
Abstract
In this work, graphene oxide/metal–organic framework-74/Fe3O4/polytyramine (GO/MOF-74/Fe3O4/PTy) nanoporous composite was synthesized as a sustainable sorbent for magnetic dispersive micro-solid phase extraction (MD-µ-SPE) of prokinetic drugs. The new nanosacle sorbent in combination with high performance liquid chromatography–ultraviolent detection (HPLC–UV) was applied for simultaneous trace quantification of target drugs in human plasma. MOF-74 and PTy were used as surface modifiers to improve the properties of GO nanosheets, such as surface area-to-volume ratio, adsorption capacity, hydrophobic interactions and selectivity. A composite of GO/MOF-74/Fe3O4 was fabricated and then an oxidative polymerization of tyramine was performed on the surface of sorbent using horseradish peroxidaze (HRP) enzyme as a catalyst. The characterization of the hybrid sorbent was evaluated using scanning electron microscopy (SEM), energy-dispersive X-ray analysis (EDX), X-ray diffraction (XRD) and Fourier transform-infrared (FT-IR) spectroscopy. The limit of detections (LODs, S/N = 3) for domperidone (DOM) and itopride (ITP) were 0.4 and 1.1 ng mL−1, respectively. Notable linearity (0.995 ≥ r2 ≥ 0.991) and practical dynamic concentration ranges of 1.5–1100.0 ng mL−1 and 4.0–1750.0 ng mL−1 were obtained for DOM and ITP, respectively. Intra-assay (≤ 8.6%, n = 12) and inter-assay (≤ 9.0%, n = 12) precisions along with appreciable accuracies (≤ 9.3%) demonstrated satisfactory performance of the current method. Ultimately, this approach was utilized for trace monitoring of DOM and ITP in human plasma after low dose administration and some pharmacokinetic features were investigated in detail.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Recent attempts in the field of sample preparation are focused on improving the sensitivity of analytical data, removing the main interferences and launching new technological developments such as miniaturization, cost-effective presentation (Li et al. 2018), on-line coupling with measurement instrument (Kostolanska et al. 2019) and reduction of hazardous reagents (Golebiowski et al. 2017; Hagarová 2017).
Among new sample enrichment protocols, magnetic solid-phase extraction (MSPE) as an inventive and well-organized kind of solid-phase extraction (SPE) has received extensive attention and it has been widely applied for the analysis of environmental, biological, pharmaceutical and food samples (Musile et al. 2018). Magnetic nanoscale sorbents are considered as innovative and promising extractors and they revealed wide applications in MSPE for different analytical purposes (Li et al. 2017, 2018; Adlnasab et al. 2018; Corazza et al. 2017).
Graphene oxide (GO), a new class of carbon-based substance with oxygen containing functional groups, has been widely applied as an efficient nanomaterial in sample preparation methods (Georgakilas et al. 2016). GO possesses some extraordinary characteristics such as high pore volume, considerable thermal and chemical stability along with excellent ability in adsorption processes through covalent and non-covalent interactions involving π–π stacking, electrostatic interactions and hydrogen bonding (Huang et al. 2012; Liu et al. 2014).
GO has been functionalized with different materials including silica (Wang et al. 2017), TiO2 (Sheshmani and Nayebi 2017), polymers (Liu et al. 2013) and magnetic nanoparticles (Wierucka and Biziuk 2014; Asgharinezhad and Ebrahimzadeh 2016). Some magnetic particles like Fe3O4 provide significant advantages including simple magnetic separation of sorbent without any special tools and appreciable reusability of hybrid material. Furthermore, among the compounds applied for modification of GO, metal–organic frameworks (MOFs) are superior and they reveal individual possessions such as uniformity, tunable pore size and facility to selective functionalization, which make them good alternatives for fabricating hybrid materials (Zhang et al. 2016; Ahmed and Jhung 2014; Stock and Biswas 2012). MOFs are known as a novel class of highly porous materials and they are created through coordination of metal ions with bi- or multidentate organic linkers (Li et al. 2017; Ahmed and Jhung 2014; Chakraborty and Maji 2014; Doherty et al. 2014; Janiak and Vieth 2010). The combination of GO and MOFs increases the merits of sorbent such as reusability, pore volume, dispersion capability, extraction capacity, mechanical strength as well as surface area (Li and Huo 2015; Petit and Bandosz 2009a, b). To date, several composites of MOF-GO including MOF-5 (Lin et al. 2017; Karimzadeh et al. 2019), HKUST-1 (Zhou et al. 2014; Zhao et al. 2014), MIL-100 (Tang and Wang 2017; Petit and Bandosz 2011) and ZIF-8 (Zhou et al. 2016; Wang et al. 2016) have been reported for isolation and quantitation of organic and inorganic species. MOF-74 belongs to a class of MOFs, in which transition metals are coordinated with 5-dioxidoterephthalate [M2(DOT)(H2O)2 (M = Mg, Mn, Fe, Co, Ni, and Zn; DOT = 2,5-dihydroxyterephthalate)]. MOF-74 has one-dimensional channels providing different diffusion routes and higher surface area-to-volume ratio (Wang et al. 2018; Asgharinezhad and Ebrahimzadeh 2016). In this work, MOF-74 was preferred to other MOFs and applied as a modifier agent of GO.
Some polymers polythionine, polyaniline and polythiophene have been reported in the literature to improve the surface quality of carbon-based materials (Babaei et al. 2018; Mehdinia et al. 2015; Salimikia et al. 2018; Zeeb and Farahani 2018). Polytyramine (PTy) could be easily formed by an oxidative polymerization in the presence of HRP horseradish peroxidaze (HRP) enzyme as a catalyst. The alkyl group existing in tyramine facilitates the formation of macropores in the polymer which increases the rate of diffusion (Yoshimura et al. 2012; Levashov et al. 2015). Therefore, PTy seems to be an opportune for modification of MOF-GO hybrid material, and it significantly improves π-stacking length, aromatic–aromatic interactions, the steadiness of sorbent and extraction yield of compounds in various matrices.
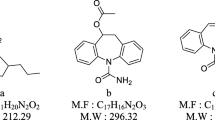
Prokinetic drugs such as itopride hydrochloride (ITP) and domperidone (DOM) are considered as newly developed drug. ITP (Fig. 1a) is able to stimulate gastrointestinal motor activity during the synergistic blockade of dopamine D2 receptor (Emmanuel et al. 1998; Ptaček et al. 2009; Singh et al. 2005; Ibrahim and Nasr 2016; Sisinthy et al. 2015). DOM (Fig. 1b) can block metabolization of enzyme acetylcholine esterase, which results an increase in stimulatory influence of acetylcholine (Sivakumar et al. 2007; Ali et al. 2006; Thanikachalam et al. 2008; Patel et al. 2007; Bose et al. 2009). As a result, combination therapy benefits from the properties of both drugs involving blocking dopamine D2 receptor and inhibiting acetylcholine esterase (Bose et al. 2009). Several analytical protocols such as spectrophotometry, potentiometry and high-performance liquid chromatography with fluorescence detection have been reported for quantification of DOM and ITP (Mohamed 2015; Abdel-Haleem et al. 2016; Ragab et al. 2015; Yoshizato et al. 2014; Abdelrahman 2013; Abdel-Ghany et al. 2015). However, these methods suffer from some limitations such as low sensitivity, tedious sample preparation, high matrix effect and lack of simultaneous quantification ability.

