
Abstract
We herein report Mannich aminomethylation of variously structural flavonoids and their biological evaluation against human breast cancer cell. Mannich reaction showed that substitution at C-6 position depends on amine basicity and C-ring feature of flavonoids. All five flavonoid substrates reacted with strong amine bases to afford the bis(6,8-aminomethyl) derivatives, while with weak amines, the different products were obtained dependently on structural characteristic of flavonoid. 3-OH and 3-O-substituted groups on the C-ring exhibited the deactivated aminomethylation at C-6 position, whereas substitution at this position was independent on bond feature at C-2 and C-3 on the C-ring. Screening anti-proliferative activity showed six flavonoids possessed activity against breast cancer cell, MDA-MB-231. Among them, the flavonoids, luteolin (2) and 3′,4′,5,7-tetrahydroxy-6,8-bis(pyrrolidin-1-ylmethyl)-3-rutinosylflavone (3a), displayed the highest anti-proliferative activity with the lowest IC50 values.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Flavonoid structures are based upon a 15-carbon skeleton consisting of two benzene rings linked via a heterocyclic pyran ring. They can be divided into a variety of classes which differ in the level of oxidation and pattern of substitution of the pyran ring (e.g., quercetin, a flavonol, and luteolin, a flavone, bearing different substitution groups at C-3 position) (Fig. 1). Individual compounds within a class differ in the pattern of substitution of the benzene rings (Kumar and Pandey 2013). Biological activities of flavonoids depend remarkably on their structural characteristics such as degree of hydroxylation and substitution. A small alteration in the chemical structure may lead to significant changes in biological activities. Hence, the flavonoids possess a wide range of bioactivities such as antioxidant (Rice-Evans et al. 1996; Rice-Evans 2001; Pietta 2000), antifungal (Tempesti et al. 2012), antiviral (Ng et al. 1997; Orhana et al. 2010; Kaul et al. 1985), anti-proliferative (Chahar et al. 2011; Kanadaswami et al. 2005; Ravishankar et al. 2013), and anti-inflammatory agent (Pan et al. 2010; Cottiglia et al. 2005; Candiracci et al. 2012).

Structures of flavonoids
In recent years, numerous studies have been conducted on the potential antimetastasis activity of natural flavonoids in various tumor cell lines. For instance, quercetin has been shown to reduce the human prostate cancer, PC-3 cells (Vijayababu et al. 2006), and kaempferol, genistin, and daidzein revealed to inhibit human breast cancer, MDA-MB-231 cells (Lee et al. 2008; Phromnoi et al. 2009). Among of the flavonoid derivatives, aminomethylated flavonoids are particularly valued due to potential activity against human cancer cells (Ha et al. 2016; Nguyen et al. 2017; Chen et al. 2015; Tugrak et al. 2015). Although Mannich aminomethylation of flavonoids has been known, most of the reported studies used C-6-substituted flavonoids as substrates (Zhang et al. 2008; Babu et al. 2008; Yusakul et al. 2016) or cyclic imines were used as the electrophilic reagents (Nguyen et al. 2011). Recently, there are several studies on the aminomethylation of natural flavonoids using in situ generated iminium ions that proceeded with formation of C-6 (Frasinyuk et al. 2015; Kukhareva et al. 2004) or C-8-monosubstituted (Buravlev et al. 2017; Gorbunov et al. 2016) or disubstituted flavonoids (Nguyen et al. 2015b; Zhurakulov et al. 2015; Nifant’ev et al. 2013). Helgen et al. (2015) have aminomethylated quercetin to afford only a 8-monoaminomethyl derivative when using an imine salt made from 1-methylpiperazine while only a 6,8-diaminomethyl derivative was produced with the imine salts of piperidine. All cases focused on the investigation of bioactivity and no systematic studies were carried out for investigating the influence of amine and iminium ions reactivity and features of flavonoid skeleton driving the Mannich aminomethylation.
In connection with our ongoing research program involving the effect of natural product structures on the synthesis (Dang et al. 2014, 2017; Nguyen et al. 2015a, 2016; Hoang et al. 2015), we have showed that alkylation and acylation of 5-hydroxyflavonoids significantly depended on reagents as well as structural characteristic of ring C of flavonoids, and these derivatives also exhibited especial bioactivities (Hoang et al. 2015). The previously reported results prompted us to investigate this effect on the aminomethylation of flavonoids as well as their bioactive evaluation. Herein, we report that the Mannich reaction of various flavonoid substrates with iminium salts generated in situ from secondary cyclic amines and aqueous formaldehyde can provide either 8-monosubstituted or 6,8-disubstituted derivatives depending on amine reagents and substrate features.
Results and discussion
Chemistry
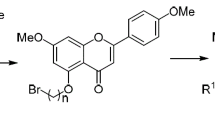
The general synthetic routes are illustrated in Scheme 1. Our strategy for the synthesis of the aminomethylated derivatives at C-6 and C-8 of A-ring of flavonoids relied upon electrophilic substitution via the Mannich reaction carried out between flavonoids with formaldehyde and secondary cyclic amines at room temperature in ethanol. Three cyclic amines with different basicity, pyrrolidine (pKa = 11.27), 1-methylpiperazine (pKa = 9.14), and morpholine (pKa = 8.36) were converted in situ into the respective electrophilic iminium ions. To get an insight into influence of flavonoid structures on the substitution at various positions, five natural flavonoids with structural variations were used as the substrates.

