
Abstract
1,3-Dipolar cycloaddition of nitrones to oxanorbornadienes is an important method for the enantioselective synthesis of highly substituted 5-membered heterocycles such as furans and isoxazolidines, which have high utility in the chemical and pharmaceutical industries. The mechanism of the reaction and the effects of substituents on the (3 + 2) cycloaddition reactions (32CA) of C,N-dialkyl nitrones with a series of substituted oxanorbornadienes have been studied with focus on the site-selectivity (attack on the more substituted double bond of the oxanorbornadiene derivatives versus attack on the less substituted double bond), enantioselectivity and stereo-selectivity using density functional theory calculations at the M06/6-311++G(d,p) of theory. The results showed that the addition step to form the bicyclic isoxazolidines cycloadducts has generally low barriers compared to the cycloreversion step which converts the cycloadducts into furans and monocyclic isoxazolidines. Generally, electron-withdrawing substituents favour the nitrone attack on the highly substituted double bond, while electron-donating substituents favour the attack on less substituted double bond. The R enantiomers are generally favoured over the S enantiomers, and exo stereo-isomers are generally favoured over the endo stereo-isomers, irrespective of substituents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
1,3-Dipolar cycloaddition is one of the classic reactions of synthetic organic chemistry which generates regio- and stereo-chemically defined heterocycles of vital importance for both academic and industrial researches [1,2,3,4,5,6]. Reactions between nitrones, nitrile imines and alkenes leading to the formation of isoxazolidines and pyrazolines are well-known examples of this kind [7]. The products of these reactions have found wide applications in almost every area of organic synthesis and drug design and as intermediates for the synthesis of a variety of compounds after cleavage of the N–O bond [7].
These reactions, like Diels–Alder cycloadditions, depend on the electrophilic and nucleophilic characters of the reagents, and the reaction is influenced by substituent effects of dipoles and dipolarophiles [8]. Synthesis of isoxazolidines by dipolar cycloadditions of nitrones to alkenes and alkynes has been extensively explored. A lot of experimental studies have been devoted to the regio- and stereo-selective synthesis of these reactive intermediates. In cycloadditions of nitrones with alkenes, the nitrone can approach in an endo or an exo mode giving rise to two different diastereomers. The endo/exo selectivity in the 1,3-dipolar cycloaddition reaction is primarily controlled by the structure of the reactants or by a catalyst [9]. Although nitrone-alkene cycloaddition reactions have been utilized in the synthesis of many isoxazolidines including those which form the cores of potential antifungal and antibacterial agents, some aspects of this chemistry would benefit from detailed molecular-level understanding [10]. This versatile pericyclic reaction, which involves a range of electron-poor, electron-rich and neutral dipolarophiles, offers the possibility of generating isoxazolidines with up to three new contiguous stereo-centres (regio-, stereo- and enantioselectivities). This stereochemistry can thus be controlled by either choosing the appropriate reactants or by controlling the reaction by a metal complex acting as a catalyst [11]. Agarwal et al. [6] found that sulfoxide-based ketene thioacetals are very good chiral controllers of (3 + 2) cycloadditions and reported on the further application of sulfoxide-based ketene thioacetals as dipolarophiles in an intramolecular nitrone cycloaddition and its application to a short synthesis of the antifungal antibiotic cispentacin. Nie et al. [2] also reported the use of recyclable tetraarylphosphonium-supported imidazolidinone catalyst to control the dipolar cycloaddition of nitrones with unsaturated aldehydes to provide isoxazolidine aldehydes in good yields with excellent diastereo- and enantioselectivities. Cristina et al. [12] reported the reactions of a series of 1,3-dipoles with norbornadiene derivatives. Depending on the type of dipole used, ratios of varying selectivities were obtained (see Scheme 1).

