition, as mirtrons biogenesis requires expression of their host genes and intron splicing, they are tissue and/or developmental time dependent.
Prediction of Cassava miRNA Target Gene
The knowledge on target function of the identified cassava miRNA will help us to gain insight into the important function and regulation of miRNAs in this plant. Plant miRNAs are known to perfectly or near-perfectly match to their target mRNAs and help regulate post-transcriptional gene expression by binding to mRNAs and promoting mRNA degradation, or binding and inhibiting protein translation [1]. Based on the complementarities between miRNAs and their target mRNAs, we employed this feature to facilitate the prediction of cassava miRNA target genes. Using the newly identified miRNAs in cassava as query sequences to blast against cassava genome database in Phytozome coupled with using psRNA-target, we were able to predict various miRNA target genes in cassava such as genes involved in transcription, translation, stress response, structural component, development, and metabolism (Table 4). Consistent with previous finding, well-conserved cassava miRNAs also have conserved miRNA target sites on specific target genes in divergent plant species such as maize [14], tomato [16], potato [18], tobacco [26], and switchgrass [27]. For instance, squamosa promoter-binding protein, a plant-specific family of transcription factor regulating the early flower development and vegetative phase changes, is considered to be a conserved target of miR156 families in tomato [16] and potato [18]. We also found this squamosa promoter-binding protein gene represented as one of the three target genes of miR156/157 in cassava. The previous studies have been reported that miR156/157 also targeted liguleless1 protein, a nuclear-localized protein involved in ligules and auricle development [50], and sigma factor B regulation protein, which suggested that one miRNAs can regulate more than one target gene [51]. Although miRNAs are well conserved in long evolutionary timescales, some of their sequences have changed and display variations in a few nucleotide positions which may provide the chance for some miRNAs to base pair with other target mRNAs, exhibiting species-specific regulatory pattern [52]. Furthermore, miR156/157 and miR529 share the same target gene, liguleless1 protein, suggesting the network of multiple miRNAs may regulate the activities of one target gene [51, 53].
MYB transcription factors, a large family of proteins functionally diverse in plants, represent key regulators for controlling plant development, metabolism as well as responses to biotic and abiotic stress response [54]. In our study, myb81 and R2R3-myb transcription factor were identified as miR159 targeted genes in cassava. The R2R3-myb transcription factor (MYB with two repeats) was involved in the regulation of anthocyanin biosynthesis in Rosaceae [55]. However, the precise function of this transcription factor has not yet been known in cassava. Phytohormone auxin plays crucial roles, many of which are mediated by members of the auxin response transcription factor (ARF) family [56]. Our study showed that miR160 was complementary to ARF which was consistent with previous results in other plants. The plant-specific transcription factor, the floral homeotic protein APETALA2 (AP2) is known to be involved in flowering time and floral organ identities [57, 58]. In this study, AP2 was also predicted as a target of miR172.
In addition to transcription factor, some of newly identified cassava miRNAs also targeted various kinds of enzymes such as sulfate adenylyltransferase, cinnamoyl-CoA reductase, laccase, multicopper oxidases, 5-methyltetrahydrofolate : homocysteine methyltransferase, short chain alcohol dehydrogenase, and acyl-protein thiosterase that played critical roles in various metabolism. However, the biological functions of some target genes have not been known yet in cassava. In addition, some miRNA families such as miR166, miR393, miR394, miR403, miR408 were failed to identify their target genes. This may probably be the result of incomplete cassava sequence databases.
Cis-Regulatory Elements in Cassava miRNA Promoter Regions
The upstream sequences up to 1,000 bp from the 5′-end of the entire hairpin structure of miRNA precursor sequence of the cassava miRNA genes were analyzed by using PlantCARE to reveal known cis-regulatory elements that could regulate the expression [39]. As expected, promoter regions of numerous miRNA genes have been found to contain a TATA-box (a core promoter sequence served for initiation of the transcription process) and a CAAT-box (a common cis-element in promoter and enhancer regions) (Supplement Table S2), confirming that miRNA genes were transcribed by RNA Polymerase II similar to other protein-coding genes [7, 8]. The cis-regulatory element involved in light responsiveness were found to be more prevalent among the regions found on miRNA genes suggesting the important role of light on miRNA gene expression. Table 5 showed known stress-responsive elements [22, 31], such as ABRE (cis-regulatory element involved in abscisic acid responsiveness), MBS (a MYB binding site involved in drought-inducibility) and other stress-relevant elements [59] such as P-box and TATC-box, (cis-element involved in gibberellin responsiveness), GCN4 and Skn-1 (cis-element involved in endosperm expression, but also has been reported to possibly involve in abiotic stress).