Synthetic routes of flavonoids. Reagents. (i) amines (10 equiv.), HCHO (37%, 10 equiv.), ethanol, rt
The aminomethylation of quercetin with these bases and aqueous formaldehyde afforded products 1a–1c in over 60% yields. The result showed that aminomethylation of flavonoids was strongly affected by basicity of amines. The substitution with pyrrolidine occurred at both C-6 and C-8 positions. This is evident from 1H NMR of 1a which protons at C-6 (δ 6.20 ppm) and C-8 (δ 6.40 ppm) positions of quercetin disappeared. Interestingly, only C-8 monosubstituted products were obtained when flavonoids were treated with an excess of 1-methylpiperazine and morpholine to give 1b and 1c, respectively. This is attributed to the lower basicity of these amines. Similar results were reported by Lis and Marisca (1987) for the other substrates under the same condition.
The position of the aminomethyl groups in products 1b and 1c was unambiguously determined by HMBC NMR spectroscopy. The aminomethylene protons of 1b and 1c (δ 3.87 and 3.82 ppm, respectively), which were well resolved, were coupled long-range to respective C-9 carbons at δ 153.63 and 153.94 ppm. This allows for the assignment of the aminomethyl group to the C-8 position of the quercetin derivatives.
To determine influence of 3-OH group in flavonoids, luteolin (2) was used in comparison with quercetin. As expected, the aminomethylation of 2 with the weakest base afforded the bis(6,8-morpholin) product (2a) in 63% yield. In comparison with formation of 1c, this result reflected that 3-OH group obstructed to electrophilic substitution at C-6 in the flavonoid skeleton. Although reason of the effect has not been described previously, it may be because the electron pair of oxygen at C-3 position stabilized the non-covalent six-membered ring (Hoang et al. 2015) which is attributed to reduction of electron density at C-6 position of quercetin.
In a similar fashion, rutin (3) was treated with all bases. Unfortunately, conversion occurred in very poor yields and no pure aminomethylation products were obtained when treating with either 1-methylpiperazine or morpholine under the same reaction conditions. However, the aminomethylation of 3 with pyrrolidine was observed more easily to afford bis(6,8-pyrrolidine) product (3a) in 54% yield, indicating that 3-O-substituted group is also an important factor in this transformation.
Finally, to understand the influence of the double bond of C-ring on the flavonoid aminomethylation, hesperetin (4) was employed in comparison with substitution of diosmetin (5). Similar to luteolin, the reaction of 4 with morpholine was converted into bis(6,8-morpholine) derivative (4a) in a good yield (81%). In addition, conversion of 5 with both pyrrolidine and morpholine gave diaminomethylated derivatives 5a and 5b, respectively in over 60% yields. It showed that bond feature at C-2 and C-3 was independent on the Mannich aminomethylation at the C-6 position. All the synthesized flavonoids, including new derivatives 1a, 2a, 3a, 4a, 5a, and 5b, were determined physical properties and identified the structure by HRMS and NMR spectra.
Anti-proliferative activity
The anti-proliferative activity of all flavonoid derivatives was tested in the breast cancer cell line MDA-MB-231 (Denizot and Lang 1986). A screening program revealed that several flavonoids exhibited anti-proliferative activity at 100 μM, as depicted in Table 1. Six compounds 1, 1c, 2, 2a, 3a, and 5a showed good anti-proliferative activity (inhibition more than 50%) which would be further used to test IC50 values. It is noteworthy that the disubstituted derivatives of rutin (3a) and hesperetin (5c) possessed the activity greater than the parent flavonoids, while all derivatives of quercetin exhibited the lower activity than the parent compounds.
IC50 values against MDA-MB-231 cell are shown in Table 2. Although the IC50 values of the flavonoids are higher than that of the standard drug (paclitaxel), most of the compounds displayed a significant anti-proliferative activity against MDA-MB-231 cell. Among them, flavonoids 2 and 3a exhibit the highest anti-proliferative activity with IC50 of 7.29 and 7.57 μmol/mL, respectively.
Conclusions
Our studies on Mannich aminomethylation of flavonoids were suggested as follows: (1) amine basicity was critical to design the aminomethylated products and iminium ions prepared from strong bases attracted to both C-6 and C-8 positions of flavonoid. (2) With weak bases, 3-OH and 3-O-substituted flavonoids could obstruct electrophilic substitution at C-6 of flavonoid. (3) Bond feature at C-2 and C-3 did not almost affect the substitution at C-6 even the weak bases.
All the compounds were evaluated for cytotoxicity of breast cancer cell, MDA-MB-231. Six compounds 1, 1c, 2, 2a, 3a, and 5a showed good anti-proliferative activity. Among them, the flavonoids 2 and 3a displayed the lowest IC50 values.