32CA reactions of 1,3-dipoles with norbornadiene derivatives. However, to the best of our knowledge, no theoretical studies of the 32CA of these 1,3-dipoles to oxanorbornadienes have been described in the literature so far. Thus, the impact of varying substituents and solvents on these reactions is completely unknown. Herein we fill this gap by reporting the regio-, stereo- and enantioselectivities in the 1,3-dipolar cycloadditions reactions of oxanorbornadiene with phenyl nitrones and dimethyl nitrilimine (Schemes 2, 3, 4 and 5). DFT calculations have been performed to delineate the impact of substituents, particularly electron-donating and electron-withdrawing substituents on the dipolarophile reactants on the selectivity of the reaction
2 Computational details and methodology
We performed all the DFT calculations with the Spartan’14 [13] and Gaussian 09 [14] quantum chemistry software packages at the M06/6-311++G(d,p) level of theory. The M06 functional developed by Zhao and Truhlar [15] has been found to be effective at computing thermochemistry and kinetics of reactions [16,17,18]. In the Minnesota hybrid meta-generalized gradient approximations (meta-GGA) suite of density functionals, M06 is among the best performing in geometry optimizations and energy calculations [19].
The initial guess structures of the considered molecules were constructed using the Spartan’s graphical model builder and minimized interactively using the Sybyl force field [20]. Transition-state structures were computed by first obtaining guess input structures by constraining specific internal coordinates of the molecules (bond lengths, bond angles, dihedral angles) while fully optimizing the remaining internal coordinates. This procedure gives appropriate guess transition-state input geometries, which are then submitted for full transition-state calculations without any geometry or symmetry constraints. Using the polarizable continuum model (PCM), benzene was employed to compute solvation effects in the reactions [21]. The full optimization calculations were carried out with the Gaussian 09 package. Full harmonic vibrational frequency calculations were carried out to ensure that all transition-state structures have a Hessian matrix with only a single negative eigenvalue, characterized by an imaginary vibrational frequency along the respective reaction coordinates. Intrinsic reaction coordinate calculations were then performed to ensure that each transition state smoothly connects the reactants and products along the reaction coordinate [22,23,24,25,26,27].
The global electrophilicities (ω) and maximum electronic charge (ΔNmax) of the various oxanorbornadiene and both dimethyl nitrilimine and nitrone derivatives are calculated using Eqs. (1) [28] and (2) [29, 30]. The electrophilicity index measures the ability of a reactant to accept electrons [31], and it has been found to be a function of the electronic chemical potential, μ = (EHOMO + ELUMO)/2 and chemical hardness, η = (ELUMO − EHOMO) as defined by Pearson’s acid–base concept [28]. Hence, species with large electrophilicity values are more reactive towards nucleophiles. These equations are based on the Koopmans theory [32] originally established for calculating ionization energies from closed-shell Hartree–Fock wavefunctions, but have since been adopted as acceptable approximations for computing electronic chemical potential and chemical hardness.
The maximum electronic charge transfer (ΔNmax) measures the maximum electronic charge that the electrophile may accept. Thus, species with large ΔNmax index would be the best electrophile given a series of compounds.
The global electrophilic (\( P_{K}^{ + } \)) and nucleophilic (\( P_{K}^{ - } \)) Parr functions were obtained through the analysis of the Mulliken and natural bond orbital (NBO) atomic spin densities (ASD) of the radical anion and the radical cation by single-point energy calculations over the optimized neutral geometries using the unrestricted UM06 formalism for the radical species [33].
3 Results and discussion
3.1 32CA reaction of nitrones with oxanorbornadiene (ONBD)
This section begins with a minimally substituted model system where the reactant ONBD is unsubstituted. The goal here is to examine the intrinsic reactivity of the two species in the absence of any substituents for later comparison with more realistic models. The reactions follow a two-step mechanism involving 1,3-dipolar reaction between unsubstituted oxanorbornadiene and phenyl nitrone to form the bicyclic intermediate, followed by Diels–Alder cycloreversion to form five-membered heterocyclic furans and isoxazolidine.