Chemical structure of itopride (a) and domperidone (b)
In this work, GO was functionalized with MOF-74 and then it was magnetized by Fe3O4 particles through a co-precipitation method. The fabrication process of sorbent was followed by an oxidative polymerization of tyramine in the presence of HRP enzyme to immobilize polytyramine on the surface. The newly designed hybrid material was applied as a sustainable and recyclable nanosorbent in magnetic dispersive micro-solid phase extraction (MD-µ-SPE). Two prokinetic drugs including domperidone (DOM) and itopride (ITP) were chosen as the model analytes to demonstrate the applicability of the fabricated sorbent for simultaneous extraction and enrichment prior to quantitation by HPLC–UV. Finally, the feasibility of the current approach was investigated by analyzing major pharmacokinetic data of DOM and ITP in human plasma of healthy volunteers after oral administration of fixed-dose combination tablets.
Experimental
Chemicals
All chemicals used in this work were obtained from analytical grade and used without extra purification. 2,5-Dihydroxy triphetalic acid, N,N-dimethylformamide, tyramine, horseradish peroxidaze and graphite powder (mesh of 100) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Iron (III) chloride hexahydrate (FeCl3·6H2O), iron (II) chloride tetrahydrate (FeCl2·4H2O), sodium nitrate (NaNO3), potassium permanganate (KMnO4), sulfuric acid (H2SO4, 98%), nickel (II) nitrate hexahydrate [Ni(II) (NO3)2·6H2O], sodium hydroxide (NaOH), Hydrochloric acid (HCl 5%), hydrogen peroxide (H2O2, 30%), ethanol (C2H5OH) and triethylamine were obtained from Merck Chemicals (Darmstadt, Germany). The standards of DOM and ITP drugs were supplied from Kusum Healthcare (Punjab, India). In all experiments, ultrapure water was used (Millipore, Bedford, MA, USA). Iranian blood transfusion organization (Tehran, Iran) provided fresh blood plasma sample and all biological samples were stored at − 18 °C and they were thawed just before use. HPLC grades of methanol, acetonitrile, acetone and potassium dihydrogen phosphate were obtained from Merck (Darmstadt, Germany). A fixed-dose combination form tablets (50 mg itopride/20 mg domperidone) were purchased from Darupakhsh Company (Tehran, Iran).
Instrumentation
Scanning electron microscopy (SEM) images and energy Dispersive X-ray (EDX) spectra were recorded via a TESCAN-Vega 3 machine (TESCAN, Czech Republic) and Ziess Sigma VP machine (Jena, Germany). The X-ray diffraction (XRD) spectra were recorded by applying Kα radiation (λ = 1.54 Å) provided by Cu element on a D8 Advance X-ray diffractometer (Bruker, Germany). Fourier transform infrared (FT-IR) spectral studies were done using Perkin Elmer FTIR spectrometer (RXI, Germany).
Chromatographic condition
Chromatographic analyses were carried out using a waters alliance e2695 (Massachusetts, USA) under the following conditions: A waters 2487 dual wavelength detector and C18 TMS end capping/reversed phase column (luna 5 µm C18 100A HPLC column 250 × 4.6 mm id, phenomenex Co, Torrance, CA) at 40 °C temperature. The elution process was performed in an isocratic mode while mobile phase was composed of acetonitrile–phosphate buffer (KH2PO4, pH 4) (40:60, V/V). Flow rate of pump was adjusted at 1.5 mL min−1 for all analyses. The mobile phase was filtered by applying a 0.2 µm membrane filter made of polytetrafluoroethylene (PTFE) (Millipore, Bedford, MA, USA) and it was degassed each day before separation. UV wavelength and the injection volume were set at 220 nm and 20 µL, respectively.
Synthesis of GO
Nanosheets of GO were simply synthesized by Hummer’s method with some minor modifications (Marcano et al. 2010). In this modified procedure, 3 g graphite powder was mixed with 1.5 g NaNO3 and 69 mL H2SO4 and the resultant mixture was placed in a bath of ice water and cooled to 0 °C. Then, 9 g KMnO4 as an oxidizing agent was added to the above mixture and its temperature was raised up to 20 °C. The obtained solution was kept constant for 5 min and after that it was heated to 35 °C and stirred for 60 min. The resultant material was poured into a beaker containing 400 mL ice and 3 mL H2O2 30%. Afterwards, for phase separation, it was centrifuged within 5 min at rate of 4000 rpm. The upper solution was poured off and the resulting solid phase at bottom of the sample vessel was washed twice with HCl 5% and water and afterwards ultrasonic irradiation was utilized to obtain particle uniform distribution. Finally, to separate GO particles from the supernatant, centrifugation was applied again and the remaining compound was dried at 100°C (Fig. S1).
Synthesis of GO/MOF-74
0.25 g GO, 0.23 g 2,5-dihydroxytriphetalic acid and 1.13 g Ni(NO3)2·6H2O (Nickel (II) nitrate hexahydrate) were mixed together and then it was added to a mixed solvent composed of N,N-dimethylformamide-ethanol-deionized water (1:1:1, v/v/v %, 100 mL). To make a suspension, ultrasonic irradiation was applied for 10 min (Ahmed and Jhung 2014). The mixture solution was transferred into an autoclave and kept there for 24 h at 100 °C and then it was cooled at room temperature (approximately 1.1 g). To remove the remaining impurities, the supernanent was taken away and the sediment was washed 4 times per 2 days with methanol.
Synthesis of GO/MOF-74/Fe3O4
To fabricate GO/MOF-74/Fe3O4, 0.8 g FeCl3·6H2O and 0.3 g of FeCl2·4H2O were dissolved in 25 mL water. The previously synthesized GO/MOF-74 was added to the above solution under nitrogen flow and the pH was adjusted at ten using ammonia. The mixture solution was transferred into an autoclave and kept there for 24 h at 100 °C and then it was cooled at room temperature. To eliminate the residual impurities, the supernanent was taken away and the sediment was washed 4 times per 2 days with methanol. The resultant material was placed into an autoclave and kept there at 250 °C for 2 h, to obtain a brown powder (Fig. S1).
Synthesis of GO/MOF-74/Fe3O4/Polytyramine
For preparing GO/MOF-74/Fe3O4/PTy, an oxidative polymerization of tyramine was carried out on the surface of GO/MOF-74/Fe3O4 using HRP enzyme as a catalyst. HRP is a hemoprotein containing two different kinds of metal center (usually referred as the ‘heme group’) iron (III) protoporphyrin IX and two calcium atoms. Figure 2a, b shows the structure of HRP (a) and its catalytic reaction with tyramine (b), respectively, where decomposition of hydrogen peroxide is catalyzed. Synthesis procedure was as follow: 200 mg GO/MOF-74/Fe3O4, 160 mg tyramine, 1 mg HRP, 8 mL acetone and 4 mL phosphate buffer (0.1 M, pH 7) were dissolved together. 240 µL hydrogen peroxide was added to the mixture at a temperature of 30 °C. The precipitated were filtered and dried in the oven (Fig. S1) [8]. A schematic diagram of synthesis routes is provided in Fig. 3.

