
Abstract
A new capillary electrophoresis (CE) method was presented for the determination of asenapine, an atypical antipsychotic drug, in pharmaceuticals. Box-Behnken design, an experimental design method, was used to investigate the effects of run buffer pH, run buffer concentration and applied potential on the separation of asenapine and granisetron (IS). The optimum conditions were phosphate buffer (15 mM, pH: 3.1) with 25.7 kV voltage and 20 ℃ capillary temperature. The method was validated according to ICH guideline. A good linearity was obtained in the concentration range of 0.27–6.4 µg/mL with LOD and LOQ values of 0.07 and 0.24 μg/mL, respectively. The precision and accuracy of the method were satisfying with intra and interday recovery values of 97.8–100.8% and RSD less than 2%. The proposed CE method was applied to asenapine tablets successfully, for the first time.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Schizophrenia is a well-known mental disorder which is characterized by auditory hallucinations, delusions, disorganized thoughts, lack of motivation, social withdrawal and also memory impairments (Volk and Lewis 2015).
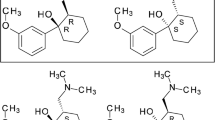
Asenapine (ASE) is an antipsychotic drug used in the treatment of schizophrenia. It is also effective in treating manic and mixed episodes of bipolar disorder. The molecule contains two chiral centers as seen in Fig. 1 and can exist as four stereoisomers. Two trans isomers have higher binding affinity to receptors than the cis isomers, therefore ASE has been approved in the form of a racemate of R,R- and S,S-enantiomers (Protti et al. 2018). It has been commercially available in sublingual tablet form since 2009 in the USA (Citrome 2014). ASE has affinity on serotonin receptors, dopamine receptors, adrenergic receptors, histamine (H1) receptors and also moderate affinity on histamine (H2) receptors. (Gonzalez et al. 2011).

Chemical structure of ASE
Several methods were reported for ASE determination in biological samples using liquid chromatography (Kovatsi et al. 2015; Protti et al. 2018), liquid chromatography-mass spectrometry (de Boer et al. 2012a,b ; Ansermot et al. 2013; Reddy et al. 2013; Patteet et al. 2014; Sempio et al. 2014; Sistik et al. 2016; Patel et al. 2018) and gas chromatography-mass spectrometry (Miller et al. 2013).
Also, there are several papers including spectrometry (Borkar and Gaikwad 2016), HPLC (Chhalotiya et al. 2012; Aneesh and Rajasekaran 2012; Govindarajan et al. 2012; Managuli et al. 2016; Karaca and Ugur 2017) and HPTLC methods (Patel et al. 2015; Ramadan et al. 2017) reported for the quantitation of ASE in pharmaceutical preparations (Table 1).
CE has become a widely applied separation technique in pharmaceutical analyses. It has numerous advantages like simplicity, rapid analysis, automation and low cost (Ahuja 2011). This technique offers better separation, shorter analysis times and lower reagent consumption compared to HPLC. Low consumption of reagents in CE is due to the low internal volume of capillary and the ability to use the same buffer vials for electrophoretic runs. In addition, residues produced in CE are mainly composed of salts that are naturally disposable after neutralization. As a result, CE is a healthier and more environmentally friendly method than HPLC which produces larger amounts of residues that require special handling. On the other hand, CE has some limitations such as lower sensitivity and reproducibility compared to HPLC (Bizzotto et al. 2013). Lower sensitivity limitation can be improved by preconcentration techniques or alternative detection techniques (Hempel 2000). Another disadvantage of CE is its limited use for preparative separations due to narrow capillaries and complicated adaptation of analytical CE setups (Kašicka 2020). Considering all these advantages and disadvantages, it can be concluded that CE is a valuable counterpart and/or complement of HPLC method.
Although many HPLC methods were developed for ASE quantitation in pharmaceuticals (Table 1), no CE method was reported for this purpose. Szabó et al. (2016) developed the only CE method for chiral separation of ASE enantiomers and they characterized cyclodextrin complexes by mass spectrometry, NMR spectroscopy and molecular modeling. The developed method was validated, only for the parameters of range, linearity, precision and accuracy. Although it has the advantage of wide linear range, LOD and LOQ values (2 µg/mL and 5 µg/mL, respectively) are higher than the proposed method.