The cis-regulatory elements for phytohormone responses also existed in promoter region of various miRNA genes in cassava (Table 6). Several studies reported that miR159 generally targeted GAMYB-related proteins (a gibberellin-specific transcription factor) which specifically bound to gibberellins-responsive element (GARE) located in the promoter region of the affected gene [60]. Gibberellins (GA) has been demonstrated to positively regulate the expression of miR159, and miR159 modulates GA-mediated developmental regulation via its effects on GAMYB activity [60]. Interestingly, we found that mes-miR159b contains putative GARE in its promoter region. This observation hinted that mes-miR159b might regulate itself through the feedback circuit, in order to mediate the balancing between the level of transcription factor and miRNA expression, which is consistent with previous reports in Arabidopsis [38, 39]. In this study, many putative cis-regulatory elements have been identified in cassava miRNA gene promoters. The results of this study present a set of putative transcription factor binding site that may be further investigated for evidence of miRNA regulation in cassava. These sites may be used to search for a functional connection between transcription factors and the targets of the miRNAs they potentially regulate.
We preliminary confirmed the existence of some candidate miRNAs by using experimental approaches. RNA were extracted from the two main types of cassava root system (fibrous and storage root) and subsequently cDNA was synthesized for PCR to amplify the mature miRNA by using miRNA-specific primer. This method allows detection and validation of specific miRNAs in cassava root system. The amplified fragments were further analyzed by cloning and sequencing. We were able to identify three cassava miRNAs, namely miR159, miR164, and miR167. The qRT-PCR assay showed that miR159 was down-regulated in fibrous root while it was up-regulated in storage root (Supplement Fig. S2). Further analysis of relationship between miRNAs and their target gene expression should be studied to unraveling of the role of miRNAs in cassava storage root development. The better understandings of the miRNAs in storage root development would be beneficial in helping to develop cassava variety with desired properties. It should be noted that this study used mainly a computational approach, which provide benefit information for experimental design, therefore further experiments need to be performed to verify the function of these miRNAs and their target genes in cassava. Taken together, the knowledge gained from this research will provide an insight into cassava miRNAs-mediated genes network which would undoubtedly be hugely beneficial for the cassava variety improvement schemes in the future.
References
Bartel, D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell, 116, 281–297.
Carthew, R. W., & Sontheimer, E. J. (2009). Origins and mechanisms of miRNAs and siRNAs. Cell, 136, 642–655.
Jones-Rhoades, M. W., Bartel, D. P., & Bartel, B. (2006). MicroRNAs and their regulatory roles in plants. Annual Review plant Biology, 57, 19–53.
Zhang, B. H., Pan, X. P., Cobb, G. P., & Anderson, T. A. (2006). Plant microRNA: a small regulatory molecule with big impact. Developmental Biology, 289, 3–16.
Dugas, D. V., & Bartel, B. (2004). MicroRNA regulation of gene expression in plants. Current Opinion in Plant Biology, 7, 512–520.
Chen, X. (2005). MicroRNA biogenesis and function in plants. FEBS Letters, 579, 5923–5931.
Cui, X., Xu, S. M., Mu, D. S., & Yang, Z. M. (2009). Genomic analysis of rice microRNA promoters and clusters. Gene, 431, 61–66.
Lee, Y., Kim, M., Han, J., Yeom, K., Lee, S., et al. (2004). MicroRNA genes are transcribed by RNA polymeraseII. EMBO Journal, 23, 4051–4060.
Reinhart, B. J., Weinstein, E. G., Rhoades, M. W., Bartel, B., & Bartel, D. P. (2002). Micro-RNAs in plants. Genes and Development, 16, 1616–1626.
Papp, I., Mette, M. F., Aufsatz, W., Daxinger, L., Schauer, S. E., et al. (2003). Evidence for nuclear processing of plant microRNA and short interfering RNA precursors. Plant Physiology, 132, 1382–1390.
Park, M. Y., Wu, G., Gonzalez-Sulser, A., Vaucheret, H., & Poethig, R. S. (2005). Nuclear processing and export of microRNAs in Arabidopsis. Proceedings National Academy Sciences, 102, 3691–3696.
Voinnet, O. (2009). Origin, biogenesis, and activity of plant microRNAs. Cell, 136, 669–687.
Adai, A., Johnson, C., & Mlotshwa, S. (2005). Computational prediction of miRNAs in Arabidopsis thaliana. Genome Research, 15, 78–91.
Zhang, B. H., Pan, X. P., & Anderson, T. A. (2006). Identification of 188 conserved maize microRNAs and their targets. FEBS Letters, 580, 3753–3762.
Zhang, B. H., Wang, Q. L., Wang, K. B., Pan, X. P., Liu, F., et al. (2007). Identification of cotton microRNAs and their targets. Gene, 397, 26–37.