Structure of iron heme group in horseradish peroxidase (a) and the catalytic reaction of horseradish peroxidase with tyramine (b)

a Schematic diagram of the fabrication of GO/MOF-74/Fe3O4/PTy. b Schematic diagram of the MD-µ-SPE-HPLC method in extraction and quantification of DOM and ITP
Standard solutions and quality control samples of DOM and ITP
Two stock solutions of DOM and ITP were prepared independently at a concentration level of 200.0 mg L−1 by dissolving the calculated amount of each prokinetic drug in HPLC grade methanol. To avoid decomposition of these drugs, they were prepared once a week. The stock solutions were diluted with deionized water step by step to prepare desirable concentration levels of working standard solutions. Human plasma samples were spiked with different concentrations of the working standard solutions, to obtain standard calibration samples and subsequently plot calibration curve. To assess accuracy and reproducibility of the analytical method, quality control samples of target drugs at concentration values of 20, 200, 750 and 1000 ng mL−1 were prepared and kept in dark sample vessel at a temperature of − 18 °C.
Protein precipitation of human plasma
Frozen human plasma samples were thawed at room temperature and transferred into a sample tube. Acetonitrile was applied as a deproteinizing agent to precipitate all proteins prior to the extraction process. For this purpose, 1.9 mL of plasma was spiked with 100.0 µL of working standard solutions to obtain the proper concentration level of each drug and then 2.0 mL acetonitrile was added to the sample. The resultant real sample was vortexed for 2 min and centrifuged for 6 min at 4000 rpm. The existing acetonitrile in the upper phase was evaporated using a nitrogen flow and the acetonitrile-free sample was poured into 4.0 mL buffer solution at pH 5.0 to obtain approximately a total volume of 6.0 mL. 5.0 mL of the obtained sample was analyzed using MD-µ-SPE followed by HPLC–UV.
The procedure of MD-µ-SPE-HPLC–UV
The experimental arrangement of MD-µ-SPE-HPLC–UV for extraction, preconcentration and trace determination of target drugs is illustrated in Fig. 3. To perform the extraction process, firstly 5.0 mL of deproteinized plasma sample was moved into a glass sample tube and second 15.0 mg of GO/MOF-74/Fe3O4/PTy was added to the sample. Then, ultrasonic irradiation was applied for 5 min for complete dispersing of magnetic nanosorbent throughout the sample media and subsequent extracting of the analytes. The sample vessel exposed to a strong magnet composed of neodymium (Nd), iron (Fe) and boron (B) (0.8 T) to isolate and separate the drug-loaded nanosorbent from the solution. Afterwards, the sample solution was decanted and the sorbent was washed with 1.5 mL (0.5 mL in each elution) while ultrasonic irradiation was applied within 2 min. The Nd–Fe–B–Nd magnet was utilized again and the solution containing the desorbed drugs was collected and heated under a stream of nitrogen to obtain dried residue. Remaining materials was dissolved in 100.0 µL of the applied mobile phase of HPLC and finally 20.0 µL of the sample was injected into HPLC column for further separations and analyses.
Results and discussion
Characterizations
Figure 4a–c presents the FTIR spectra of GO, GO/MOF-74, GO/MOF-74/Fe3O4/PTy, respectively, within the range of 4000–400 cm−1. Due to the presence of oxygen-containing functional groups and aromatic carbon bonds in the hybrid material, the FTIR spectrum of sorbent exhibits some absorption peaks located at 1100–1200 cm−1, 1650–1680 cm−1 and 1410–1450 cm−1 which can be assigned to stretching vibrations of epoxy C–O, C=O and aromatic C=C, respectively. Furthermore, broad peak at 3200–3648 cm−1 shows the stretching vibrations of hydroxyl groups (O–H) in the GO structure. The peak located around 500–580 cm−1 is attributed to Fe–O stretching vibration, which displays that GO has been appropriately modified with magnetic Fe3O4 particles and hydrogen bonds have been attached to iron oxide bonds. In addition, two sharp peaks around 850–950 cm−1 can be assigned to the (OH)− mode, corresponding to the µ-hydroxo groups presents at the corner-sharing hexagonal units of MOF-74 (Adhikari and Lin 2013). The FTIR spectrum of GO/MOF-74/Fe3O4/PTy reveals vibration peaks at 1210–1250 cm−1 and 1550–1570 cm−1, which can be corresponded to the C–N and N–H bonds. The latter results show that proper immobilization of PTy has been conducted on the surface of GO/MOF-74/Fe3O4 and the monomer conversion to polymer has been done perfectly.

The FT-IR spectra of GO (a), GO/MOF-74 (b) and GO/MOF-74/Fe3O4/PTy nonporous composite (c)
A comparison between the XRD patterns of GO, MOF-74, GO/MOF-74, GO/MOF-74/Fe3O4 and GO/MOF/Fe3O4/PTy has been provided in Fig. 5. The result of this comparison exhibited the appearance of a diffraction peak (101) of GO at 2θ = 11/28° in XRD patterns indicating successful synthesis of GO. Moreover, the XRD patterns of Fig. 5 represent MOF-74 as a unique crystalline framework, observed in all patterns except for GO, with X-ray reflections indexed to (110) to (300) planes and diffractions peaks of 2θ = 7°, 12°, which notably denotes the crystallite structure of MOF-74. The XRD patterns of GO/MOF-74/Fe3O4 and GO/MOF-74/Fe3O4/PTy presented in Fig. 5 exhibit some observed characteristic X-ray diffraction peaks at 2θ = 30.3°, 35.7°, 43.3°, 57.3° and 62.9° indexed to (220), (311), (400), (511) and (400), which showed the formation of Fe3O4. Furthermore, other diffraction peaks at 33.2°, 40.8° and 35.6° can be assigned to the (104), (110), (113) planes of Fe2O3, respectively (Ahmadi Daryakenary and Zeeb 2017; Zhou et al. 2017). These results revealed that both Fe3O4 and Fe2O3 have been created through magnetization procedure. The obtained data are in agreement with the Joint Committee on Powder Diffraction Standards (JCPDS card). By comparing these two patterns, it can be concluded that magnetic properties of the sorbent has been remained approximately unchanged during surface modifications. In the XRD pattern of GO/MOF-74/Fe3O4/PTy, a compound called naphthalimide composed of an aromatic core along with N-imide was observed. This compound has been probably formed due to the interaction between MOF-74 and PTy (Gopikrishna et al. 2018). To analyze elements and confirm the successful modification of the nanosorbent, EDX spectrum of GO/MOF-74/Fe3O4/PTy was recorded (Fig. S2). This spectrum illustrated the existence of N, Fe, O, Ni, and C confirming proper adjustment of GO with MOF-74, Fe3O4 and PTy. Furthermore, the morphology and particle size of the nanoporous composite was evaluated using SEM images. Figure 6 shows SEM images of GO (a, b), MOF-74 (c), GO/MOF-74 (d), GO/MOF-74/Fe3O4 (e) and GO/MOF-74/Fe3O4/PTy (f). As it can be seen in Fig. 6a, GO nanosheets have been clearly formed with lamellar morphology. Also, in Fig. 6b, the robust agglomeration of graphene sheets are observed with wrinkles and folds, which consists of multiple stacked layers of graphene, mainly driven by the strong interactions of surface groups on the graphene-like layers. When MOF-74 is attached to GO, as shown in Fig. 6d, crystalline MOF-74 (Fig. 6c) can be clearly seen to be evenly dispersed over the surface of GO sheets. When the SEM image of MOF-74 is compared with GO-MOF-74 image, i.e. Figure 6c, d we notice a great similarity. To confirm the successful immobilization of MOF-74 upon the process, EDX analyses of SEM images of MOF-74 and GO/MOF-74 were performed and the obtained data proved the homogenous distribution of element contents of Ni, C and O in both groups, confirming the successful attachment of MOF-74 with GO layers. The mass percentage of the carbon material is increased from 31.2 to 49.2%, which proves the presence of GO in GO/MOF-74 composite. Thus, the obtained results determine that MOF-74 has been prosperously dispersed over the GO sheets. Figure 6e exhibits that crystalline MOF-74 and Fe3O4 particles have grown on the nanosheet of GO and resulted sphere-like morphologies with appropriate distribution on the GO surface. Figure 6f shows SEM image of GO/MOF-74/Fe3O4 modified with PTy, as it can be seen the layers of polymer have been created on the surface of sorbent and a significant increase in thickness can be observed revealing suitable coating of GO/MOF-74/Fe3O4 with the modifier polymer.