Experimental design approach has been replaced traditional optimization process because of its advantages including reliable and valid data and effort, time, resource saving with a reduced number of experiments. It uses multivariate data and establishes a linear or quadratic empirical function among the interacting data to have information about a system (Hibbert 2012). Application of experimental design can be classified as screening designs and response surface designs. Screening designs aim to determine which factors have significant effects on a process. These techniques are important especially when the important factors, their ranges, and interactions are not yet known (Hanrahan et al. 2008). Response surface designs are used for discovering the best values of factors. The most frequently used designs in analytical method development are central composite and Box–Behnken designs. (Hanrahan et al. 2008).
The aim of the current work is to propose a novel, fast and simple CE method for the determination of ASE in its pharmaceuticals using experimental design method, for the first time. The optimum method was fully validated according to related ICH guideline (2005).
Materials and methods
Materials
ASE (≥ 98%) was purchased from Sigma Aldrich (India) and granisetron (GRA) was obtained from LGC Standards (Germany). H3PO4 (85%) was from VWR Chemicals (France). KH2PO4 (99.5–100.5%) and NaOH (≥ 97%) were supplied from Merck (Germany). The other chemicals were HPLC grade from Sigma Aldrich (Germany).
Preparation of standard solutions and quality control (QC) samples
The stock solutions of ASE and GRA were dissolved in methanol and distilled water, respectively, at a concentration of 1 mg/mL. The last dilutions to prepare working solutions were conducted with 1/10 diluted background electrolyte (BGE). BGE was 15 mM phosphate buffer (pH: 3.1). The calibration solutions and QC samples were prepared by adding diluted GRA solution to serially diluted ASE solutions. Concentrations of the calibration solutions were in the range of 0.27–6.4 μg/mL ASE with 5 μg/mL GRA. Also, QC solutions including 0.42, 1.68 and 5.6 μg/mL ASE with 5 μg/mL GRA were analyzed.
Instrumentation and electrophoretic conditions
Electrophoretic experiments were conducted by an Agilent 7100 CE system (Agilent Technologies, USA) with a photodiode array detector. The compounds were monitored at 220 nm. Separations were achieved in a 75 µm i.d. capillary of 40 cm effective and 48 cm total length. The analytical balance and pH-meter were from Mettler-Toledo (Switzerland). The solutions were sonicated in an ultrasonic bath (Bandelin Electronic RK510H, Germany). Velp Scientifica ZX3 model vortex (Italy) and Eppendorf 5810R model centrifuge (Germany) were also used. All solutions were filtered by a Macherey–Nagel Chromafil PET-45/25 polyester 0.45 µm filter (Germany) before injection.
New fused-silica capillary was activated by flushing with 1.0 M NaOH for 30 min. Each day, consecutive 10 min conditioning steps with 0.1 M NaOH, water and BGE were applied. The samples were then injected by vacuum injection for 1 s. Between runs, the capillary was flushed with 0.1 M NaOH and water for 2 min, respectively, and then 2 min with BGE. At the end of each day, the capillary was rinsed with 0.1 M NaOH and water for 10 min and left with aspirated air. The voltage was set at 25.7 kV during the analysis.
Sample preparation
A Sycrest® tablet (10 mg ASE) was weighed as 30.71 mg. It was powdered and the amount of 3.1 mg was weighed, dissolved in methanol and sonicated for 10 min. The solution was centrifuged at 4000 rpm for 10 min and the supernatant was separated as tablet stock solution including 1.0 mg/mL ASE. It was daily diluted by methanol and 1/10 diluted BGE, respectively.
Method validation
The validation of the presented method was performed according to ICH guideline (2005). Accuracy, LOD, LOQ, linearity, precision, specificity, robustness and stability of ASE solutions were investigated.
The precision and accuracy were investigated by six replicated analyzes of QC solutions (0.42, 1.68 and 5.6 μg/mL ASE) on three consecutive days. Also, accuracy in drug product was determined by the analysis of tablet extracts spiked with standard ASE solutions at the same concentrations.
The specificity was evaluated by the analysis of ASE solutions after treatments of 0.1 M HCl, 0.1 M NaOH or 3% H2O2 at room temperature for 30 min.
The stability was tested using ASE solutions after storage under three conditions. One of them is keeping ASE solutions at room temperature for 24 h to investigate short term stability, another is keeping at − 20 ℃ for two weeks to investigate long term stability. And as the last one, three freeze–thaw cycles were applied to the ASE solution for testing freeze–thaw stability. After analyzing the solutions in three replicates, RSD and recovery were evaluated.
The linearity was investigated with six concentrations in the range of 0.27–6.4 μg/mL. Calibration sets were injected for three consecutive days.