Yin, Z., Li, C., Han, X., & Shen, F. (2008). Identification of conserved microRNAs and their target genes in tomato (Lycopersicon esculentum). Gene, 414, 60–66.
Song, C., Fang, J., Li, X., Liu, H., & Chao, C. T. (2009). Identification and characterization of 27 conserved microRNAs in citrus. Planta, 230, 671–685.
Xie, F., Frazier, T. P., & Zhang, B. (2011). Identification, characterization and expression analysis of microRNAs and their targets in the potato (Solanum tuberosum). Gene, 473, 8–22.
Dhandapani, V., Ramchiary, N., Paul, P., Kim, J., Choi, S. H., et al. (2011). Identification of potential microRNAs and their targets in Brassica rapa L. Molecules and Cells, 32, 21–37.
Li, B., Qin, Y., Duan, H., Yin, W., & Xia, X. (2011). Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. Journal of Experimental Botany. doi:10.1093/jxb/err051.
Meyers, B. C., Axtell, M. J., Bartel, B., Bartel, D. P., Baulcombe, D., et al. (2008). Criteria for annotation of plant microRNAs. Plant Cell, 20, 3186–3190.
Zhang, B. H., Pan, X. P., Wang, Q. L., Cobb, G. P., & Anderson, T. A. (2005). Identification and characterization of new plant microRNAs using EST analysis. Cell Research, 15, 336–360.
Sunkar, R., & Jagadeeswaran, G. (2008). In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biology, 8, 37.
Feng, D. J., Jun, W. Y., Feng, F. X., Xia, C. J., Liang, Z., et al. (2010). Prediction of sorghum miRNAs and their targets with computational methods. Chinese Science Bulletin, 55, 1263–1270.
Lu, Y., & Yang, X. (2010). Computational identification of novel microRNAs and their targets in Vigna unguiculata. Comparative and Functional Genomics, 2010, 1–17.
Frazier, T. P., Xie, F., Freistaedter, A., Burklew, C. E., & Zhang, B. (2010). Identification and characterization of microRNAs and their target genes in tobacco (Nicotiana tabacum). Planta, 232, 1289–1308.
Xie, F., Frazier, T. P., & Zhang, B. (2010). Identification and characterization of microRNAs and their targets in the bioenergy plant switchgrass (Panicum virgatum). Planta, 232, 417–434.
Bonnet, E., Wuyts, J., Rouze, P., & Van de Peer, Y. (2004). Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics, 20, 2911–2917.
Rhoades, M. W., Reinhart, B. J., Lim, L. P., Burge, C. B., Bartel, B., et al. (2002). Prediction of plant microRNA targets. Cell, 110, 513–520.
Schwab, R., Palatnik, J. F., Riester, M., Schommer, C., Schmid, M., et al. (2005). Specific effects of microRNAs on the plant transcriptome. Developmental Cell, 8, 517–527.
Lewis, R., Mendu, V., Mcnear, D., & Tang, G. (2010). Roles of microRNAs in plant abiotic stress. In S. M. Jain & D. S. Brar (Eds.), Molecular techniques in crop improvement (2nd ed., pp. 357–372). Cambridge, MA: Springer.
Griffiths-Jones, S. (2006). miRBase: the microRNA sequence database. Methods in Molecular Biology, 342, 129–138.
Griffiths-Jones, S., Saini, H. K., Van Dongen, S., & Enright, A. J. (2008). miRBase: tools for microRNA genomics. Nucleic Acids Research, 36, D154–D158.
Hofacker, I. L. (2003). Vienna RNA secondary structure server. Nucleic Acids Research, 31, 3429–3431.
Zuker, M. (2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Research, 31, 3406–3434.
Zuker, M., & Stiegler, P. (1981). Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Research, 9, 133–148.
Dai, X., & Zhao, P. X. (2011). psRNATarget: a plant small RNA target analysis server. Nucleic Acids Research, 39, W155–W159.
Megraw, M., Baev, V., & Rusinov, V. (2006). MicroRNA promoter element discovery in Arabidopsis. RNA, 12, 1612–1619.
Meng, Y., Shao, C., & Chen, M. (2011). Toward microRNA-mediated gene regulatory networks in plants. Brief Bioinformatics. doi:10.1093/bib/bbq091.
Zhou, L., Liu, Y., Liu, Z., Kong, D., Duan, M., et al. (2010). Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. Journal of Experimental Botany, 61, 4157–4168.
Lescot, M., Dehais, P., Thijs, G., Marchal, K., Moreau, Y., et al. (2002). PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Research, 30, 325–327.
Higo, K., Ugawa, Y., Iwamoto, M., & Korenaga, T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Research, 27, 297–300.
Zeng, C., Wang, W., Zheng, Y., Chen, X., Bo, W., et al. (2009). Conservation and divergence of microRNAs and their functions in Euphorbiaceous plants. Nucleic Acids Research, 38, 981–995.