Gas-phase energies and energies corrected with benzene solvation were computed for the reactions of ONBD with C-phenyl N-methyl nitrone. Details of the energies are shown in Fig. 1. Due to the symmetric nature of the ONBD, for which addition across either double bond results in the same structure, the addition can take place along four reaction channels since regioselectivity in the reaction is lost. The remaining selectivity arises from one pair of stereo-isomers and one pair of enantiomers as depicted in Schemes 2 and 3. Clearly, from the results, the most favourable pathway for the reaction is through TS1-ex/S. The difference in activation energies between TS1-ex/S and TS2-en/S is 4.6 kcal/mol and that between TS1-ex/S and TS1-ex/R is 0.7 kcal/mol, and thus, the predicted trend favours exo/S stereo-/enantioselectivity. In Fig. 1, inclusion of solvent (benzene) effects using the PCM method only increases the barrier by 1 kcal/mol and the trends in the stereo- and enantioselectivities remain unchanged. In the Diels–Alder cycloreversion step (thermolytic cleavage step), higher activation barriers are recorded along all the isomeric transition states computed in this study. In this step, the TS with the least energy barrier is TS2A-en/S (34.2 kcal/mol in gas phase as well as 32.4 kcal/mol in benzene). Also, considering the most favoured reaction pathway (TS1-ex/S), it is observed that upon formation of I1 with a reaction energy of 51.7 kcal/mol (gas phase), the cycloreversion step requires an activation barrier of 42.4 kcal/mol to form the thermolytic adducts. Hence, considering these energetic demands it can easily be concluded that the reaction will terminate at the first step. These observations are in total agreement with studies [12, 23] that have shown that in the reaction of norbornadiene derivatives with nitrones, bicyclic intermediates are isolated as the final products as our results show that the barriers leading to the formation of the five-membered rings are generally too high.

Free energy profile of the reaction of methyl nitrone with oxanorbornadiene in gas phase at the M06/6-311++G(d,p) level of theory. Results for computations in benzene at 298.15 K are in parenthesis

The reaction of phenyl nitrone with substituted oxanorbornadiene to form bicyclic isoxazolidine cycloadducts

Diels–Alder cycloreversion of bicyclic isoxazolidine cycloadducts to form derivatives of furan and isoxazolidines
3.2 Substituent effects on the reaction
In order to analyse electronic effects on the reaction, we will present and discuss, in two different sections, the energy profiles corresponding to the eight reactive channels as depicted previously in Schemes 2 and 3. In the first section, the effects of electron-withdrawing substituents on the ONBD reactant are discussed. In the other section, the effects of electron-donating substituents on the ONBD are discussed.
In the first step of the two-step reaction process, the dipole adds to the dipolarophile to form eight bicyclic intermediates I1–I8 arising from the unsymmetrical nature of the substituted ONBD through transition states TS1-ex/S, TS1-ex/R, TS2-en/S, TS2-en/R, TS3-ex/S, TS3-ex/R, TS4-en/S and TS4-en/R. In the second step, the bicyclic adducts undergo Diels–Alder cycloreversion through transition states TS1A-ex/S, TS1A-ex/R, TS2A-en/S, TS2A-en/R, TS3A-ex/S, TS3A-ex/R, TS4A-en/S and TS4A-en/R to form five-membered heterocycles (furans and isoxazolidines) P1–P6.
3.2.1 The reaction of C-phenyl, N-methyl nitrone with substituted oxanorbornadienes
Examination of the energetics of the cycloadditions of ester-substituted ONBD shown in Fig. 2 leads to the conclusion that the most favoured pathway for the reaction is through TS1-ex/S. The activation energy of the reaction through this transition state is 2.2 kcal/mol. The transition state of the corresponding enantiomer (TS1-ex/R) is not significantly different (5.0 kcal/mol) in energy than TS1-ex/S, and thus, a mixture of enantiomers can be expected in this reaction, which is in complete agreement with yields experimentally obtained by Christina et al. [12]. The remaining six transition states leading to the formation of the different cycloadducts, however, are relatively higher. The subsequent Diels–Alder cycloreversion steps have relatively higher activation barriers, and hence, the reactions are likely to terminate at the intermediate stage. This observation again was made experimentally [12]. The energy profiles corresponding to the eight reaction pathways at the DFT M06/6-311++G(d,p) level of theory are displayed in Fig. 2. We also repeated the cycloadditions of ester-substituted ONBD with the phenyl nitrone in solvent (benzene) in order to mimic the actual experimental conditions, and the results are shown in Table S1 in supporting information. As it can be seen from Table S1, although there are some slight variations in the magnitude of the energies, the trends remain the same as that observed for the gas-phase calculations. Hence, on the basis of this we can conclude that the gas-phase calculations are sufficient for the study of the systems under consideration. The optimized geometries of the transitions states as well as relevant geometrical parameters involved in the 32CA of ester-substituted oxanorbornadiene to C-phenyl, N-methyl nitrone are shown in Fig. 3.