The robustness was evaluated by inspecting the counter plots. The flat area around the optimal conditions means the values of the responses near this point are stable (Jancic-Stojanovic et al. 2009). In addition, analyzes using 1.68 μg/mL ASE were carried out by conducting deliberate changes in run buffer pH (3.0, 3.2), run buffer concentration (13, 17 mM), voltage (25.2, 26.2 kV) or wavelength (218, 222 nm). Recovery values under these altered conditions were compared with those in optimum conditions.
Software
Experimental design studies and validation calculations were conducted using the programs of Minitab 19 and GraphPad Prism 6, respectively. In addition to these programs, Microsoft Excel was used.
Results and discussion
Optimization of the method
In this study, choosing factors with a preliminary study was preferred instead of a screening study because CE parameters influencing the analysis were predictable according to our previous experiences in this area. The Box-Behnken design was used as a response surface design because of its advantages like avoiding the extreme levels of factors and requiring less analysis compared to central composite designs (Hibbert 2012). Applied potential, concentration and pH of run buffer were selected as factors for this study because of their high influences on electrophoretic separation. (Table S1). Other parameters such as capillary, capillary temperature, internal standard and type of run buffer were optimized in a preliminary research. A capillary of 75 µm i.d and 40 cm effective length (48 cm total) was used at 20 ℃. Phosphate buffer was used as a run buffer and GRA was chosen as the internal standard because it has a detector response similar to that of ASE. Determination wavelength was 220 nm. The expectations from the planned analysis and restrictive factors of CE were taken into consideration during the selection of responses. Migration time of ASE and resolution between ASE and GRA were selected to ensure the effective separation of analyte and internal standard in a short time. Peak area of ASE was selected to see the highest signal, therefore to develop a suitable method for the analysis at low concentrations. And finally generated current was selected because all factors were effective on the current and the increasing current value was restrictive due to the joule heating. Briefly, migration time of ASE, peak area of ASE, generated current and resolution between ASE and GRA were selected as responses.
A solution including ASE and GRA (both are 10 μg/mL) was analyzed for three times in random order according to the experimental plan using Box-Behnken design (Table S2). Minitab 19 was used for data analysis and quadratic response models as shown in Eq. 1 were found for each response.
Y is the response; b values are the coefficients and x1, x2, x3 are the factors. If the coefficients differ significantly from zero and the p value is lower than 0.050, it is considered as a factor affecting the response (Jancic-Stojanovic et al. 2009). The significant responses are marked with * as seen in Table 2.
By the Derringer`s desirability function, the challenge of optimizing all answers at the same time has been overcome. Using Eqs. 2–4, each response was transformed to a desirability function (between 0 and 1). 1 means a completely desired response and 0 means an undesirable response. If the value of response is to be maximum, Eq. 2 is applied, if the value of response is to be minimum Eq. 3 is applied and if a response target is a good type, Eq. 4 is preferred (Jancic-Stojanovic et al. 2009).
Li exhibits the lower value of the response, Ui shows the upper value of the response and Ti is the target value of the response Yi (Table S3). The weight factors (s and t) can be chosen according to attribute importance level to the target value. When the weight factors are equal to 1, the desirability function is linear (Jancic-Stojanovic et al. 2009). The overall desirability (D) value is obtained from the geometric average of the desirability values found for each variable (Table S4). Another quadratic response model was prepared using D as only response (Table 2).
R2 and \({R}_{a}^{2}\) values are the determination coefficient and adjusted determination coefficient, respectively. They show the suitability of the system. R2 value which is approximately 1 indicates the closeness of measured and predicted values (Mason et al. 2003). The desirability function shows the goodness of the model as the values of R2 and \({R}_{a}^{2}\), that were 0.8937 and 0.7023, respectively.
The optimal conditions were obtained using the last quadratic response model which is found with D value. In Fig. 2, surface and counterplots are presented. Optimum conditions were found as phosphate run buffer (15 mM, pH: 3.1) and 25.7 V applied voltage. An analysis was made in these optimum conditions and appropriate migration time, peak area, resolution and current were obtained (Table S5). An electropherogram of ASE and GRA in tablet extract solution is shown in Fig. 3.