Experimental
Chemistry
All the materials were purchased from Merck (Germany) or Aldrich. The other solvents were purchased from Fluka and used without further purification. Melting points were measured with an Electrothermal Model 9200 (England). UV–Vis spectra were measured using a Shimadzu, Model UV-1650PC spectrophotometer, and reported as λmax in nm (abs.). FTIR spectra were obtained with an Equinox 55 IR—Bruker (Germany) spectrometer; absorption bands are recorded in wave number (cm−1). The ESI–MS were performed on a VG Zab Spec (70 eV) instrument. 1H (500 MHz) and 13C (125 MHz) NMR were recorded on a BRUKER AVANCE 500 NMR spectrometer using DMSO-d6 as solvent and tetramethylsilane (TMS) as an internal standard. Chemical shifts are reported in δ relative to TMS.
General procedure for aminomethylation of flavonoids
To a stirred solution of flavonoid (3.31 mmol) in ethanol (50 mL) was slowly added amines (33.3 mmol) and a 37% formaldehyde solution (2.48 g, 35 mmol). The mixture was stirred at room temperature for 4 h, monitoring by TLC. The residue was filtered off, washed with 50 mL of ethanol, and then dried in the vacuum.
2-(3,4-Dihydroxyphenyl)-3,5,7-trihydroxy-6,8-bis(pyrrolidin-1-ylmethyl)-4H-chromen-4-one (1a)
Yellow powder, m.p. 223–225 °C. Yield 68%. FTIR (KBr) ν/cm−1 3376, 2968, 1651, 1560, 1260, 1069. UV–Vis λmax (nm) (abs.) 238 (0.15), 246 (0.16), 287 (0.09), 321 (0.11), 341 (0.10), 268 (0.12). 1H NMR (500 MHz, DMSO-d6, δ, ppm): 7.69 (1H, d, J 2.0 Hz, H-2′),7.44 (1H, dd, J 8.5, 2.0 Hz, H-6′), 6.85 (1H, d, J 8.5 Hz, H-5′), 3.92 (2H, s, C-8-CH2), 3.88 (2H, s, C-6-CH2), 2.72–2.62 (8H, m, N(CH2CH2)2), 1.76–1.67 (8H, m, N(CH2CH2)2). 13C NMR (125 MHz, DMSO-d6, δ, ppm): 175.35 (C-4), 168.00 (C-7), 156.92 (C-5), 153.52 (C-9), 147.61 (C-4′), 145.50 (C-2), 145.14 (C-3′), 135.26 (C-3), 122.42 (C-1′), 119.67 (C-6′), 115.56 (C-5′), 114.89 (C-2′),103.83 (C-10), 100.85 (C-6), 100.30 (C-8), 52.82 (C-8-CH2), 52.73 (C-6-CH2), 48.68 (N(CH2CH2)2), 46.90 (N(CH2CH2)2), 23.12 (N(CH2CH2)2), 23.05 (N(CH2CH2)2). HRMS (m/z): 469.1965 [M+H]+; 469.1975 calcd [M+H]+ for C25H29N2O7.
2-(3,4-Dihydroxyphenyl)-3,5,7-trihydroxy-8-((4-methylpiperazin-1-yl)methyl)-4H-chromen-4-one (1b)
Yellow powder, m.p. 233–235 °C (233–235 °C, Helgen et al. 2015). Yield 66%. FTIR (KBr) ν/cm−1 3296, 2975, 1655, 1555, 1201, 1043. UV–Vis λmax (nm) (abs.) 229 (0.78), 246 (1.09), 277 (0.36), 363 (1.16). 1H NMR (500 MHz, DMSO-d6, δ, ppm) (Zhang et al. 2008; Helgen et al. 2015): 12.52 (1H, brs, OH) 7.70 (1H, d, J 2.0 Hz, H-2′), 7.57 (1H, dd, J 8.5, 2.0 Hz, H-6′), 6.89 (1H, d, J 8.5 Hz, H-5′), 6.17 (1H, s, H-6), 3.87 (2H, s, C-8-CH2), 2.63–2.58 (4H, m, CH3-N-(CH2CH2)2), 2.38–2.36 (4H, m, CH3-N-(CH2CH2)2), 2.16 (3H, s, N-CH3). 13C NMR (125 MHz, DMSO-d6, δ, ppm): 175.90 (C-4), 164.15 (C-7), 159.54 (C-5), 153.63 (C-9), 147.66 (C-4′), 146.45 (C-2), 145.08 (C-3′), 135.62 (C-3), 122.17 (C-1′), 119.89 (C-6′), 115.61 (C-5′), 114.97 (C-2′), 102.73 (C-8), 99.74 (C-10), 98.12 (C-6), 54.43 (CH3-N-(CH2CH2)2), 51.95 (CH3-N-(CH2CH2)2), 51.18 (C-8-CH2), 45.45 (N-CH3). HRMS (m/z): 415.1471 [M+H]+; 415.1505 calcd [M+H]+ for C21H23N2O7.