Amiteye, S., Corral, J. M., & Sharbel, T. F. (2011). Overview of the potential of microRNAs and their target gene detection for cassava (Manihot esculenta) improvement. African Journal of Biotechnology, 11, 2562–2573.
Unver, T., Parmaksız, İ., & Dündar, E. (2010). Identification of conserved micro-RNAs and their target transcripts in opium poppy (Papaver somniferum L.). Plant Cell Reports, 29, 757–769.
Mi, S., Cai, T., Hu, Y., Chen, Y., Hodges, E., et al. (2008). Sorting of small RNAs into Arabidopsis Argonaute complexes is directed by the 5′ terminal nucleotide. Cell, 133, 116–127.
Zhang, B. H., Pan, X. P., & Stellwag, E. J. (2008). Identification of soybean microRNAs and their targets. Planta, 229, 161–182.
Okamura, K., Hagen, J. W., Duan, H., Tyler, D. M., & Lai, E. C. (2007). The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell, 130, 89–100.
Ruby, J. G., Jan, C. H., & Bartel, D. P. (2007). Intronic microRNA precursors that bypass Drosha processing. Nature, 448, 83–86.
Moreno, M. A., Harper, L. C., Krueger, R. W., Dellaporta, S. L., & Freeling, M. (1997). Liguleless1 encodes a nuclear-localized protein required for induction of ligules and auricles during maize leaf organogenesis. Genes and Development, 11, 616–628.
Peter, M. E. (2010). Targeting of mRNAs by multiple miRNAs: the next step. Oncogene, 29, 2161–2164.
Yin, Z., & Shen, F. (2010). Identification and characterization of conserved microRNAs and their target genes in wheat (Triticum aestivum). General Molecular Research, 9, 1186–1196.
Wu, S., Huang, S., Ding, J., Zhao, Y., Liang, L., et al. (2010). Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene, 29, 2302–2308.
Dubos, C., Stracke, R., Grotewold, E., Weisshaar, B., Martin, C., et al. (2010). MYB transcription factors in Arabidopsis. Trends in Plant Science, 15, 573–581.
Lin-Wang, K., Bolitho, K., Grafton, K., Kortstee, A., Karunairetnam, S., et al. (2010). An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biology, 10, 50.
Wang, D., Pei, K., Fu, Y., Sun, Z., Li, S., et al. (2007). Genome-wide analysis of the auxin response factors (ARF) gene family in rice (Oryza sativa). Gene, 394, 13–24.
Jofuku, K. D., Boer, B., Montagu, M. V., & Okamuro, J. K. (1994). Control of Arabidopsis flower and seed development by the Homeotic Gene APETALA2. Plant Cell, 6, 1211–1225.
Mlotshwa, S., Ynag, Z., Kim, Y., & Chen, X. (2006). Floral patterning defects induced by Arabidopsis APETALA2 and microRNA172 expression in Nicotiana benthamiana. Plant Molecular Biology, 61, 781–793.
Rizhsky, L., Davletova, S., Liang, H., & Mittlers, R. (2004). The zinc finger protein zat12 is required for cytosolic Ascorbate peroxidase 1 expression during oxidative stress in Arabidopsis. Journal of Biological Chemistry, 279, 11736–11743.
Achard, P., Herr, A., Baulcombe, D. C., & Harberd, N. P. (2004). Modulation of floral development by a gibberellin-regulated microRNA. Development, 131, 3357–3365.
Acknowledgments
This research was financially supported by Mahidol University. OP was partially supported by Center of Excellence on Agricultural Biotechnology, Science and Technology Postgraduate Education and Research Development Office, Commission on Higher Education, Ministry of Education (AG-BIO/PERDO-CHE), Thailand.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
12033_2012_9521_MOESM3_ESM.pdf
Supplement Figure S1 Predicted secondary hairpin structure for 169 cassava miRNAs identified in this study. (PDF 2073 kb)
12033_2012_9521_MOESM4_ESM.tif
Supplement Figure S2 Relative transcript abundance of miR159 in the fibrous and storage root. The relative expression corresponds to the ratio of the transcript abundance of miR159 gene/transcript abundance of the small RNA U6 control gene. Data are means of three independent experiments and SE (n = 9). Different letters indicate values that are significantly different between the transcripts of the fibrous and storage root cDNA samples (Tukey’s test, one-way ANOVA; P < 0.05). (TIFF 677 kb)
Rights and permissions
About this article
Cite this article
Patanun, O., Lertpanyasampatha, M., Sojikul, P. et al. Computational Identification of MicroRNAs and Their Targets in Cassava (Manihot esculenta Crantz.). Mol Biotechnol 53, 257–269 (2013). https://doi.org/10.1007/s12033-012-9521-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-012-9521-z