Counter and surface plots: (a, d) D = f(x2, x1); (b, e) D = f(x3, x1); (c, f) D = f(x3, x2). D overall desirability, x1 run buffer pH, x2 run buffer concentration, x3 voltage

An electropherogram of ASE in tablet extract solution (spiked with 1.68 μg/mL ASE and 5 μg/mL GRA). Conditions: Capillary of 75 µm i.d and 40 cm effective length (48 cm total); run buffer of phosphate buffer (15 mM, pH: 3.1). The voltage was 25.7 kV with detection wavelength of 220 nm and capillary temperature of 20 ℃
Method validation
The accuracy and precision of the method were investigated as described in the experimental section and intra- and interday RSD and recovery values were given in Table 3. Method was found accurate and precise due to recovery values in the range of 97.8–100.8% and RSD values which are less than 2%.
The specificity was investigated by the analysis of 1.68 μg/mL ASE after three stress treatments of base hydrolysis (0.1 M NaOH), acid hydrolysis (0.1 M HCl) and oxidation (3% H2O2) for 30 min. Following base hydrolysis, low recovery was obtained with no interference peak. Recovery values were given in Table 4.
The stability of ASE solutions was investigated by analysis of 0.42, 1.68 and 5.6 μg/mL ASE after storage under three different conditions as described in the experimental section. High recoveries as seen in Table 4, demonstrate that ASE is stable under all working and storage conditions.
The results of linear regression analysis were given in Table 5. The y-intercepts of the linear equations were found statistically insignificant (p > 0.05). Also, calculated p-values which are higher than 0.05 show that the residuals have a normal distribution (Table 5). LOD and LOQ were obtained based on signal-to-noise approach. LOD, the concentration corresponding to a signal-to-noise of 3:1, was determined as 0.07 μg/mL. LOQ, the concentration corresponding to a signal to noise ratio of 10:1, was found as 0.24 μg/mL. According to these results, the present method is more sensitive (Aneesh and Rajasekaran 2012; Govindarajan et al. 2012; Ramadan et al. 2017; Karaca and Ugur 2017) and also, more rapid (Chhalotiya et al. 2012; Managuli et al. 2016; Karaca and Ugur 2017) than many other reported methods (Table 1).
The counterplots of desirability given in Fig. 2 were demonstrated the robustness of the developed method. The flat area around the optimal conditions means that the values of responses (migration time of ASE, the peak area of ASE, generated current and resolution between ASE and GRA) are stable near this point. This represents the robustness of the optimal conditions. The robustness of the method was confirmed with the analyzes carried out in slightly changed experimental conditions (run buffer pH, run buffer concentration, voltage or wavelength). No significant changes in recovery values were observed in changed conditions as seen in Table 6.
Tablet analysis
The proposed method was used for the determination of ASE in Sycrest® tablets (10 mg ASE). Tablet solutions (0.42, 1.68 and 5.6 μg/mL ASE) were analyzed in six replicates. ASE content was obtained as 10.10 ± 0.16 mg with the recovery of 100.9% and RSD of 1.54%. Also, the electropherograms with no interference support that the presented method can be successfully used for the determination of ASE in tablets.
Conclusion
CE has many advantages over other methods including rapid analysis time, low solvent consumption, high efficiency and selectivity especially for pharmaceutical analysis. A fast and simple CE method was presented for the determination of ASE in pharmaceuticals. Experimental design approach was employed to obtain a faster, easier and more reliable optimization process with less number of experiments. The method was fully validated according to precision, linearity, accuracy, robustness, LOD, LOQ and specificity parameters and all parameters were found appropriate. The present method was successfully used for the determination of ASE in pharmaceuticals, confirming its applicability in routine analysis.