2-(3,4-Dihydroxyphenyl)-3,5,7-trihydroxy-8-(morpholinomethyl)-4H-chromen-4-one (1c)
Yellow powder, m.p. 253–257 °C, yield 65%. FTIR (KBr) ν/cm−1 3320, 2970, 1654, 1552, 1201, 1072. UV–Vis λmax (nm) (abs.) 229 (0.20), 246 (0.28), 276 (0.09), 363 (0.30). 1H NMR (500 MHz, DMSO-d6, δ, ppm) (Zhang et al. 2008): 7.73 (1H, d, J 2.0 Hz, H-2′), 7.61 (1H, dd, J 8.5, 2.0 Hz, H-6′), 6.91 (1H, d, J 8.5 Hz, H-5′), 6.22 (1H, s, H-6), 3.82 (2H, C-8-CH2), 3.59 (4H, m, N-(CH2CH2)2O), 2.53 (4H, m, N-(CH2CH2)2O). 13C NMR (125 MHz, DMSO-d6, δ, ppm): 175.95 (C-4), 163.48 (C-7), 159.54 (C-5), 153.94 (C-9), 147.68 (C-2), 146.58 (C-4′), 145.08 (C-3′), 135.64 (C-3), 122.21 (C-1′), 119.93 (C-6′), 115.60 (C-5′), 115.05 (C-2′), 102.87 (C-10), 100.17 (C-8), 97.94 (C-6), 66.08 (N-(CH2CH2)2O), 52.70 (N-(CH2CH2)2O), 50.96 (C-8-CH2). HRMS (m/z): 402.1189 [M+H]+; 402.1189 calcd [M+H]+ for C20H20NO8.
2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-6,8-bis(morpholinomethyl)-4H-chromen-4-one (2a)
Yellow powder, m.p. 230–233 °C, yield 63%. FTIR (KBr) ν/cm−1 3260, 2963, 1649, 1583, 1213, 1072. UV–Vis λmax (nm) (abs.) 236 (0.65), 247 (0.68), 252 (0.68), 289 (0.38). 1H NMR (500 MHz, DMSO-d6, δ, ppm): 7.44 (1H, dd, J 9.0, 2.0 Hz, H-6′), 7.42 (1H, d, J 2.0 Hz, H-2′), 6.91 (1H, d, J 9.0 Hz, H-5′), 6.68 (1H, s, H-3), 3.78 (2H, s, C-8-CH2), 3.73 (2H, s, C-6-CH2), 3.60–3.56 (8H, m, N-(CH2CH2)2O), 2.54–2.49 (8H, m, N-(CH2CH2)2O). 13C NMR (125 MHz, DMSO-d6, δ, ppm): 181.95 (C-4), 164.37 (C-7), 163.61 (C-2), 158.16 (C-5), 154.43 (C-9), 149.75 (C-4′),145.79 (C-3′), 121.75 (C-1′), 118.97 (C-6′), 113.43 (C-5′), 116.06 (C-2′), 103.71 (C-10), 102.76 (C-6), 102.55 (C-3), 100.90 (C-8), 66.15 (N-(CH2CH2)2O), 65.97 (N-(CH2CH2)2O), 52.78 (N-(CH2CH2)2O), 52.36 (N-(CH2CH2)2O), 51.43 (C-6-CH2), 50.36 (C-8-CH2). HRMS (m/z): 485.1920 [M+H]+; 485.1924 calcd [M+H]+ for C25H29N2O8.
2-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-6,8-bis(pyrrolidin-1-ylmethyl)-3-(((2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-((((2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)methyl)tetrahydro-2H-pyran-2-yl)oxy)-4H-chromen-4-one (3a)
Orange powder, m.p. 154–156 °C, yield 57%. FTIR (KBr) ν/cm−1 3479, 2983, 1754, 1629, 1503, 1218, 1073. UV–Vis λmax (nm) (abs.) 229 (0.56), 243 (0.64), 269 (0.31), 299 (0.53). 1H NMR (500 MHz, DMSO-d6, δ, ppm): 7.62–7.60 (2H, m, H-5′,6′), 6.83 (1H, d, J 9.0 Hz, H-2′), 5.30 (1H, d, J 7.5 Hz, H-1-glucose), 4.33 (1H, s, H-1-rhamnose), 3.94 (2H, s, C-8-CH2), 3.84 (2H, s, C-6-CH2), 3.70 (2H, d, J 10.5 Hz, H-6-glucose), 3.30–3.19 (5H, m, H-2,3,4,5-glucose, H-5-rhamnose), 3.09–3.02 (3H, m, H-2,3,4-rhamnose), 2.81 (2H, m, N(CH2CH2)2), 2.69 (2H, m, N(CH2CH2)2), 1.81 (2H, m, N(CH2CH2)2), 1.74 (2H, m, N(CH2CH2)2), 1.00 (3H, d, J 6.5 Hz, H-6-rhamnose). 13C NMR (125 MHz, DMSO-d6, δ, ppm): 176.64 (C-4), 162.30 (C-7), 157.50 (C-2), 155.34 (C-9), 153.98 (C-5), 148.79 (C-3), 148.65 (C-4′), 144.85 (C-3′), 132.75 (C-1′), 121.44 (C-6′), 116.15 (C-5′), 115.15 (C-2′), 103.88 (C-10), 103.19 (C-6), 101.46 (C-1-glucose), 100.94 (C-1-rhamnose), 100.73 (C-8), 76.57 (C-3-glucose), 75.93 (C-5-glucose), 74.05 (C-2-glucose), 71.90 (C-2-rhamnose), 70.52 (C-4-rhamnose), 70.25 (C-4-glucose, C-3-rhamnose), 68.21 (C-5-rhamnose), 66.90 (C-6-glucose), 53.25 (N(CH2CH2)2), 52.70 (N(CH2CH2)2), 48.93 (C-8-CH2), 47.03 (C-6-CH2), 23.10 (N(CH2CH2)2), 23.04 (N(CH2CH2)2), 17.72 (C-6-rhamnose). HRMS (m/z): 777.3099 [M+H]+; 777.3082 calcd [M+H]+ for C37H49N2O16.