XRD patterns of GO, MOF-74, GO/MOF-74, GO/MOF-74/Fe3O4 and GO/MOF-74/Fe3O4/PTy

SEM images of GO (a, b), MOF-74 (c), GO/MOF-74 (d) and GO/MOF-74/Fe3O4 (e) and GO/MOF-74/Fe3O4/PTy (f)
Effect of magnetic nanosorbent amount
In extraction methods based on nanocomposite materials, the dosage of sorbent is a critical parameter, which significantly affects the extraction performance and subsequent analytical features (Badragheh et al. 2018). To obtain the best sensitivity and reproducibility in the analysis of DOM and ITP, different dosages of nanosorbent ranging from 2.0 to 35.0 mg were subjected to MD-µ-SPE. As it can be seen in Fig. 7a, peak area increases rapidly as the amount of nanoscale sorbent varies from 2.0 to 15.0 mg and after that it decreases. Due to the nanoporous structure of GO/MOF-74/Fe3O4/PTy and its large surface area, the maximum extraction efficiency is obtained at approximately low amounts of extractor (15 mg), which can be considered as an important advantage of the current enrichment method. However, by expanding the dosage of GO/MOF-74/Fe3O4/PTy from 15.0 mg the extraction yields of DOM and ITP declines faintly. The later results are due to this fact that at higher dosages, separation and isolation of nanosorbent may not be performed effectively and they remained suspended in sample media. In addition, due to the aggregation of the sorbent at higher dosages, the available surface for nanosorbent–analyte interactions decreases, thus a significant reduction in extraction efficiency is observed (Farajvand et al. 2018). Consequently, 15.0 mg of GO/MOF-74/Fe3O4/PTy was chosen as the optimum value and utilized for the rest of the work.

a Impact of GO/MOF-74/Fe3O4/PTy composite amount, experimental conditions: concentration level of each drug 250.0 ng mL−1; pH 4.0; extraction time 5 min; eluting solvent acetonitrile; desorption time 2 min. b Impact of sample pH, experimental conditions: concentration level of each drug 250.0 ng mL−1; sorbent amount 15.0 mg; extraction time 5 min; eluting solvent acetonitrile; desorption time 2 min. c Impact of extraction time, experimental conditions: concentration level of each drug 250.0 ng mL−1; sorbent amount 15.0 mg; pH 4.0; eluting solvent acetonitrile; desorption time 2 min. d Impact of ACN volume, experimental condition: concentration level of each drug 250.0 ng mL−1; sorbent amount 15.0 mg; pH 4.0; extraction time 5 min; desorption time 2 min. e Impact of ionic strength, experimental conditions: concentration level of each drug 250.0 ng mL−1; sorbent amount 15.0 mg; pH 4.0; extraction time 5 min; eluting solvent acetonitrile; desorption time 2 min
Effect of pH
The pH value of sample solution in one of the most important variables managing the kind of interactions and following adsorption of analytes on the surface of sorbent (Aghaei et al. 2017; Roto et al. 2016).The effect of pH solution on the extraction and quantitation of ITP and DOM was investigated in the range of 2.0–12.0 using 0.01 M HCl and NaOH. The obtained data are summarized in Fig. 7b. As it can be seen, analytical sensitivities were approximately steady in the range of 4–7 and then decreased at higher pH values. The fabricated sorbent has hydrophobic characteristics, so the dissociated forms of drugs at different pH values should be considered according to their acid dissociation constants (pKa values). ITP and DOM are ionizable compounds and they have acid dissociation constant (pKa) above 7 (Khan et al. 2016), consequently they can be considered as weak acids. By changing the pH value of sample media the molar ratio of uncharged species to charged species varies resulting a significant change in hydrophobic properties of drugs. As the pH decreases the uncharged forms of ITP and DOM (molecular forms) become prevalent and hydrophobic–hydrophobic interaction between drugs and GO/MOF-74/Fe3O4/PTy happen, which it causes a noteworthy improvement in extraction yield. Ultimately, according to these criteria, pH 5.0 was selected as the optimum value in all extraction and measurement steps to obtain a compromise between stability and sensitivity of signals.
Effect of extraction time
The ultrasonic irradiation was applied as a disperser means for the entire dispersion of nanoscale sorbent throughout the sample media, and its time was defined as the extraction time. Extraction time is a notable variable affecting mass transfer performance and extraction yield (Asgharinezhad et al. 2015). The impact of the current variable on the peak area of ITP and DOM was investigated from 0 to 10 min (Fig. 7c). Enrichment of analytes showed a notable improvement by rising the extraction time from 0 to 5 min. Based on these data, 5 min seems to be proper for complete dispersion of nanosorbent and subsequent adsorption equilibrium. Rapid adsorption of target drugs takes places because of the application of ultrasonic irradiation, and high surface area of nanosorbent. By increasing the extraction time a negative effect on analytical signals was observed which it may due to this fact that some parts of analytes come back to sample media during higher time values, so 5 min is enough to obtain desirable extraction efficiency.
Desorption condition
To obtain an optimum desorption condition, some eluting solvents including methanol, acetonitrile, acetone, ethanol, propanol and chloroform were subjected to the extraction method. Among these organic agents, acetonitrile exhibited a different and powerful eluting capability and it causes a higher extraction yield of the drugs, so it was preferred to other organic solvents. The impact of volume of eluting agent was taken to the account and it was assessed in the range of 0.5–5.0 mL. The data obtained in this investigation showed that by growing the volume of acetonitrile up to 1.5 mL enrichment efficiency amplified, which is due to the sufficient desorption and dispersion in this volume of solvent (Fig. 7d). At higher amounts of eluting agent, no considerable improvement was observed. In further evaluations, it was indicated that three times elution (0.5 mL in each time) caused better extraction recoveries and provided more steady signals, so three times elution with total volume of 1.5 mL was selected as the optimum condition. Furthermore, the impact of desorption time on elution of drugs was evaluated within the range of 1–7 min and better presentation of elution was achieved in 2 min.