Free energy profile for the 32CA of ester-substituted oxanorbornadiene to C-phenyl, N-methyl nitrone at the M06/6-311++G(d,p) level of theory. Relative energies in kcal/mol

Optimized geometries of the transitions states involved in the 32CA of ester-substituted oxanorbornadiene to C-phenyl, N-methyl nitrone at the M06/6-311++G(d,p) level of theory. All bond distances are measured in Å. Hydrogen atoms are ignored for brevity. Atomic colour code (red = oxygen, blue = nitrogen, grey = carbon)
In view of the observed energetic trends, we extended the studies by replacing the ester groups on the dipolarophile to investigate the effects on product outcomes. Results of the reaction of the substituted oxanorbonadiene with the phenyl nitrone are shown in Tables 1 and 2. In the case of bromo-substituted oxanorbornadiene reaction with the C-phenyl, N-methyl nitrone, energetic trends analogous to the ester-substituted ONBD are observed where the TS1-ex/S pathway is the most favoured with an activation barrier of 4.6 kcal/mol. TS2-en/S is found to be the least favoured with an energy barrier of 15.9 kcal/mol. In the reaction of amide-substituted oxanorbornadiene reaction with the C-phenyl, N-methyl nitrone, TS1-ex/R is found to be the most favoured route with an activation barrier of 0.4 kcal/mol. However, for cyano-substituted oxanorbornadiene reaction with the C-phenyl, N-methyl nitrone, TS2-en/R is found to be the most favoured with an activation barrier of 0.2 kcal/mol. In all cases, the intermediates formed from the (3 + 2) cycloaddition are very stable. The stability of the 3 + 2 adducts and the high barriers of the cycloreversion steps make the cycloreversion step unfavourable. Therefore, there are no doubts that the cycloreversion step will not occur. These observations are consistent with some recent studies that have found the thermolytic cleavage step in these kinds of reactions as highly unfavourable due to high barrier constraints [22, 24]. The energies of the cycloreversion step as well as the reaction energies for all the substituents considered in the reaction of substituted ONBD and C-phenyl, N-methyl nitrone are shown in Table 2.
3.3 The reaction of methyl nitrone with CH3 −-, OMe−-, OH−- and NH2-substituted ONBD
This section discusses the effects of electron-donating substituents (CH3, OMe, OH, NH2) on the energetics of the reaction of ONBD with methyl nitrones. Methyl nitrone has been used as a surrogate of phenyl nitrone as recent studies on similar organic reactions have shown that methyl nitrone can be used as a reduced model for phenyl nitrone used in experiments [23, 24]. The activation and reaction energies of the various electron-donating substituents considered in this section of our study are given in Tables 3 and 4. Generally, it has been found that substituting the R groups on the dipolarophile with electron-donating groups generally favours the attack of the nitrone on the least hindered side which is contrary to the reaction of the phenyl nitrone with the substituted oxanorbornadiene. It turns out from the calculated activation energies in the case of amine-, methyl- and hydroxyl-substituted ONBD given in Table 3 that the most favoured reaction channel corresponds to the formation of the I2 cycloadduct through TS1-ex/R. These favoured TSs are located 0.9 kcal/mol (amine-substituted ONBD), 1.1 kcal/mol (methyl-substituted ONBD) and 1.2 kcal/mol (hydroxy-substituted ONBD) above the reactants. The remaining seven transition states leading to the formation of the different cycloadducts in each case, however, are relatively higher. On the other hand, in the reaction of OMe-substituted ONBD with methyl nitrone it found that the formation of the I8 via TS4-n/R is preferred.