5,7-Dihydroxy-2-(3-hydroxy-4-methoxyphenyl)-6,8-bis(morpholinomethyl)-4H-chromen-4-one (4a)
Pale yellow powder, m.p. 278–280 °C, yield 81%. FTIR (KBr) ν/cm−1 3420, 2961, 1649, 1513, 1217, 1069. UV–Vis λmax (nm) (abs.) 250 (0.29), 262 (0.34), 294 (0.18), 343 (0.31). 1H NMR (500 MHz, DMSO-d6, δ, ppm): 7.57 (1H, dd, J 8.5, 2.5 Hz, H-6′), 7.47 (1H, d, J 2.5 Hz, H-2′), 7.12 (1H, d, J 8.5 Hz, H-5′), 6.76 (1H, s, H-3), 3.87 (3H, s, OCH3), 3.79 (2H, s, C-6-CH2), 3.75 (2H, s, C-8-CH2), 3.62–3.57 (8H, m, N-(CH2CH2)2O), 2.54–2.50 (8H, m, N-(CH2CH2)2O). 13C NMR (125 MHz, DMSO-d6, δ, ppm): 181.97 (C-4), 164.45 (C-7), 163.21 (C-2), 158.12 (C-5), 154.46 (C-9), 151.17 (C-4′), 146.84 (C-3′), 123.22 (C-1′), 118.64 (C-6′), 112.99 (C-2′), 112.22 (C-5′),103.68 (C-6), 103.19 (C-10), 102.80 (C-3), 100.99 (C-8), 66.14 (N-(CH2CH2)2O), 65.94 (N-(CH2CH2)2O), 55.77 (OCH3), 52.81 (N-(CH2CH2)2O), 52.33 (N-(CH2CH2)2O), 51.47 (C-8-CH2), 50.33 (C-8-CH2). HRMS (m/z): 499.2077 [M+H]+; 499.2080 calcd [M+H]+ for C26H31N2O8.
5,7-Dihydroxy-2-(3-hydroxy-4-methoxyphenyl)-6,8-bis(pyrrolidin-1-ylmethyl)chroman-4-one (5a)
Pale yellow powder, m.p. 178–180 °C, yield 64%. FTIR (KBr) ν/cm−1 3418, 2958, 1643, 1537, 1257, 1060. UV–Vis λmax (nm) (abs.) 239 (0.20), 248 (0.21), 259 (0.19), 283 (0.27). 1H NMR (500 MHz, DMSO-d6, δ, ppm): 6.97 (1H, d, J 2.0 Hz, H-2′), 6.89 (1H, dd, J 8.0, 2.0 Hz, H-6′), 6.94 (1H, d, J 8.0 Hz, H-5′), 5.38 (1H, dd, J 12.0, 3.0 Hz, H-2), 4.01 (2H, s, C-8-CH2), 3.98 (2H, s, C-6-CH2), 3.77 (3H, s, OCH3), 3.17–3.12 (8H, m, N-(CH2CH2)2), 3.01 (2H, dd, J 17.0, 12.0 Hz, H-3a), 2.65 (1H, dd, J 17.0, 3.0 Hz, H-3b), 1.90–1.87 (8H, m, N-(CH2CH2)2). 13C NMR (125 MHz, DMSO-d6, δ, ppm): 191.65 (C-4), 177.57 (C-7), 163.11 (C-5), 160.91 (C-9), 147.68 (C-4′), 146.54 (C-3′), 131.85 (C-1′), 117.20 (C-6′), 113.88 (C-2′), 112.03 (C-5′), 98.88 (C-6), 98.64 (C-8), 96.03 (C-10), 77.64 (C-2), 55.67 (OCH3), 52.50 (N-(CH2CH2)2), 52.31 (N-(CH2CH2)2), 47.83 (C-8-CH2), 47.69 (C-6-CH2), 41.37 (C-3), 22.68 (N-(CH2CH2)2). HRMS (m/z): 469.2345 [M+H]+; 469.2339 calcd [M+H]+ for C26H33N2O6.