The effect of ionic strength
In this study, the effect of salt content of sample media on the quantitation of ITP and DOM by means of MD-µ-SPE-HPLC was tested within the range of 0–10% w/v (Fig. 7e). NaCl salt was selected as an electrolyte to adjust the ionic strength of the sample. As the salt content of sample raises the sensitivity of signals increases, which is due to a known phenomena called salting out effect (Shen and Lee 2002). But here different results were observed and by increasing the ionic strength a significant reduction in analytical signals was obtained. It seems that at higher salt concentrations, the viscosity of sample media increases and it makes difficult mass transfer of analytes towards the nanosorbent. As a result, no salt was used for the rest of the work.
Reusability
To reduce the cost of analysis and provide better stability of the extraction process, it is essential that the fabricated nanosorbent shows a considerable reusability. So, the reusability of GO/MOF-74/Fe3O4/PTy was examined in detail and after the extraction of analytes, the sorbent was washed with 1.5 mL acetonitrile and 2.0 mL double-distilled water while sonication was applied within 5 min. After that, the sorbent was dried at room temperature and applied again through MD-µ-SPE. The extraction recoveries of analytes were assessed and it was indicated that after 15 cycles of enrichment procedure, the extraction recovery decreased around 10% which exhibited a minor loss of the adsorption capability.
Analytical feature
Main analytical figures of merits involving linear dynamic range (LDR), correlation coefficient (r2), limit of detection (LOD), limit of quantification (LOQ), extraction recovery (ER) along with enrichment factor (EF) for trace quantification of DOM and ITP by MD-µ-SPE-HPLC–UV were investigated (Table 1). The spiked human plasma samples with different amounts of drugs were subjected to the proposed method to plot the calibration curves of DOM and ITP. EF value of each drug was calculated based on the slope ratio of calibration curves before and after extraction method. Furthermore, ER was defined according to the following equation (Asadi et al. 2015):
The limit of detections (LODs, S/N = 3) for DOM and ITP were 0.4 and 1.1 ng mL−1, respectively. Remarkable linearity (0.995 ≥ r2 ≥ 0.991) and convenient dynamic concentration ranges of 1.5–1100.0 and 4.0–1750.0 ng mL−1 were obtained for DOM and ITP, respectively. A calibration curve using spiked human plasma samples was plotted and the obtained data showed that the calibration curve plotted from spiked samples did not exhibit an acceptable correlation compared to that for aqueous standard samples. Hence, the quantitation of DOM and ITP in real samples could not be performed using calibration curve of aqueous standard solutions at the same instrumental conditions. This fact is due to the difference between plasma matrix and aqueous standard solution matrix. Figure 8 illustrates the HPLC chromatograms for blank sample and two different spiked levels of human plasma. Furthermore, extraction recoveries of DOM and ITP at the concentration level of 250.0 0 ng mL−1 without application of MOF-74 were examined and significant reductions of 14.6% and 19.0% were observed for DOM and ITP, respectively.

HPLC–UV chromatograms of DOM and ITP in human plasma: blank (a); spiked human plasma with concentration level of each prokinetic drug at b 150.0 ng mL−1, c 500.0 ng mL−1 and d 750.0 ng mL−1
Precision and accuracy
For recoveries assessments, desirable practice is to define recovery values at three or four levels (QC samples) indicated as close to the LOQ, a middle value and a high value with respect to the requirements of the analysis. Different quality control (QC) samples (20, 200, 750 and 1000 ng mL−1) were subjected to the current method to assay intra-day (within 1 day) and inter-day (within 3 days) precisions and accuracies. Relative standard deviations (RSDs) of intra-day and inter-day as well as corresponding accuracies are summarized in Table 2. Intra-assay (≤ 8.6%, n = 12) and inter-assay (≤ 9.0%, n = 12) precisions along with noticeable accuracies (≤ 9.3%) confirmed reasonable performance of the current method for trace quantification of prokinetic drugs in complex matrices.
Applicability of the method
The validated method was utilized to quantify DOM and ITP concentrations in six healthy volunteers within the age range of 25–40 years after oral administration of a fixed dose combination of tablet (50 mg ITP/20 mg DOM) to assess main pharmacokinetic data. Blood samples were collected into ethylenediaminetetraacetic acid (EDTA) tubes at 0, 1, 2, 3, 4, 5, 6, 8, 10, 12 and 24 h after drug administration. Each blood sample was centrifuged at 4000 rpm for 7 min to obtain human plasma and then they were transferred into polypropylene tubes and stored at − 18 °C. All examinations were conducted and approved by “The Committee for Research Ethics” of Department of Pharmacy and Pharmaceutical Sciences Research Center, Tehran University of Medical Sciences. Deproteinized plasma samples were analyzed using the proposed protocol and the curve involving the mean concentration level of each drug versus time was plotted. Some major pharmacokinetic characteristics, such as Tmax, Cmax, AUC0–t, AUC0–∞, and T½ were tested (Table 3). Successful application of the MD-µ-SPE followed by HPLC–UV to pharmacokinetic study demonstrated the appropriate capability of the method for simultaneous quantitation of drugs after low dose administration.
Comparison with other methods
Major analytical features of the current method were compared with some of the superior techniques for the extraction and determination DOM and ITP (Table 4). This comparison exhibits several notable advantages of the method including low LODs, wide dynamic linearity ranges, comparable RSDs and reasonable recoveries of target drugs. Many reported methods in literature have been designed for quantification of only one drug and the lack of simultaneous determination capability could be considered as negative point of these methods. Furthermore, it is notable that the fabricated hybrid material benefited from not only proper extraction capacity of GO, but also noteworthy extraction characteristics of MOF and PTy, which resulted higher sensitivity, acceptable reusability of sorbent, short extraction time and successful interferences removal.
Conclusion
In this study, a promising hybrid material based on GO/MOF-74/Fe3O4/PTy was successfully fabricated and applied as a new and effective sorbent for MD-µ-SPE. Surface modification of GO nanosheets with MOF-74, PTy and iron oxide particles merged all unique properties of these materials which results noteworthy improvements in π-stacking length, diffusion routes, aromatic–aromatic interactions and pore volume. Furthermore, the newly designed nanosorbent revealed some other significant merits including simple magnetic separation without any special tools, appreciable reusability (at least 15 cycles) and good enrichment factors (EF). After evaluation of the extraction conditions, MD-µ-SPE was followed with HPLC–UV for simultaneous quantification of DOM and ITP, as prokinetic drugs, in human plasma. Notable accuracies and precisions were obtained through pharmacokinetic analyses and recovery experiments while the sample matrices had no considerable interferences with the performance of GO/MOF-74/Fe3O4/PTy. The advantages show that this technique has remarkable potential in trace screening of drugs in complex matrices and it is a good alternative for toxicodynamic and bioequivalence studies.