3.4 The reaction of methyl nitrone with CN−-, Br−-, CO2Me−- and SO2H-substituted ONBD
In this section, we present a discussion on the effects of electron-withdrawing substituents (CN, Br, CO2Me and SO2H) on the energetics of the reaction of ONBD with methyl nitrones. Herein, the results shown in Tables 3 and 4 suggest that CN-substituted ONBD, CO2Me-substituted ONBD and SO2H-substituted ONBD proceed in a disparate fashion to the electron-donating groups (EDGs) reactivity where in all cases the TS1-ex/S is the most preferred reaction channel. The calculated activation barriers are 0.1 kcal/mol for CN, 0.4 kcal/mol for CO2Me and 0.8 kcal/mol for SO2H, respectively. However, for bromo-substituted ONBD, the TS1-ex/R is found to be the most favoured route with an activation barrier of 0.1 kcal/mol, which is in agreement with the established trend in the EDGs reactivity. Here again, the cycloreversion step involved in the reaction of the methyl nitrone and the various derivatives of oxanorbornadiene are found to be highly unfavourable. The energies of the cycloreversion step as well as the reaction energies for all the substituents considered in the reaction of substituted ONBD and C-methyl, N-methyl nitrone are shown in Table 4. It should be noted that in Tables 3 and 4, some transition states could not be located although it is conceivable they may exist.
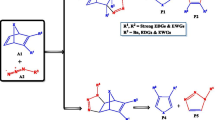
3.5 The reaction of dimethyl nitrilimine with methyl-substituted ONBD
Intrigued by the outcomes of the study so far, we extended our studies to the reactivity of methyl-substituted ONBD with a different dipole (dimethyl nitrilimine). In this case, the 1,3-dipolar cycloaddition reaction of the dimethyl nitrilimine with ONBD can take place along four reaction channels corresponding to the regioselectivity arising from whether the approach of the dipolarophile by the dipole is via path ‘a’ or path ‘b’, and the various stereo-selectivities arising from whether the group is syn or anti to the bridging atom as shown in Schemes 4 and 5. From the calculated relative energies (Fig. 4), TS4-ex is favoured kinetically in comparison with the other approaches; in addition, the formation of products P1 + P2 is favoured thermodynamically. Therefore, the expected isolable products in these reactions are the 5-membered heterocycles (furans and pyrazolines).

The reaction of methyl-substituted oxanorbornadiene with dimethyl nitrilimine

Diels–Alder cycloreversion of bicyclic pyrazoline cycloadducts to form derivatives of furan and monocyclic pyrazolines

Free energy profile for 32CA of a methyl-substituted oxanorbornadiene to dimethyl nitrilimine. All relative energies in kcal/mol
3.6 Global reactivity indices
In recent times, the global reactivity index has become a popular reactivity descriptor in computational organic chemistry. In view of this, we calculated the electrophilicity indices (ω) and maximum electronic charge transfer (ΔNmax) of the various derivatives of the oxanorbonadienes considered in this work. Within the context of the global reactivity index, reactants with large ω values are more reactive towards nucleophiles. Inspection of the tabulated results (Table 5) will reveal that the trends in the electrophilicity indices follow the order nitro > CN = aldehyde > ester > parent > methyl = hydroxy > amine > methoxy. Thus, given the series of the substituted oxanorbonadiene considered in this present study, nitro-substituted oxanorbonadiene will be the most reactive towards a nucleophile, whereas methoxy-substituted oxanorbonadiene is expected to be the least reactive. This observation is in total agreement with the trends in activation energies computed for the reactions of the various oxanorbonadiene derivatives with the nitrones considered in this work.