5,7-Dihydroxy-2-(3-hydroxy-4-methoxyphenyl)-6,8-bis(morpholinomethyl)chroman-4-one (5b)
Pale yellow powder, m.p. 175–180 °C, yield 62%. FTIR (KBr) ν/cm−1 3450, 2948, 1622, 1535, 1259, 1027. UV–Vis λmax (nm) (abs.) 244 (0.21), 279 (0.86). 1H NMR (500 MHz, DMSO-d6, δ, ppm): 13.26 (1H, br.s, OH), 9.15 (1H, br.s, OH), 6.96 (1H, m, H-2′), 6.94 (1H, m, H-5′), 6.91 (1H, dd, J 8.0, 2.0 Hz, H-6′), 5.46 (1H, dd, J 12.0, 3.0 Hz, H-2), 4.04 (2H, s, C-8-CH2), 4.00 (2H, s, C-6-CH2), 3.78 (3H, s, OCH3), 3.78 (8H, m, N-(CH2CH2)2O), 3.14 (8H, dd, J 17.0, 12.0 Hz, H-3a), 3.07 (8H, m, N-(CH2CH2)2O), 2.75 (1H, dd, J 17.0, 3.5 Hz, H-3b). 13C NMR (125 MHz, DMSO-d6, δ, ppm): 193.91 (C-4), 162.76 (C-5), 161.08 (C-9), 147.79 (C-4′), 146.53 (C-3′), 131.28 (C-1′), 117.37 (C-6′), 113.95 (C-2′), 112.04 (C-5′), 98.21 (C-6), 97.47 (C-8), 89.16 (C-10), 78.05 (C-2), 63.81–63.13 (N-(CH2CH2)2O), 55.64 (OCH3), 50.96–50.85 (N-(CH2CH2)2O), 50.62 (C-8-CH2), 50.16 (C-6-CH2), 41.37 (C-3). HRMS (m/z): 501.2234 [M+H]+; 501.2237 calcd [M+H]+ for C26H33N2O8.
Cell Culture
Human breast adenocarcinoma (MDA-MB-231) cell line (ATCC® HTB-26™) was provided by School of Medicine, Sungkyunkwan University, Korea. MDA-MB-231 cells were cultured in DMEM (Gibco, USA), containing 10% FCS (Gibco, USA), 2 mM l-glutamine, 100 IU/mL penicillin, and 100 µg/mL streptomycin (Gibco, USA) in a humidified atmosphere of 5% CO2 at 37 °C to attain confluency.
Anti-proliferative activity assay
The samples were dissolved in DMSO at 10 mM and then diluted with culture medium to get tested concentrations. Paclitaxel (Anzatax®, Mayne Pharma, New Zealand) was used as the reference compound. DMSO at different concentrations was used as a blank control. The cells were then trypsinized, harvested, and counted using trypan blue (Sigma-Aldrich, USA), and seeded in 96-well plates at 104 cells/well. After 24 h incubation at 37 °C, 5% CO2, cells were treated with culture medium containing tested compounds at 100 µM for 72 h. After 72 h treatment, cell viability was evaluated as mitochondrial succinate dehydrogenase (SDH) activity, a marker of viable cells using MTT test as described by Denizot and Lang (1986).
Briefly, SDH activity was detected after 3 h incubation in culture medium without serum containing 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), which was converted into formazan crystals dissolved in acidified isopropanol. The produced purple solution was spectrophotometrically measured at 570 nm on Multikan™ microplate reader. Each tested concentration was performed in triplicate. The percentage of proliferation inhibition was calculated as follows:
The compounds with significant potential against MDA-MB-231 cells (percent of proliferation inhibition at 100 μM more than 50%) were then tested for their toxicity at five different concentrations (2–100 μM). Concentrations inducing a 50% inhibition of cell growth (IC50) were deduced by exponential regression of the inhibitory percentage − tested concentration. Values were mean ± SD from three independent experiments.