References
Abdel-Ghany MF, Abdel-Aziz O, Mohammed YY (2015) Validation of four different spectrophotometric methods for simultaneous determination of Domperidone and Ranitidine in bulk and pharmaceutical formulation. Spectrochim Acta A 149:30–40. https://doi.org/10.1016/j.saa.2015.04.021
Abdel-Haleem FM, Madbouly A, El Nashar RM, Abdel-Ghani NT (2016) Molecularly imprinted polymer-based bulk optode for the determination of itopride hydrochloride in physiological fluids. Biosens Bioelectron 85:740–742. https://doi.org/10.1016/j.bios.2016.05.081
Abdelrahman MM (2013) Simultaneous determination of Cinnarizine and Domperidone by area under curve and dual wavelength spectrophotometric methods. Spectrochim Acta A 113:291–296. https://doi.org/10.1016/j.saa.2013.04.120
Adhikari AK, Lin K (2013) Synthesis, fine structural characterization, and CO2 adsorption capacity of metal organic frameworks-74. J Nanosci Nanotechno 13:1–9. https://doi.org/10.1166/jnn.2013.8621
Adlnasab L, Ezoddin M, Shojaei RA, Aryanasab F (2018) Ultrasonic-assisted dispersive micro solid-phase extraction based on melamine-phytate supermolecular aggregate as a novel bio-inspired magnetic sorbent for preconcentration of anticancer drugs in biological samples prior to HPLC-UV analysis. J Chromatogr B 1095:226–234. https://doi.org/10.1016/j.jchromb.2018.08.001
Aghaei E, Alorro RD, Encila AN, Yoo K (2017) Magnetic adsorbents for the recovery of precious metals from leach solutions and wastewater. Metals 7:529–561. https://doi.org/10.3390/met7120529
Ahmadi Daryakenary M, Zeeb M (2017) Trace determination of chlorpheniramine in human plasma using magnetic dispersive solid-phase extraction based on a graphene oxide/Fe3O4@polythionine nanocomposite combined with high-performance liquid chromatography. RSC Adv 7:53210–53218. https://doi.org/10.1039/C7RA09707G
Ahmed I, Jhung SH (2014) Composites of metal–organic frameworks: preparation and application in adsorption. Mater Today 17:136–146. https://doi.org/10.1016/j.mattod.2014.03.002
Ali MS, Ghori M, Roshanali Khatri A (2006) Stability indicating simultaneous determination of domperidone (DP), methylparaben (MP) and propylparaben by high performance liquid chromatography (HPLC). J Pharm Biomed Anal 41:358–365. https://doi.org/10.1016/j.jpba.2005.12.026
Asadi M, Dadfarnia S, Haji Shabani AM, Abbasi B (2015) Simultaneous extraction and quantification of lamotrigine, phenobarbital, and phenytoin in human plasma and urine samples using solidified floating organic drop microextraction and high-performance liquid chromatography. J Sep Sci 38:2510–2516. https://doi.org/10.1002/jssc.201500237
Asgharinezhad AA, Ebrahimzadeh H (2016) Poly (2-aminobenzothiazole)-coated graphene oxide/magnetite nanoparticles composite as an efficient sorbent for determination of non-steroidal anti-inflammatory drugs in urine sample. J Chromatogr A 1435:18–29. https://doi.org/10.1016/j.chroma.2016.01.027
Asgharinezhad AA, Karami S, Ebrahimzadeh H, Shekari N, Jalilian N (2015) Polypyrrole/magnetic nanoparticles composite as an efficient sorbent for dispersive micro-solid-phase extraction of antidepressant drugs from biological fluids. Int J Pharm 494:102–112. https://doi.org/10.1016/j.ijpharm.2015.08.001
Babaei A, Zeeb M, Es-haghi A (2018) Magnetic dispersive solid-phase extraction based on graphene oxide/Fe3O4@polythionine nanocomposite followed by atomic absorption spectrometry for zinc monitoring in water, flour, celery and egg. J Sci Food Agric 9:3571–3579. https://doi.org/10.1002/jsfa.8873
Badragheh S, Zeeb M, Talei Bavil Olyai MR (2018) Silica-coated magnetic iron oxide functionalized with hydrophobic polymeric ionic liquid: a promising nanoscale sorbent for simultaneous extraction of antidiabetic drugs from human plasma prior to their quantitation by HPLC. RSC Adv 8:30550–30561. https://doi.org/10.1039/c8ra02109k
Bayyari MA, Tutunji LF, Al-Bayyari N, Tutunji MF (2015) Liquid chromatography tandem mass spectrometry method for determination of domperidone in human plasma. J Chem Pharm Res 7:980–985. https://doi.org/10.1365/s10337-009-1032-0
Bose A, Bhaumik U, Ghosh A, Chatterjee B, Chakrabarty US, Sarkar AK, Pal TK (2009) LC–MS simultaneous determination of itopride hydrochloride and domperidone in human plasma. Chromatographia 69:1233–1241. https://doi.org/10.1365/s10337-009-1032-0
Chakraborty A, Maji TK (2014) Mg-MOF-74@SBA-15 hybrids: synthesis, characterization, and adsorption properties. APL Materials 2:124107–124114. https://doi.org/10.1063/1.4902816
Corazza G, Merib J, Magosso HA, Bittencourt OR, Carasek E (2017) A hybrid material as a sorbent phase for the disposable pipette extraction technique enhances efficiency in the determination of phenolic endocrine-disrupting compounds. J Chromatogr A 1513:42–50. https://doi.org/10.1016/j.chroma.2017.07.050
Doherty CM, Buso D, Hill A, Furukawa S, Kitagawa S, Falcaro P (2014) Using functional nano- and microparticles for the preparation of metal organic framework composites with novel properties. Acc Chem Res 47:396–405. https://doi.org/10.1021/ar400130a
Emmanuel AV, Kamm MA, Roy AJ, Antonelli K (1998) Effect of a novel prokinetic drug, R093877, on gastrointestinal transit in healthy volunteers. Gut 42:511–516. https://doi.org/10.1136/gut.42.4.511
Farajvand M, Farajzadeh K, Faghani GH (2018) Synthesis of graphene oxide/polyaniline nanocomposite for measuring cadmium(II) by solid phase extraction combined with dispersive liquid-liquid microextraction. Mater Res Express 5:10. https://doi.org/10.1088/2053-1591/aace3a
Georgakilas V, Tiwari JN, Kemp KC, Perman JA, Bourlinos AB, Kim KS, Zboril R (2016) Noncovalent functionalization of graphene and graphene oxide for energy materials, biosensing, catalytic, and biomedical applications. Chem Rev 9:5464–5519. https://doi.org/10.1021/acs.chemrev.5b00620
Gołębiowski M, Stepnowski P, Leszczyńska D (2017) Application of carbon nanotubes as solid-phase extraction sorbent for analysis of chlorophenols in water samples. Chem Pap Chem Zvesti 71:831–839. https://doi.org/10.1007/s11696-016-0098-z
Gopikrishna P, Meher N, Iyer PK (2018) Functional 1,8-Naphthalimide AIE/AIEEgens: recent advances and prospects. ACS Appl Mater Inter 10:12081–12111. https://doi.org/10.1021/acsami.7b14473