References
Ahuja S (2011) Overview of capillary electrophoresis in pharmaceutical analysis. In: Ahuja S, Jimidar MI (eds) Capillary Electrophoresis methods for pharmaceutical analysis. Academic Press, USA, pp 1–8
Aneesh TP, Rajasekaran A (2012) Stress degradation studies and development and validation of RP-HPLC method for the estimation of asenapine maleate. Int J Pharm Pharm Sci 4(4):448–451
Ansermot N, Brawand-Amey M, Kottelat A, Eap CB (2013) Fast quantification of ten psychotropic drugs and metabolites in human plasma by ultra-high performance liquid chromatography tandem mass spectrometry for therapeutic drug monitoring. J Chromatogr A 1292:160–172. https://doi.org/10.1016/j.chroma.2012.12.071
Bizzotto CS, Meinhart AD, Ballus CA, Ghiselli G, Godoy HT (2013) Comparison of capillary electrophoresis and high performance liquid chromatography methods for caffeine determination in decaffeinated coffee. Food Sci Technol 33(1):186–191. https://doi.org/10.1590/S0101-20612013005000013
de Boer T, Meulman E, Meijering H, Wieling J, Dogterom P, Lass H (2012a) Quantification of asenapine and three metabolites in human plasma using liquid chromatography-tandem mass spectrometry with automated solid-phase extraction: application to a phase I clinical trial with asenapine in healthy male subjects. Biomed Chromatogr 26(2):156–165. https://doi.org/10.1002/bmc.1640
de Boer T, Meulman E, Meijering H, Wieling J, Dogterom P, Lass H (2012b) Development and validation of automated SPE-HPLC-MS/MS methods for the quantification of asenapine, a new antipsychotic agent, and its two major metabolites in human urine. Biomed Chromatogr 26(12):1461–1463. https://doi.org/10.1002/bmc.2722
Borkar AA, Gaikwad NJ (2016) UV spectrophotometric and RP-HPLC estimation of drug asenapine in tablet dosage form. Int J Pharm Sci Res 7(7):3080–3084. https://doi.org/10.13040/IJPSR.0975-8232.7(7).3080-84
Chhalotiya UK, Bhatt KK, Shah DA, Patel JR (2012) Stability-indicating liquid chromatographic method for the quantification of the new antipsychotic agent asenapine in bulk and in pharmaceutical formulation. Sci Pharm 80(2):407–417. https://doi.org/10.3797/scipharm.1112-07
Citrome L (2014) Asenapine review, part II: clinical efficacy, safety and tolerability. Expert Opin Drug Saf 13(6):803–830. https://doi.org/10.1517/14740338.2014.908183
Gonzalez JM, Thompson PM, Moore TA (2011) Review of the safety, efficacy, and side effect profile of asenapine in the treatment of bipolar 1 disorder. Patient Prefer Adher 5:333–341. https://doi.org/10.2147/PPA.S10968
Govindarajan N, Koulagari S, Methuku A, Podhuturi S, Venkata RB (2012) Method development and validation of RP-HPLC method for determination of new antipsychotic agent asenapine maleate in bulk and in pharmaceutical formulation. Der Pharmacia Lettre 4(6):1805–1810
Hanrahan G, Montes R, Gomez FA (2008) Chemometric experimental design based optimization techniques in capillary electrophoresis: a critical review of modern applications. Anal Bioanal Chem 390:169–179. https://doi.org/10.1007/s00216-007-1619-y
Hempel G (2000) Strategies to improve the sensitivity in capillary electrophoresis for the analysis of drugs in biological fluids. Electrophoresis 21:691–698. https://doi.org/10.1002/(SICI)1522-2683(20000301)21:4<691:AID-ELPS691>3.0.CO;2-U
Hibbert DB (2012) Experimental design in chromatography: a tutorial review. J Chromatogr B Analyt Technol Biomed Life Sci 910:2–13. https://doi.org/10.1016/j.jchromb.2012.01.020
ICH (2005) International Conference on Harmonization. Guideline Q2(R1): Validation of analytical procedures. https://www.ich.org/products/guidelines/quality/quality-single/article/analytical-procedure-development-and-revision-of-q2r1-analytical-validation-copy-1.html. Accessed 15 April 2019
Jancic-Stojanovic B, Malenovic A, Ivanovic D, Rakic T, Medenica M (2009) Chemometrical evaluation of ropinirole and its impurity’s chromatographic behavior. J Chromatogr A 1216(8):1263–1269. https://doi.org/10.1016/j.chroma.2008.10.059
Karaca SA, Ugur DY (2017) A stability indicating ion-pair LC method for the determination of asenapine in pharmaceuticals. J Chil Chem Soc 62(1):3325–3329. https://doi.org/10.4067/S0717-97072017000100004
Kašicka V (2020) Recent developments in capillary and microchip electroseparations of peptides (2017–mid 2019). Electrophoresis 41:10–35. https://doi.org/10.1002/elps.201900269