References
Babu TH, Rao VRS, Tiwari AK, Babu KS, Srinivas PV, Ali AZ, Rao JM (2008) Synthesis and biological evaluation of novel 8-aminomethylated oroxylin A analogues as α-glucosidase inhibitors. Bioorg Med Chem Lett 18:1659–6662. https://doi.org/10.1016/j.bmcl.2008.01.055
Buravlev EV, Shevchenko OG, Chukicheva IY, Kutchin AV (2017) Synthesis and membrane-protective properties of aminomethyl derivatives of quercetin at the C-8 position. Chem Pap. https://doi.org/10.1007/s11696-017-0272-y
Candiracci M, Piatti E, Dominguez-Barragan M, Garcia-Antras D, Morgado B, Ruano D, Gutierrez JF, Parrado J, Castano A (2012) Anti-inflammatory activity of a honey flavonoid extract on lipopolysaccharide-activated N13 microglial cells. J Agric Food Chem 60:12304–12311. https://doi.org/10.1021/jf302468h
Chahar MK, Sharma N, Dobhal MP, Joshi YC (2011) Flavonoids: a versatile source of anticancer drugs. Pharmacogn Rev 5:1–12. https://doi.org/10.4103/0973-7847.79093
Chen Y, Cass SL, Kutty SK, Yee EMH, Chan DSH, Gardner CR, Vittorio O, Pasquier E, Black DS, Kumar N (2015) Synthesis, biological evaluation and structure–activity relationship studies of isoflavene based Mannich bases with potent anti-cancer activity. Bioorg Med Chem Lett 25:5377–5383. https://doi.org/10.1016/j.bmcl.2015.09.027
Cottiglia F, Casu L, Bonsignore L, Casu M, Floris C, Sosa S, Altinier G, Loggia RD (2005) Topical anti-inflammatory activity of flavonoids and a new xanthone from Santolina insularis. Z Naturforsch 60c:63–66. https://doi.org/10.1515/znc-2005-1-212
Dang CH, Nguyen CH, Nguyen TD, Im C (2014) Synthesis and characterization of N-acyl-tetra-O-acyl glucosamine derivatives. RSC Adv 4:6239–6245. https://doi.org/10.1039/C3RA46007J
Dang CH, Le VD, Nguyen CH, Nguyen TD (2017) A facile synthesis of aggregation pheromones of Rhinoceros beetle and Rhychophorus weevil. ARKIVOC 2017; 2017(v):187–195. https://doi.org/10.24820/ark.5550190.p010.271
Denizot F, Lang R (1986) Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 89:271–277. https://doi.org/10.1016/0022-1759(86)90368-6
Frasinyuk MS, Mrug GP, Bondarenko SP, Sviripa VM, Zhang W, Cai X, Fiandalo MV, Mohler JL, Liu C, Watt DS (2015) Application of Mannich bases to the synthesis of hydroxymethylated isoflavonoids as potential antineoplastic agents. Org Biomol Chem 13:11292–11301. https://doi.org/10.1039/c5ob01828e
Gorbunov EB, Rusinov GL, Ulomskii EN, Eltsov OS, Rusinov VL, Kartsev VG, Charushin VN, Khalymbadzha IA, Chupakhin ON (2016) Direct modification of quercetin by 6-nitroazolo[1,5-a]pyrimidines. Chem Nat Compd 52:708–710. https://doi.org/10.1007/s10600-016-1749-6
Ha L, Qian Y, Zhang S, Ju X, Sun S, Guo H, Wang Q, Li K, Fan Q, Zheng Y, Li H (2016) Synthesis and biological evaluation of scutellaria flavone cyclaneaminol Mannich base derivatives as novel CDK1 inhibitors. Anticancer Agents Med Chem 16:914–924. https://doi.org/10.2174/1871520615666150928114425
Helgen TR, Sciotti RJ, Lee P, Duffy S, Avery VM, Igbinoba O, Akoto M, Hagen TJ (2015) The synthesis, antimalarial activity and CoMFA analysis of novel aminoalkylated quercetin analogs. Bioorg Med Chem Lett 25:327–332. https://doi.org/10.1016/j.bmcl.2014.11.039
Hoang TKD, Huynh TKC, Nguyen TD (2015) Synthesis, characterization, anti-inflammatory and anti-proliferative activity against MCF-7 cells of O-alkyl and O-acyl flavonoid derivatives. Bioorg Chem 63:45–52. https://doi.org/10.1016/j.bioorg.2015.09.005
Kanadaswami C, Lee LT, Lee PP, Hwang JJ, Ke FC, Huang YT, Lee MT (2005) The antitumor activities of flavonoids. In Vivo 19:895–910
Kaul TN, Middleton JMDE, Ogra PL (1985) Antiviral effect of flavonoids on human viruses. J Med Virol 15:71–79. https://doi.org/10.1002/jmv.1890150110
Kukhareva TS, Krasnova VA, Koroteev MP, Kaziev GZ, Kuleshova LN, Korlyukov AA, Antipin MY, Nifant’ev EE (2004) Electrophilic substitution in the dihydroquercetin system. Aminomethylation. Russ J Org Chem 40(8):1190–1193. https://doi.org/10.1023/B:RUJO.0000045904.63368.06
Kumar S, Pandey AK (2013) Chemistry and biological activities of flavonoids: an overview. Sci World J. https://doi.org/10.1155/2013/162750 article ID 162750
Lee WJ, Chen WK, Wang CJ, Lin WL, Tseng TH (2008) Apigenin inhibits HGF promoted invasive growth and metastasis involving blocking PI3K/Akt pathway and beta 4 integrin function in MDA-MB-231 breast cancer cells. Toxicol Appl Pharmacol 226:178–191. https://doi.org/10.1016/j.taap.2007.09.013
Lis R, Marisca AJ (1987) Methanesulfonanilides and the Mannich reaction. J Org Chem 52:4377–4379. https://doi.org/10.1021/jo00228a041