Hagarová I (2017) Cloud point extraction utilizable for separation and preconcentration of (ultra)trace elements in biological fluids before their determination by spectrometric methods: a brief review. Chem Pap Chem Zvesti 71:869–879. https://doi.org/10.1007/s11696-016-0014-6
Huang X, Qi X, Boeyab F, Zhang H (2012) Graphene-based composites. Chem Soc Rev 41:666–686. https://doi.org/10.1039/C1CS15078B
Ibrahim F, Nasr JJ (2016) Fourth-derivative synchronous spectrofluorimetry and HPLC with fluorescence detection as two analytical techniques for the simultaneous determination of itopride and domperidone. Luminescence 31:255–263. https://doi.org/10.1002/bio.2955
Imran A, Gupta VK, Singh P, Negi U (2014) Spe-HPLC techniques for separation and identification of domperidone in human plasma. J Liq Chromatogr Relat Technol 37:2587–2597. https://doi.org/10.1080/10826076.2013.853307
Janiak C, Vieth JK (2010) MOFs, MILs and more: concepts, properties and applications for porous coordination networks (PCNs). New J Chem 34:2366–2388. https://doi.org/10.1039/c0nj00275e
Karimzadeh Z, Javanbakht S, Namazi H (2019) Carboxymethylcellulose/MOF-5/Graphene oxide bio-nanocomposite as antibacterial drug nanocarrier agent. Bioimpacts 9:5–13. https://doi.org/10.15171/bi.2019.02
Khan A, Iqbal Z, Khadra I, Ahmad L, Khan A, Khan MI, Ullah Z (2016) Simultaneous determination of domperidone and Itopride in pharmaceuticals and human plasma using RP-HPLC/UV detection: method development, validation and application of the method in in vivo evaluation of fast dispersible tablets. J Pharm Biomed Anal 121:6–12. https://doi.org/10.1016/j.jpba.2015.12.036
Kostolanska K, Novotna L, Taborska E, Pes O (2019) Online solid-phase extraction liquid chromatography–mass spectrometry of hair cortisol using a surrogate analyte. Chem Pap Chem Zvesti 73:151–158. https://doi.org/10.1007/s11696-018-0560-1
Levashov PA, Ovchinnikova ED, Afanaseva MI, Frida DA, Az’muko AA, Adamova IY, Pokrovskii SN (2015) Tyramine and tryptamine as ligands for medical and biotechnological affinity sorbents. J Chem Eng Jpn 41:23–30. https://doi.org/10.1134/s1068162015010112
Li S, Huo F (2015) Metal-organic framework composites: from fundamentals to applications. Nanoscale 7:7482–7501. https://doi.org/10.1039/C5NR00518C
Li Z, Qi M, Tu C, Wang W, Chen J, Wang AJ (2017) Magnetic metal-organic framework/graphene oxide-based solid-phase extraction combined with spectrofluorimetry for the determination of enrofloxacin in milk sample. Food Anal Methods 10:4094–4103. https://doi.org/10.1007/s12161-017-0971-6
Li N, Jiang HL, Wang X, Wang X, Xu G, Zhang B, Wang L, Zhao RS, Lin JM (2018) Recent advances in graphene-based magnetic composites for magnetic solid-phase extraction. TrAC, Trends Anal Chem 102:60–74. https://doi.org/10.1016/j.trac.2018.01.009
Lin LC, Paik D, Kim J (2017) Understanding gas adsorption in MOF-5/graphene oxide composite material. Phys Chem Chem Phys 19:11639–11644. https://doi.org/10.1039/C7CP00066A
Liu Y, Ma J, Wu T, Wang X, Huang G, Liu Y, Qiu H, Li Y, Wang W, Gao J (2013) Cost-effective reduced graphene oxide-coated polyurethane sponge as a highly efficient and reusable oil-absorbent. ACS Appl Mater Inter 5:10018–10026. https://doi.org/10.1021/am4024252
Liu JW, Zhang Y, Chen XW, Wang JH (2014) Graphene oxide—rare earth metal—organic framework composites for the selective isolation of hemoglobin. ACS Appl Mater Inter 6:10196–10204. https://doi.org/10.1021/am503298v
Marcano DC, Kosynkin DV, Berlin JM, Sinitskii A, Sun Z, Slesarev A, Alemany LB, Lu W, Tour JM (2010) Improved synthesis of graphene oxide. ACS Nano 4:4806–4814. https://doi.org/10.1021/nn1006368
Mehdinia A, Khodaee N, Jabbari A (2015) Fabrication of graphene/Fe3O4@polythiophene nanocomposite and its application in the magnetic solid-phase extraction of polycyclic aromatic hydrocarbons from environmental water samples. Anal Chim Acta 868:1–9. https://doi.org/10.1016/j.aca.2014.12.022
Mohamed HM (2015) A study of selective spectrophotometric methods for simultaneous determination of Itopride hydrochloride and rabeprazole sodium binary mixture: resolving sever overlapping spectra. Spectrochim Acta A 136:1308–1315. https://doi.org/10.1016/j.saa.2014.10.018
Musile G, Cenci L, Piletska E, Gottardo R, Bossi AM, Bortolotti F (2018) A novel in-house mixed-mode solid-phase extraction of urine for the determination of 16 basic drugs by HPLC-Ion Trap MS. J Chromatogr A 1550:10–18. https://doi.org/10.1016/j.chroma.2018.05.019
Patel B, Patel M, Patel J, Suhagia B (2007) Simultaneous determination of omeprazole and domperidone in capsules by RP-HPLC and densitometric HPTLC. J Liq Chromatogr R T 30:1749–1762. https://doi.org/10.1080/10826070701360368
Petit C, Bandosz TJ (2009a) MOF–graphite oxide composites: combining the uniqueness of graphene layers and metal–organic frameworks. Adv Mater 21:4753–4757. https://doi.org/10.1002/adma.200901581
Petit C, Bandosz TJ (2009b) MOF–graphite oxide nanocomposites: surface characterization and evaluation as adsorbents of ammonia. J Mater Chem 19:6521–6528. https://doi.org/10.1039/B908862H
Petit C, Bandosz TJ (2011) Synthesis, characterization, and ammonia adsorption properties of mesoporous metal-organic framework (MIL(Fe))–graphite oxide composites: exploring the limits of materials fabrication. Adv Funct Mater 21:2108–2117. https://doi.org/10.1002/adfm.201002517
Ptaˇcek P, Klima J, Macek J (2009) Optimized method for the determination of itopride in human plasma by high-performance liquid chromatography with fluorimetric detection. J Chromatogr B 877:842–846. https://doi.org/10.1016/j.jchromb.2009.02.023
Ragab MT, Abd El-Rahman MK, Ramadan NK, El-Ragehy NA, El-Zeany BA (2015) Novel potentiometric application for the determination of pantoprazole sodium and itopride hydrochloride in their pure and combined dosage form. Talanta 138:28–35. https://doi.org/10.1016/j.talanta.2015.01.045
Roto R, Yusran Y, Kuncaka A (2016) Magnetic adsorbent of Fe3O4@SiO2core-shell nanoparticles modified with thiol group for chloroauric ion adsorption. Appl Surf Sci 377:30–36. https://doi.org/10.1016/j.apsusc.2016.03.099