3.7 Local reactivity indices of oxanorbornadiene and phenyl nitrone
In attempt to rationalize the regioselectivity of the reactions, we invoked the global electrophilic (\( P_{K}^{ + } ) \) and nucleophilic (\( P_{K}^{ - } ) \) Parr functions. Within the conceptual density functional theory (CDFT) proposed by Domingo et al. [33], electron density always fluxes from the nucleophile to the electrophile in polar organic reactions. Therefore, atomic centres with the largest magnitude of the electrophilic and nucleophilic Parr functions will be the point of attack during the cycloaddition. We have selected the reaction of the ester-substituted oxanorbonadiene and the phenyl nitrone as representative model for this study. Consequently, we have presented the various atomic labels for the ester-substituted oxanorbonadiene and the phenyl nitrone in Fig. 5 for brevity sake.

Atomic labels of the ester-substituted oxanorbonadiene (dipolarophile) and phenyl nitrone (dipole)
The Mulliken and NBO atomic spin densities (ASD) at the cycloaddition centres give a quantitative measure of their electron density. Therefore, atomic centres with the largest Mulliken and NBO spin densities in a given molecule will be the most favoured point of attack during the cycloaddition reaction.
Results obtained from the Mulliken and NBO atomic spin densities calculations are summarized in Table 6 in the supporting information. Some recent studies [23, 24] have found that in this type of cycloaddition reaction, the dipolarophile assumes an electrophilic character while the nitrone manifests nucleophilic behaviour. Therefore, the appropriate atomic spin density to describe the dipolarophile and the dipole is the cationic and anionic spin densities, respectively. Inspection of the results shows that, for the electrophilic Mulliken spin densities, C1 = 0.103, C2 = 0.087, C4 = −0.056 and C5 = −0.036. Hence, in the cycloaddition reaction the nitrone should be expected to selectively add across carbons C4 and C5, which is consistent with the trends established from the activation energies as well as the experimentally obtained results.
Similarly, analysis of the electrophilic NBO spin densities reveal that C1 = 0.071, C2 = 0.012, C4 = −0.094 and C5 = −0.076. Based on these results, it is expected that in the cycloaddition reaction the nitrone will preferentially add across carbons C4 and C5, which is consistent with the trends established from the activation energies as well as the experimental product outcomes reported.
For the purposes of reproducing our results, we have reported the Cartesian coordinates for all the relevant structures involved in the reaction of the dimethyl nitrile imine with the dimethyl-substituted oxanorbonadiene in Table S3. Additionally, the Cartesian coordinates for all the structures involved in the reaction of the phenyl nitrone with the diester-substituted oxanorbonadiene are also reported in Table S4.
4 Conclusion
In the reactions of C,N-disubstituted nitrones with oxanorbonadiene derivatives, the activation barriers for the formation of the R enantiomer are very low compared to the barriers for the formation of S enantiomer. The formation of the exo stereo-isomers is generally favoured, and this selectivity is as a result of steric factors in the transition states. In the reaction of the parent oxanorbonadiene with the phenyl nitrone, formation of the exo stereo-selective S isomer (TS1-ex/S to I1) is the most favoured. Generally, electron-withdrawing substituents on the oxanorbornadiene favour the nitrone attack on the highly substituted double bond, while electron-donating substituents on the oxanorbornadiene favour the attack on less substituted double bond. Results obtained from the Parr functions reveal that the cycloaddition occurs between atomic centres with the largest Mullikan and NBO atomic spin densities.
In the reactions of dimethyl nitrilimine to methyl-substituted ONBD, the reaction proceeds via addition across the least substituted double bond of the ONBD reactant (path A). The five-membered pyrazolines and furans are formed because, not only are the barriers for the cycloreversion steps relatively lower, the products are relatively more stable than the intermediates.
5 Supporting information
The Supporting Information file provides Cartesian coordinates of all optimized geometries and absolute energies for all reactants, intermediates and transition states considered in this study.