Ng TB, Huang B, Fong WP, Yeung HW (1997) Anti-human immunodeficiency virus (anti-HIV) natural products with special emphasis on HIV reverse transcriptase inhibitors. Life Sci 61:933–949. https://doi.org/10.1016/S0024-3205(97)00245-2
Nguyen TB, Wang Q, Gueritte F (2011) An efficient one-step synthesis of piperidin-2-yl and pyrrolidin-2-yl flavonoid alkaloids through phenolic Mannich reactions. Eur J Org Chem 2011:7076–7079. https://doi.org/10.1002/ejoc.201101312
Nguyen TD, Nguyen CH, Im C, Dang CH (2015a) Synthesis of corn rootworm pheromones from commercial diols. Chem Pap 69(2):380–384. https://doi.org/10.1515/chempap-2015-0027
Nguyen VS, Shi L, Luan FQ, Wang QA (2015b) Synthesis of kaempferide Mannich base derivatives and their antiproliferative activity on three human cancer cell lines. Acta Biochim Pol 6:547–552. https://doi.org/10.18388/abp.2015_992
Nguyen TD, Nguyen CH, Im C, Dang CH (2016) A facile synthesis of sex pheromone of the Cabbage Looper, Trichoplusia ni. Chem Nat Compd 52(5):877–879. https://doi.org/10.1007/s10600-016-1800-7
Nguyen VS, Shi L, Wang SC, Wang QA (2017) Synthesis of icaritin and β-anhydroicaritin Mannich base derivatives and their cytotoxic activities on three human cancer cell lines. Anticancer Agents Med Chem 17:137–142. https://doi.org/10.2174/1871520616666160404111210
Nifant’ev EE, Mosyurov SE, Kukhareva TS, Vasyanina LK (2013) N, N’-diethyl-1,3-propanediamine in the dihydroquercetin aminomethylation reaction. Dokl Chem 451(2):197–200. https://doi.org/10.1134/S0012500813080016
Orhana DD, Ozcelik B, Ozgen S, Ergun F (2010) Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiol Res 165:496–504. https://doi.org/10.1016/j.micres.2009.09.002
Pan MH, Lai CS, Ho CT (2010) Anti-inflammatory activity of natural dietary flavonoids. Food Funct 1:15–31. https://doi.org/10.1039/C0FO00103A
Phromnoi K, Yodkeeree S, Anuchapreeda S, Limtrakul P (2009) Inhibition of MMP-3 activity and invasion of the MDAMB-231 human invasive breast carcinoma cell line by bioflavonoids. Acta Pharmacol Sin 30:1169–1176. https://doi.org/10.1038/aps.2009.107
Pietta PG (2000) Flavonoids as antioxidants. J Nat Prod 63:1035–1042. https://doi.org/10.1021/np9904509
Ravishankar D, Rajora AK, Greco F, Osborn HMI (2013) Flavonoids as prospective compounds for anti-cancer therapy. Int J Biochem Cell Biol 45:2821–2831. https://doi.org/10.1016/j.biocel.2013.10.004
Rice-Evans CA (2001) Flavonoid antioxidants. Curr Med Chem 8:797–807. https://doi.org/10.2174/0929867013373011
Rice-Evans CA, Miller NJ, Paganga G (1996) Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radic Biol Med 20:933–956. https://doi.org/10.1016/0891-5849(95)02227-9
Tempesti TC, Alvarez MG, Araujo MFD, Junior FEAC, de Carvalho MG, Durantini EN (2012) Antifungal activity of a novel quercetin derivative bearing a trifluoromethyl group on Candida albicans. Med Chem Res 21:2217–2222. https://doi.org/10.1007/s00044-011-9750-x
Tugrak M, Yamali C, Sakagami H, Gul HI (2015) Synthesis of mono Mannich bases of 2-(4-hydroxybenzylidene)-2,3-dihydroinden-1-one and evaluation of their cytotoxicities. J Enzyme Inhib Med Chem 31:818–823. https://doi.org/10.3109/14756366.2015.1070263
Vijayababu MR, Arunkumar A, Kanagaraj P, Venkataraman P, Krishnamoorthy G, Arunakaran J (2006) Quercetin downregulates matrix metalloproteinases 2 and 9 proteins expression in prostate cancer cells (PC-3). Mol Cell Biochem 287:109–116. https://doi.org/10.1007/s11010-005-9085-3
Yusakul G, Sakamoto S, Juengwatanatrakul T, Putalun W, Tanaka H, Morimoto S (2016) Preparation and application of a monoclonal antibody against the isoflavone glycoside daidzin using a Mannich reaction-derived hapten conjugate. Phytochem Anal 27:81–88. https://doi.org/10.1002/pca.2604
Zhang S, Ma J, Bao Y, Yang P, Zou L, Li K, Sun X (2008) Nitrogen-containing flavonoid analogues as CDK1/cyclin B inhibitors: synthesis, SAR analysis, and biological activity. Bioorg Med Chem 16:7127–7132. https://doi.org/10.1016/j.bmc.2008.06.055
Zhurakulov SN, Babkin VA, Chernyak EI, Morozov SV, Grigor’ev IA, Levkovich MG, Vinogradova VI (2015) Aminomethylation of 1-aryl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolines by dihydroquercetin. Chem Nat Compd 51(1):57–61. https://doi.org/10.1007/s10600-051-1203-1
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hoang, T.KD., Huynh, T.KC., Do, T.HT. et al. Mannich aminomethylation of flavonoids and anti-proliferative activity against breast cancer cell. Chem. Pap. 72, 1399–1406 (2018). https://doi.org/10.1007/s11696-018-0402-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-018-0402-1