Salimikia I, Heydari R, Yazdankhah F (2018) Polyaniline/graphene oxide nanocomposite as a sorbent for extraction and determination of nicotine using headspace solid-phase microextraction and gas chromatography–flame ionization detector. J Iran Chem Soc 7:1593–1601. https://doi.org/10.1007/s13738-018-1357-4
Shen G, Lee HK (2002) Hollow fiber-protected liquid-phase microextraction of triazine herbicides. Anal Chem 74:648–654. https://doi.org/10.1021/ac010561o
Sheshmani S, Nayebi M (2017) Modification of TiO2 with graphene oxide and reduced graphene oxide; enhancing photocatalytic activity of TiO2 for removal of remazol black B. Polym Compos 40:210–216. https://doi.org/10.1002/pc.24630
Singh SS, Jain M, Sharma K, Shah B, Vyas M, Thakkar P, Shah R, Singh S, Lohray B (2005) Quantitation of itopride in human serum by high-performance liquid chromatography with fluorescence detection and its application to a bioequivalence study. J Chromatogr B 818:213–220. https://doi.org/10.1016/j.jchromb.2004.12.035
Sisinthy SP, Duraipandi S, Rao NK, Rao MB (2015) Determination and validation of a HPLC method with UV detection of itopride hydrochloride in human. Int J Pharm Pharm Sci 7:246–249
Sivakumar T, Manavalan R, Muralidharan C, Valliappan K (2007) Multi-criteria decision making approach and experimental design as chemometric tools to optimize HPLC separation of domperidone and pantoprazole. J Pharm Biomed Anal 43:1842–1848. https://doi.org/10.1016/j.jpba.2006.12.007
Stock N, Biswas S (2012) Synthesis of metal-organic frameworks (MOFs): routes to various MOF topologies, morphologies, and composites. Chem Rev 112:933–969. https://doi.org/10.1021/cr200304e
Tang J, Wang J (2017) Fe-based metal organic framework/graphene oxide composite as an efficient catalyst for Fenton-like degradation of methyl orange. RSC Adv 7:50829–50837. https://doi.org/10.1039/c7ra10145g
Thanikachalam S, Rajappan M, Kannappan V (2008) Stability-indicating HPLC method for simultaneous determination of pantoprazole and domperidone from their combination drug product. Chromatographia 67:41–47. https://doi.org/10.1365/s10337-007-0452-y0009-5893/08/01
Wang X, Qin F, Jing L, Zhu Q, Li F, Xiong Zh (2013) Development and validation of UPLC-MS/MS method for determination of domperidone in human plasma and its pharmacokinetic application. Biomed Chromatogr 27:371–3766. https://doi.org/10.1002/bmc.2801
Wang J, Wang Y, Zhang Y, Uliana AA, Zhu J, Liu JD, Bruggen BVD (2016) Zeolitic imidazolate framework/graphene oxide hybrid nanosheets functionalized thin film nanocomposite membrane for enhanced antimicrobial performance. ACS Appl Mater Inter 8:25508–25519. https://doi.org/10.1021/acsami.6b06992
Wang X, Li G, Ho Row K (2017) Graphene and graphene oxide modified by deep eutectic solvents and ionic liquids supported on silica as adsorbents for solid-phase extraction. Bull Korean Chem Soc 38:251–257. https://doi.org/10.1002/bkcs.11074
Wang X, Xiao H, Li A, Li Zh, Liu Sh, Zhang Q, Gong Y, Zheng L, Zhu Y, Chen Ch, Wang D, Peng Q, Gu L, Han X, Li J, Li Y (2018) Constructing NiCo/Fe3O4 heteroparticles within MOF-74 for efficient oxygen evolution reactions. J Am Chem Soc 140:15336–15341. https://doi.org/10.1021/jacs.8b08744
Wierucka M, Biziuk M (2014) Application of magnetic nanoparticles for magnetic solid-phase extraction in preparing biological, environmental and food samples. TrAC, Trends Anal Chem 59:50–58. https://doi.org/10.1016/j.trac.2014.04.007
Yoshimura Y, Khunathai K, Nozoe A, Ohto K, Kawakita H (2012) Precious metal recovery using poly(tyramine) prepared by radical polymerization with horseradish peroxidase. J Chem Eng Jpn 45:178–181. https://doi.org/10.1252/jcej.11we125
Yoshizato T, Tsutsumi K, Kotegawa T, Imai H, Nakano S (2014) Determination of domperidone in human plasma using high performance liquid chromatography with fluorescence detection for clinical application. J Chromatogr B 961:86–90. https://doi.org/10.1016/j.jchromb.2014.05.004
Zeeb M, Farahani H (2018) Graphene oxide/Fe3O4@polythionine nanocomposite as an efficient sorbent for magnetic solid-phase extraction followed by high-performance liquid chromatography for the determination of duloxetine in human plasma. Chem Pap Chem Zvesti 72:15–27. https://doi.org/10.1007/s11696-017-0253-1
Zhang S, Yang Q, Wang W, Wang C, Wang Z (2016) Covalent bonding of metal-organic framework-5/grapheme oxide hybrid composite to stainless steel fiber for solid-phase microextraction of triazole fungicides from fruit and vegetable samples. J Agric Food Chem 64:2792–2801. https://doi.org/10.1021/acs.jafc.5b05831
Zhao Y, Seredych M, Jagiello J, Zhong Q, Bandosz TJ (2014) Insight into the mechanism of CO2 adsorption on Cu–BTC and its composites with graphite oxide or aminated graphite oxide. Chem Eng J 239:399–407. https://doi.org/10.1016/j.cej.2013.11.037
Zhou H, Liu X, Zhang J, Yan X, Liu Y, Yuan A (2014) Enhanced room-temperature hydrogen storage capacity in Pt-loaded graphene oxide/HKUST-1 composites. Int J Hydrogen Energ 39:2160–2167. https://doi.org/10.1016/j.ijhydene.2013.11.109
Zhou Y, Zhou L, Zhang X, Chen Y (2016) Preparation of zeolitic imidazolate framework-8/graphene oxide composites with enhanced VOCs adsorption capacity. Microporous Mesoporous Mater 225:488–493. https://doi.org/10.1016/j.micromeso.2016.01.047
Zhou N, An Q, Xiao Z, Zhai S, Shi Z (2017) Solvothermal synthesis of three-dimensional, Fe2O3 NPs-embedded CNT/N-doped graphene composites with excellent microwave absorption performance. RSC Adv 7:45156–45169. https://doi.org/10.1039/c7ra06751h
Acknowledgements
The authors wish to state their thanks to Islamic Azad University South Tehran Branch, for supporting this work. The authors also deeply appreciate Miss Parisa Sadr and Dr Armin Rastboud for assisting to carry out HPLC analyses and all volunteers for helping us to evaluate pharmacokinetic data.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pourbahman, F., Zeeb, M., Monzavi, A. et al. Simultaneous trace monitoring of prokinetic drugs in human plasma using magnetic dispersive micro-solid phase extraction based on a new graphene oxide/metal–organic framework-74/Fe3O4/polytyramine nanoporous composite in combination with HPLC. Chem. Pap. 73, 3135–3150 (2019). https://doi.org/10.1007/s11696-019-00855-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-019-00855-1