References
Wanapun D, Van KA, Mosey NJ, Kerr MA, Woo TK (2005) The mechanism of 1,3-dipolar cycloaddition reactions of cyclopropanes and nitrones A theoretical study. Can J Chem. https://doi.org/10.1139/V05-182
Nie X, Lu C, Chen Z, Yang G, Nie J (2014) Enantioselective 1,3-dipolar cycloadditions of nitrones with unsaturated aldehydes promoted by a recyclable tetraarylphosphonium supported imidazolidinone catalyst. J Mol Catal A Chem 393:171–174. https://doi.org/10.1016/j.molcata.2014.06.015
Hashimoto T, Maruoka K (2015) Recent advances of catalytic asymmetric 1,3-dipolar cycloadditions. Chem Rev 115:5366–5412. https://doi.org/10.1021/cr5007182
Alcaide B, Almendros P, Alonso JM, Aly MF, Pardo C, Sáez E, Torres MR (2002) Efficient entry to highly functionalized β-lactams by regio- and stereoselective 1,3-dipolar cycloaddition reaction of 2-azetidinone-tethered nitrones. J Org Chem 67:7004–7013. https://doi.org/10.1021/jo025924e
Cheviet T, Dujardin G, Parrot I, Martinez J, Mousseron M, De Montpellier U, Bataillon PE (2016) Isoxazolidine: a privileged scaffold for organic and medicinal chemistry. Chem Rev. https://doi.org/10.1021/acs.chemrev.6b00543
Aggarwal VK, Roseblade SJ, Barrell JK, Alexander R (2002) Highly diastereoselective nitrone cycloaddition onto a chiral ketene equivalent: asymmetric synthesis of cispentacin. Org Lett 4:1227–1229. https://doi.org/10.1021/ol025665f
Nacereddine AK, Yahia W, Bouacha S, Djerourou A (2010) A theoretical investigation of the regio- and stereoselectivities of the 1,3-dipolar cycloaddition of C-diethoxyphosphoryl-N-methylnitrone with substituted alkenes. Tetrahedron Lett 51:2617–2621. https://doi.org/10.1016/j.tetlet.2010.03.025
Mandal S, Maiti KK, Banerji A, Prangé T, Neuman A, Acharjee N (2018) Experimental and DFT studies for substituent effects on cycloadditions of C,N-disubstituted nitrones to cinnamoyl piperidine. Ind J Chem 57:108–119
Maiuolo L, De Nino A (2015) Synthesis of isoxazolidines by 1,3-dipolar cycloaddition: recent advances. Targets Heterocycl Syst 19:299–345
Meng L, Wang SC, Fettinger JC, Kurth MJ, Tantillo DJ (2009) Controlling selectivity for cycloadditions of nitrones and alkenes tethered by benzimidazoles: combining experiment and theory. Eur J Org Chem. https://doi.org/10.1002/ejoc.200801211
Gothelf KV, Jørgensen KA (1998) Asymmetric 1,3-dipolar cycloaddition reactions. Chem Rev. https://doi.org/10.1021/CR970324E
Cristina D, De Amici M, De Micheli C, Gandolfi R (1981) Site selectivity in the reactions of 1,3-dipoles with norbornadiene derivatives. Tetrahedron 37:1349–1357. https://doi.org/10.1016/S0040-4020(01)92451-2
Wavefunction, Inc. (2013) Spartan’14. Wavefunction Inc, Irvine
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich A, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 09, revision A.02. Gaussian Inc., Wallingford
Zhao Y, Truhlar DG (2008) Density functionals with broad applicability in chemistry. Acc Chem Res 41(2):157–167. https://doi.org/10.1021/ar700111a
Pieniazek SN, Houk KN (2006) The origin of the halogen effect on reactivity and reversibility of Diels–Alder cycloadditions involving furan. Ang Chem Int Ed 45(9):1442–1445. https://doi.org/10.1002/anie.200502677
Paton RS, Mackey JL, Kim WH, Lee JH, Danishefsky SJ, Houk KN (2010) Origins of stereoselectivity in the trans Diels–Alder paradigm. J Am Chem Soc 132(27):9335–9340. https://doi.org/10.1021/ja1009162
Paton RS, Steinhardt SE, Vanderwal CD, Houk KN (2011) Unraveling the mechanism of cascade reactions of zincke aldehydes. J Am Chem Soc 133:3895–3905. https://doi.org/10.1021/ja107988b
Wheeler SE, Moran A, Pieniazek SN, Houk KN (2009) Accurate reaction enthalpies and sources of error in DFT Thermochemistry for aldol, Mannich, and α-aminoxylation reactions. J Phys Chem A 113:10376–10384. https://doi.org/10.1021/jp9058565