Kovatsi L, Titopoulou A, Tsakalof A, Samanidou V (2015) HPLC analysis of antipsychotic asenapine in alternative biomatrices: hair and nail clippings. J Liq Chrom Relat Tech 38(18):1666–1670. https://doi.org/10.1080/10826076.2015.1089894
Managuli RS, Kumar L, Chonkar AD, Shirodkar RK, Lewis S, Koteshwara KB, Reddy MS, Mutalik S (2016) Development and validation of a stability-indicating rp-hplc method by a statistical optimization process for the quantification of asenapine maleate in lipidic nanoformulations. J Chromatogr Sci 54(8):1290–1300. https://doi.org/10.1093/chromsci/bmw062
Mason RL, Gunst RF, Hess JL (2003) Linear regression with several predictor variables. statistical design and analysis of experiments. Wiley, New York, pp 496–534
Miller C, Pleitez O, Anderson D, Mertens-Maxham D, Wade N (2013) Asenapine (Saphris (R)): GC-MS method validation and the postmortem distribution of a new atypical antipsychotic medication. J Anal Toxicol 37(8):559–564. https://doi.org/10.1093/jat/bkt076
Patel NP, Sanyal M, Sharma N, Patel DS, Shrivastav PS, Patel BN (2018) Determination of asenapine in presence of its inactive metabolites in human plasma by LC-MS/MS. J Pharm Anal 8(5):341–347. https://doi.org/10.1016/j.jpha.2018.06.002
Patel RB, Naregalkar NS, Patel MR (2015) Stability-indicating HPTLC method for quantitative estimation of asenapine maleate in pharmaceutical formulations, equilibrium solubility, and ex vivo diffusion studies. J Liq Chrom Relat Tech 38(19):1731–1739. https://doi.org/10.1080/10826076.2015.1092448
Patteet L, Maudens KE, Sabbe B, Morrens M, De Doncker M, Neels H (2014) High throughput identification and quantification of 16 antipsychotics and 8 major metabolites in serum using ultra-high performance liquid chromatography-tandem mass spectrometry. Clin Chim Acta 429:51–58. https://doi.org/10.1016/j.cca.2013.11.024
Protti M, Vignali A, Blanco TS, Rudge J, Bugamelli F, Ferranti A, Mandrioli R, Mercolini L (2018) Enantioseparation and determination of asenapine in biological fluid micromatrices by HPLC with diode array detection. J Sep Sci 41(6):1257–1265. https://doi.org/10.1002/jssc.201701315
Ramadan NK, Mohamed TA, Fouad RM, Moustafa AA (2017) Stability-indicating high-performance liquid chromatography and thin-layer chromatography methods for the determination of cyclobenzaprine hydrochloride and asenapine maleate. JPC-J Planar Chromat 30(4):313–322. https://doi.org/10.1556/1006.2017.30.4.12
Reddy AVB, Venugopal N, Madhavi G (2013) Simultaneous determination of asenapine and valproic acid in human plasma using LC–MS/MS: application of the method to support pharmacokinetic study. J Pharm Anal 3(6):394–401. https://doi.org/10.1016/j.jpha.2013.04.008
Sempio C, Morini L, Vignali C, Groppi A (2014) Simple and sensitive screening and quantitative determination of 88 psychoactive drugs and their metabolites in blood through LC-MS/MS: application on postmortem samples. J Chromatogr B Analyt Technol Biomed Life Sci 970:1–7. https://doi.org/10.1016/j.jchromb.2014.08.039
Sistik P, Urinovska R, Brozmanova H, Kacirova I, Silhan P, Lemr K (2016) Fast simultaneous LC/MS/MS determination of 10 active compounds in human serum for therapeutic drug monitoring in psychiatric medication. Biomed Chromatogr 30(2):217–224. https://doi.org/10.1002/bmc.3538
Szabó ZI, Toth G, Voelgyi G, Komjati B, Hancu G, Szente L, Sohajda T, Beni S, Muntean DL, Noszal B (2016) Chiral separation of asenapine enantiomers by capillary electrophoresis and characterization of cyclodextrin complexes by NMR spectroscopy, mass spectrometry and molecular modeling. J Pharm Biomed Anal 117:398–404. https://doi.org/10.1016/j.jpba.2015.09.022
Volk DW, Lewis DA (2015) Schizophrenia A2 - Rosenberg, Roger N. In: Pascual JM (ed) Rosenberg's molecular and genetic basis of neurological and psychiatric disease. Academic Press, Boston, pp 1293–1299
Acknowledgements
Authors gratefully acknowledge the support of Research Council of Anadolu University for the funding of this study (Project No: 1302S023).
Funding
This work was supported by Research Council of Anadolu University (Project No: 1302S023).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Aliyeva, S., Atila Karaca, S., Uğur, A. et al. A novel capillary electrophoresis method for the quantification of asenapine in pharmaceuticals using Box-Behnken design. Chem. Pap. 74, 4443–4451 (2020). https://doi.org/10.1007/s11696-020-01256-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01256-5