Clark M, Cramer RD, Van Opdenbosch N (1989) Validation of the general purpose Tripos 5.2 force field. J Comput Chem 10(8):982–1012. https://doi.org/10.1002/jcc.540100804
Tomasi J, Mennucci B, Cammi R (2005) Quantum mechanical continuum solvation models. Chem Rev 105(8):2999–3094. https://doi.org/10.1021/CR9904009
Opoku E, Tia R, Adei E (2019) Quantum chemical studies on the mechanistic aspects of tandem sequential cycloaddition reactions of cyclooctatetraene with ester and nitrones. J Mol Graph Model 92:17–31. https://doi.org/10.1016/J.JMGM.2019.06.019
Arhin G, Adams AH, Opoku E, Tia R, Adei E (2019) 1,3-Dipolar cycloaddition reactions of selected 1,3-dipoles with 7-isopropylidenenorbornadiene and follow-up thermolytic cleavage: a computational study. J Mol Graph Model 92:267–279. https://doi.org/10.1016/j.jmgm.2019.08.004
Opoku E, Tia R, Adei E (2019) DFT mechanistic study on tandem sequential [4 + 2]/[3 + 2] addition reaction of cyclooctatetraene with functionalized acetylenes and nitrile imines. J Phys Org Chem. https://doi.org/10.1002/poc.3992
Roland D, Haleegoah JN, Opoku E, Tia R, Adei E (2019) Mechanistic studies on tandem cascade [4 + 2]/[3 + 2] cycloaddition of 1,3,4-oxadiazoles with olefins. J Mol Graph Model 93:107452. https://doi.org/10.1016/j.jmgm.2019.107452
Opoku E, Tia R, Adei E (2019) Computational studies on [4 + 2]/[3 + 2] tandem sequential cycloaddition reactions of functionalized acetylenes with cyclopentadiene and diazoalkane for the formation of norbornene pyrazolines. J Mol Model 25:168. https://doi.org/10.1007/s00894-019-4056-x
Opoku E, Tia R, Adei E (2016) [3 + 2] Versus [2 + 2] addition: a density functional theory study on the mechanistic aspects of transition metal-assisted formation of 1, 2-dinitrosoalkanes. J Chem. https://doi.org/10.1155/2016/4538696
Parr RG, Szentpály LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121(9):1922–1924. https://doi.org/10.1021/ja983494x
Domingo LR, Chamorro E, Pérez P (2008) An understanding of the electrophilic/nucleophilic behavior of electro-deficient 2,3-disubstituted 1,3-butadienes in polar Diels–Alder reactions. A density functional theory study. J Phys Chem A 112(17):4046–4053. https://doi.org/10.1021/jp711704m
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions: a theoretical study. J Org Chem 73:4615–4624. https://doi.org/10.1021/jo800572a
Domingo LR, José AM, Pérez P, Contreras R (2002) Quantitative characterization of the local electrophilicity of organic molecules. Understanding the regioselectivity on Diels–Alder reactions. J Phys Chem A 106(29):6871–6875. https://doi.org/10.1021/jp020715j
Koopmans T (1934) Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. Physica 1(1–6):104–113. https://doi.org/10.1016/S0031-8914(34)90011-2
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494. https://doi.org/10.1039/c2ra22886f
Acknowledgements
The authors are very grateful to the National Council for Tertiary Education, Republic of Ghana, for a research Grant under the Teaching and Learning Innovation Fund (TALIF/KNUST/3/0008/2005), and to South Africa’s Centre for High Performance Computing for access to additional computing resource on the Lengau cluster.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest whatsoever regarding the publication of this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Opoku, E., Arhin, G., Pipim, G.B. et al. Site-, enantio- and stereo-selectivities of the 1,3-dipolar cycloaddition reactions of oxanorbornadiene with C,N-disubstituted nitrones and dimethyl nitrilimines: a DFT mechanistic study. Theor Chem Acc 139, 16 (2020). https://doi.org/10.1007/s00214-019-2529-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-019-2529-8